

Abstract
We present dynamical studies of the dissociation of polycyclic aromatic hydrocarbon (PAH) radical cations in their ground electronic states with significant internal energy. Molecular dynamics simulations are performed, the electronic structure being described on-the-fly at the self-consistent-charge density functional-based tight binding (SCC–DFTB) level of theory. The SCC–DFTB approach is first benchmarked against DFT results. Extensive simulations are achieved for naphthalene 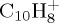 , pyrene
, pyrene 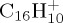 and coronene
and coronene 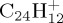 at several energies. Such studies enable one to derive significant trends on branching ratios, kinetics, structures and hints on the formation mechanism of the ejected neutral fragments. In particular, dependence of branching ratios on PAH size and energy were retrieved. The losses of H and C2H2 (recognized as the ethyne molecule) were identified as major dissociation channels. The H/C2H2 ratio was found to increase with PAH size and to decrease with energy. For
at several energies. Such studies enable one to derive significant trends on branching ratios, kinetics, structures and hints on the formation mechanism of the ejected neutral fragments. In particular, dependence of branching ratios on PAH size and energy were retrieved. The losses of H and C2H2 (recognized as the ethyne molecule) were identified as major dissociation channels. The H/C2H2 ratio was found to increase with PAH size and to decrease with energy. For 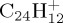 , which is the most interesting PAH from the astrophysical point of view, the loss of H was found as the quasi-only channel for an internal energy of 30 eV. Overall, in line with experimental trends, decreasing the internal energy or increasing the PAH size will favour the hydrogen loss channels with respect to carbonaceous fragments.
, which is the most interesting PAH from the astrophysical point of view, the loss of H was found as the quasi-only channel for an internal energy of 30 eV. Overall, in line with experimental trends, decreasing the internal energy or increasing the PAH size will favour the hydrogen loss channels with respect to carbonaceous fragments.
This article is part of the themed issue ‘Theoretical and computational studies of non-equilibrium and non-statistical dynamics in the gas phase, in the condensed phase and at interfaces’.
Keywords: molecular dynamics, DFTB, PAH, astrochemistry, dissociation
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) have received considerable interest since they were proposed, in the mid-1980, to be the carriers of the aromatic infrared bands, a set of infrared (IR) emission features observed in the 3−15 μm range in many regions of the interstellar medium (ISM) [1,2]. PAHs would also be candidates to carry the diffuse interstellar bands (DIBs), which are weak absorption bands measured between 0.38 and 1.3 μm on the galactic extinction curve [3]. Besides, PAHs are likely to play a role in the chemistry of the ISM, in particular a catalytic role in the formation of small molecules such as the most abundant one, H2 [4]. Despite many experimental and theoretical studies [5], yet no specific molecule has been identified. The study of the processing of PAHs (formation, destruction and reactivity) in the ISM has also aroused considerable interest. It is a current extensive research area that requires the synergy of experimental, astrophysical and chemical modelling so as to improve its understanding. This study enters this framework.
In the ISM, and in particular in photodissociation regions, PAHs are irradiated by the UV light emitted by stars. Following the absorption of a photon, four main channels to dissipate the energy enter in competition: ionization, IR radiative relaxation from vibrational modes, electronic transitions, ejection of small subfragments such as H, H2 and C2H2 [6].
Unimolecular dissociation of PAHs has received considerable interest since the mid-1990s with the pioneering work by Jochims et al. [7] through experimental and modelling studies, in order to derive photodissociation rates to be included in astrochemical models [6,8,9]. Since then, many experimental studies have been dedicated to the photodissociation of PAHs [10–16]. In the ISM, PAHs are also associated with a very harsh environment where they are likely to be destroyed through stellar winds, shock waves and hot ionized gas. The processing of PAHs in such environments, in particular their collisions with energetic ions and electrons, has been modelled [17–19], but the details of the processes are not fully understood. Erosion by hot gas would lead, in particular, to the progressive loss of C2Hn fragments (n=0,1,2) [17]. PAH dissociation induced by collision with energetic ions has been the object of several experimental studies [20–26] in which the dependence of the fragmentation patterns of the PAH on its size and on the energy and nature of the colliding particle has been investigated.
Regarding theoretical studies on the dissociation of PAHs, a few static density functional theory (DFT) studies are available [27–29], with special focus on the H and H2 losses [28,29]. To the best of our knowledge, dynamics studies regarding PAH dissociation are scarce. The dissociation of anthracene resulting from collision with H and He has recently been modelled with molecular dynamics (MD), using a force field to describe the PAH–particle interactions. This study showed that slow ion processing primarily induced a loss of C2H2 (and other CmHn fragments) rather than induced a C-atom knockout [30]. Regarding dissociation kinetics, they can be estimated with the Rice–Ramsperger–Kassel–Marcus (RRKM) theory, using the observables obtained from static calculations [11–13]. A RRKM mapping has been done for cationic naphthalene [27]. Let us mention, however, that RRKM supposes that the paths are known, whereas in the case of large PAHs, one may need to consider many possible paths. Another limitation relies on apparent non-RRKM behaviour, for instance in the case of dissociation faster than the energy redistribution. Let us mention finally that RRKM is very often used neglecting anharmonic effects of the potential energy surface (PES). To the best of our knowledge, all theoretical studies on PAH dissociation available in the literature have been achieved in their ground electronic state.
In this paper, we present dynamical studies of PAH radical cations in their ground electronic state with significant internal energy spread statistically over vibrational modes leading to isomerization and dissociation. We chose to study first the dissociation of the smallest PAH, namely naphthalene radical cation 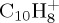 , to benchmark the approach in particular. Then the dissociation dynamics of pyrene
, to benchmark the approach in particular. Then the dissociation dynamics of pyrene 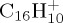 and coronene
and coronene 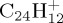 are studied, the latter being a prototype for compact PAHs of astrophysical interest. Before presenting and discussing the results of MD simulations (§4), we introduce the computational approach (§2), and some static benchmark calculations of a small portion of the PES explored during MD simulations (§3).
are studied, the latter being a prototype for compact PAHs of astrophysical interest. Before presenting and discussing the results of MD simulations (§4), we introduce the computational approach (§2), and some static benchmark calculations of a small portion of the PES explored during MD simulations (§3).
2. Computational approach
All simulations are performed in the microcanonical ensemble within the Born–Oppenheimer MD scheme, computing the potential energy on-the-fly. The nuclei obey the classical equations of motion, integrated with the velocity Verlet algorithm using a timestep of 0.1 fs so as to ensure a constant total energy (deviation less than 0.5 % of the initial kinetic energy), except in the particular case of constrained density functional tight-binding (DFTB). Initially, the kinetic (vibrational) energy is spread randomly over all the atoms of the molecular system, with fast statistical redistribution over all vibrational modes.
The scheme retained to solve the electronic problem should allow the calculation of millions of single point energies, which is mandatory to obtain statistically relevant results, and be able to describe bond breaking. In that context, the DFTB method [31–36] is particularly adapted as an approximation of DFT with a much lower computational cost. It has already proven its efficiency to describe chemical processes, such as reactivity [37]. In this work, we have used the second-order version of DFTB, also known as self-consistent charge (SCC)–DFTB [33], using the mio-set of parameters [33] and the dispersion correction as reported in reference [38]. We used a Fermi distribution (Fermi temperature between 500 and 1500 K) to determine the molecular orbital occupations. It avoids oscillation problems during the search for a self-consistent solution, often arising when computing DFTB energy for dissociated or close to dissociation systems, and allows to recover continuity in energy and gradients in the case of level crossing. We note that a Fermi temperature of 1500 K was used by Kukk et al. [39] to simulate the dissociation of the thiophene molecule SC4H4.
Modelling dissociation of charged systems with DFT, and therefore with DFTB can lead to some well-known artefacts, related to the self-interaction error [40] as for instance (i) at dissociation limit, the variational search for the electronic ground state can attribute partial charges to the fragments [41]; (ii) during the dissociation process, if the charge is spread over the two fragments, a spurious coulomb repulsion interaction appears between the two fragments. These artefacts are more important in the case of symmetric dissociations [42], but one can wonder to which extent it may affect the results of our simulations. At the dissociation limit, the problem can be cured by using the constrained DFT [43] (or its extension to DFTB [44]), in which the variational problem is solved with an additional constraint enforcing the charge to be localized on a given fragment. In practice, the DFT (or DFTB) Kohn–Sham effective hamiltonian is modified as
where P is a projector of the density on a given subfragment and λ is a Lagrange multiplier. Technically, each step of the self-consistent process includes a search of the λ parameter, which gives the correct number of electrons localized on the given subfragment. To illustrate this point, figure 1 represents the dissociation curve corresponding to the removal of an hydrogen atom from the cation 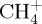 , without relaxing the fragments. The zero energy reference is taken as the sum of two independent energy calculations for the isolated fragments
, without relaxing the fragments. The zero energy reference is taken as the sum of two independent energy calculations for the isolated fragments 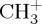 and H and corresponds to the correct dissociation energy. The DFTB dissociation curve clearly shows that (i) the lowest energy obtained at dissociation with partial charges is much lower than the correct one and that (ii) the spurious 1/R repulsion is significant and responsible for the presence of a barrier. On the opposite, enforcing the charge to be localized on the CH3 fragment with constrained DFTB leads to a correct dissociation behaviour but overestimates the energy of the molecule in its equilibrium geometry.
and H and corresponds to the correct dissociation energy. The DFTB dissociation curve clearly shows that (i) the lowest energy obtained at dissociation with partial charges is much lower than the correct one and that (ii) the spurious 1/R repulsion is significant and responsible for the presence of a barrier. On the opposite, enforcing the charge to be localized on the CH3 fragment with constrained DFTB leads to a correct dissociation behaviour but overestimates the energy of the molecule in its equilibrium geometry.
Figure 1.

Computed energy of [CH4]+ as a function of the H–CH3 distance (d), without relaxing the fragments, with DFTB (dashed lines) and with constrained DFTB (solid lines).
Therefore, for 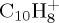 , in addition to the standard MD dissociation simulations, we performed dynamics using the following scheme: (i) we run the simulation of the molecule with standard DFTB potential (i.e. no constraint) until we detect a dissociation is about to occur. (ii) We identify the heaviest fragment. It will be naively assumed to carry the charge. For the following MD steps, the potential energy is computed from constrained DFTB, with the constraint localizing the charge on this fragment. In the case of another evaporation of this charged fragment, the process of charge localization on the heaviest fragment is repeated. We note that the idea of performing a single projection on an adiabatic surface presents some similarities to what is done in the treatment of dissociation in excited states [45]. Let us precise that we define a dissociation is occurring when the smallest distance between the atoms of two fragments is larger than a given critical distance (typically 2.5 Å).
, in addition to the standard MD dissociation simulations, we performed dynamics using the following scheme: (i) we run the simulation of the molecule with standard DFTB potential (i.e. no constraint) until we detect a dissociation is about to occur. (ii) We identify the heaviest fragment. It will be naively assumed to carry the charge. For the following MD steps, the potential energy is computed from constrained DFTB, with the constraint localizing the charge on this fragment. In the case of another evaporation of this charged fragment, the process of charge localization on the heaviest fragment is repeated. We note that the idea of performing a single projection on an adiabatic surface presents some similarities to what is done in the treatment of dissociation in excited states [45]. Let us precise that we define a dissociation is occurring when the smallest distance between the atoms of two fragments is larger than a given critical distance (typically 2.5 Å).
For a given PAH+ and for a given energy, we performed from several hundreds to 1000 trajectories. From these simulations, branching ratios and kinetic data are obtained. Two energy domains are considered: either at high energy for which all simulations present a dissociation within 100–200 ps, or minimum energy for which dissociation can be observed in reasonable timescale (5 ns maximum for 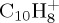 , 500 ps maximum for
, 500 ps maximum for 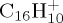 and
and 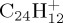 ). The number of simulations to reach convergence depends on the system and on the energy. We noted that both kinetic behaviour and qualitative trends on the branching ratios are already present with only 100–200 simulations. With 1000 simulations, convergence on kinetics and branching ratios is expected to be reached (this was checked, for instance, for
). The number of simulations to reach convergence depends on the system and on the energy. We noted that both kinetic behaviour and qualitative trends on the branching ratios are already present with only 100–200 simulations. With 1000 simulations, convergence on kinetics and branching ratios is expected to be reached (this was checked, for instance, for 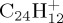 with 30 eV of internal energy). This approach also allows us to have a glance at the composition and structure of the lost neutral fragments, and on the mechanisms leading to the formation of atoms and molecules of astrophysical interest. All the MD/DFTB calculations are performed with an experimental version of the deMonNano code [46].
with 30 eV of internal energy). This approach also allows us to have a glance at the composition and structure of the lost neutral fragments, and on the mechanisms leading to the formation of atoms and molecules of astrophysical interest. All the MD/DFTB calculations are performed with an experimental version of the deMonNano code [46].
3. Benchmark of the density functional-based tight binding potential energy surface
In our simulations, structural rearrangement occurs prior to dissociation. We identified, in particular, isomerizations into H-shifted isomers, five-member ring isomers bearing a vinylidene group and opened-ring isomers with ethynyl groups. A few snapshots taken during the dynamics (prior to dissociation) are reported in figure 2.
Figure 2.

Snapshots taken during BOMD simulations for 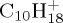 (a,b),
(a,b), 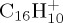 (c),
(c), 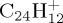 (d–f). The internal energy of the simulation is specified below each snapshot. (a) 18, (b) 18, (c) 22 eV, (d) 30 (e) 32 and (f) 32 eV. (Online version in colour.)
(d–f). The internal energy of the simulation is specified below each snapshot. (a) 18, (b) 18, (c) 22 eV, (d) 30 (e) 32 and (f) 32 eV. (Online version in colour.)
Local DFTB gradient optimizations on some particular low-lying isomers were achieved and their relative energies with respect to the most stable isomer were compared with DFT results as the benchmark. DFT calculations were performed using the B3LYP functional and the 6–311G(d,p) basis set with the Gaussian09 suite of programs [47]. The results are reported in table 1 and the corresponding structures in figures 3–5 for 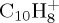 ,
, 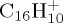 and
and 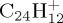 , respectively. Regarding the isomers in table 1 ((a–c) for
, respectively. Regarding the isomers in table 1 ((a–c) for 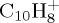 , (a–d) for
, (a–d) for 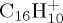 and
and 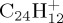 ), DFTB and DFT relative energies are in good agreement, in particular in the case of
), DFTB and DFT relative energies are in good agreement, in particular in the case of 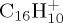 . The maximum discrepancy is of 0.19 eV for azulene versus naphthalene.
. The maximum discrepancy is of 0.19 eV for azulene versus naphthalene.
Figure 4.

DFTB optimized structures of several isomers and dissociation products of 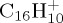 . (Online version in colour.)
. (Online version in colour.)
Table 1.
DFTB versus DFT relative energies (eV) of several isomers and dissociation fragments of 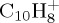 ,
, 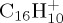 and
and 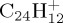 . Only the lowest spin multiplicities were considered.
. Only the lowest spin multiplicities were considered.
| species | DFT | DFTB | species | DFT | DFTB | ||
|---|---|---|---|---|---|---|---|
C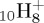
|
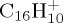 |
||||||
| naphthalene | (a) | 0.0 | 0.0 | pyrene | (a) | 0.0 | 0.0 |
| azulene | (b) | 0.81 | 1.0 | vinylidene | (b) | 1.45 | 1.45 |
| benzofulvene | (c) | 0.97 | 1.02 | H-shifted | (c) | 2.62 | 2.64 |
| ethynyl | (d) | 2.83 | 2.82 | ||||
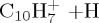 |
(d) | 5.20 | 5.48 | ||||
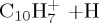 |
(e) | 5.28 | 5.58 | 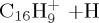 |
(e) | 5.49 | 5.95 |
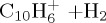 |
(f) | 4.61 | 5.81 | 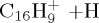 |
(f) | 5.68 | 6.00 |
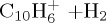 |
(g) | 4.66 | 5.89 | 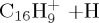 |
(g) | 5.74 | 6.02 |
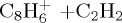 |
(h) | 4.84 | 5.64 | 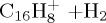 |
(h) | 4.48 | 5.65 |
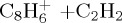 |
(i) | 4.38 | 4.76 | 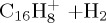 |
(i) | 4.54 | 5.74 |
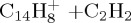 |
(j) | 7.08 | 7.65 | ||||
C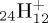
| |||||||
| coronene | (a) | 0.0 | 0.0 | ||||
| vinylidene | (b) | 1.30 | 1.46 | ||||
| H-shifted | (c) | 2.35 | 2.51 | ||||
| ethynyl | (d) | 2.48 | 2.52 | ||||
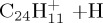 |
(e) | 5.59 | 5.99 | ||||
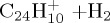 |
(f) | 4.40 | 5.58 | ||||
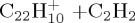 |
(g) | 7.43 | 7.96 | ||||
Figure 3.

DFTB optimized structures of several isomers and dissociation products of 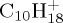 . (Online version in colour.)
. (Online version in colour.)
Figure 5.

DFTB optimized structures of several isomers and dissociation products of 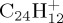 . (Online version in colour.)
. (Online version in colour.)
Regarding dissociation pathways in table 1 ((d)–(i) for 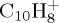 , (e)–(j) for
, (e)–(j) for 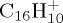 and (e)–(g) for
and (e)–(g) for 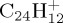 ), they are overall overestimated with respect to DFT. The most significant discrepancy concerns the loss of H2 (maximum 26% of the dissociation energy). Regarding the loss of H, we note a maximum discrepancy of 0.46 eV ((e) for
), they are overall overestimated with respect to DFT. The most significant discrepancy concerns the loss of H2 (maximum 26% of the dissociation energy). Regarding the loss of H, we note a maximum discrepancy of 0.46 eV ((e) for 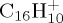 ), whereas for the loss of ethyne, the maximum difference is 0.8 eV ((h) for
), whereas for the loss of ethyne, the maximum difference is 0.8 eV ((h) for 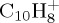 ). Regarding the relative energies of the different paths, the energy for the loss of H2 with respect to the loss of H is overestimated with DFTB versus DFT, whereas the energy difference for the loss of C2H2 versus the loss of H is similar at the DFTB and DFT levels of theory. From these energetics considerations, we may expect in the MD simulations at the DFTB level that (i) the minimum energy for PAH+ dissociation will be overestimated and (ii) the H2 path branching ratio with respect to the others will be underestimated.
). Regarding the relative energies of the different paths, the energy for the loss of H2 with respect to the loss of H is overestimated with DFTB versus DFT, whereas the energy difference for the loss of C2H2 versus the loss of H is similar at the DFTB and DFT levels of theory. From these energetics considerations, we may expect in the MD simulations at the DFTB level that (i) the minimum energy for PAH+ dissociation will be overestimated and (ii) the H2 path branching ratio with respect to the others will be underestimated.
4. Dissociation: molecular dynamics results
We studied the statistical dissociation of 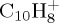 ,
, 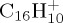 and
and 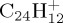 with high internal energy, either sufficient for the PAH+ parent ion abundance to be found below 1% within 100–200 ps, or with minimal energy to observe at least 10% of dissociation within reasonable time (approx. 500 ps). The influence of the energy on the fragmentation patterns and kinetics is discussed. We benchmarked the approach in the case of the smallest PAH,
with high internal energy, either sufficient for the PAH+ parent ion abundance to be found below 1% within 100–200 ps, or with minimal energy to observe at least 10% of dissociation within reasonable time (approx. 500 ps). The influence of the energy on the fragmentation patterns and kinetics is discussed. We benchmarked the approach in the case of the smallest PAH, 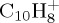 . Indeed, as specified in §2, our method essentially presents drawbacks related to self-interaction error, and we performed some benchmark with the more time-consuming charge constraint approach, which also has its own drawbacks but not those associated with self-interaction error.
. Indeed, as specified in §2, our method essentially presents drawbacks related to self-interaction error, and we performed some benchmark with the more time-consuming charge constraint approach, which also has its own drawbacks but not those associated with self-interaction error.
(a). C10H8+
The evolution of the abundance of the parent 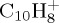 ion with 18 eV of internal energy, and of the fragment ions as a function of time are reported in figure 6. Comparing figure 6a,b (also c and d focusing on the most abundant fragments), we see that constraining the charge on the largest fragment during the dynamics does not modify the disappearance kinetics of the parent ion (liftetime τ of 30 ps in both cases, cf. table 2). Regarding the branching ratios, they are similar although not identical, the domination of the
ion with 18 eV of internal energy, and of the fragment ions as a function of time are reported in figure 6. Comparing figure 6a,b (also c and d focusing on the most abundant fragments), we see that constraining the charge on the largest fragment during the dynamics does not modify the disappearance kinetics of the parent ion (liftetime τ of 30 ps in both cases, cf. table 2). Regarding the branching ratios, they are similar although not identical, the domination of the 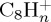 fragments being enhanced with the charge constraint (27% versus 23% after 100 ps). The dominant fragment is
fragments being enhanced with the charge constraint (27% versus 23% after 100 ps). The dominant fragment is 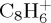 , resulting from the loss of C2H2. Interestingly, for astrophysical implication, this molecule has been identified as the ethyne molecule in more than 99% of trajectories. With a similar although overall slightly smaller ratio, appears the
, resulting from the loss of C2H2. Interestingly, for astrophysical implication, this molecule has been identified as the ethyne molecule in more than 99% of trajectories. With a similar although overall slightly smaller ratio, appears the 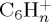 fragments resulting from the loss of C4Hn, individual fragments
fragments resulting from the loss of C4Hn, individual fragments 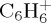 ,
, 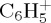 and
and 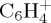 being of similar abundance. These remain minor with respect to
being of similar abundance. These remain minor with respect to 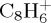 . At a lower ratio, one can find the
. At a lower ratio, one can find the 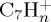 fragments, the major individual species being
fragments, the major individual species being 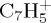 , resulting from the loss of C3H3. The ratio for the loss of H and H2 remains small (6% total at 100 ps), the latter remaining really minor. Finally, the
, resulting from the loss of C3H3. The ratio for the loss of H and H2 remains small (6% total at 100 ps), the latter remaining really minor. Finally, the 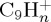 fragments ions (loss of CHn) are minor (ratio lower than 2%). Regarding the branching ratios, the predominance of the loss of C2Hn fragments, and in particular of the ethylene molecule, appears as characteristic of a statistical fragmentation process [48]. On the opposite, the loss of CHn would not be favoured, and is indeed a minor path in our trajectories.
fragments ions (loss of CHn) are minor (ratio lower than 2%). Regarding the branching ratios, the predominance of the loss of C2Hn fragments, and in particular of the ethylene molecule, appears as characteristic of a statistical fragmentation process [48]. On the opposite, the loss of CHn would not be favoured, and is indeed a minor path in our trajectories.
Figure 6.

Dissociation of 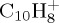 with 18 eV of internal energy. Time evolution of the parent and fragment
with 18 eV of internal energy. Time evolution of the parent and fragment 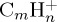 ions without (a,c) and with (b,d) charge constraint. Top: for a given number m of C atoms (we also indicate the corresponding observed numbers n of H atoms); bottom: for a selection of the most abundant fragments identified by the (m,n) pair. The lines joining the data points were drawn for convenience. (Online version in colour.)
ions without (a,c) and with (b,d) charge constraint. Top: for a given number m of C atoms (we also indicate the corresponding observed numbers n of H atoms); bottom: for a selection of the most abundant fragments identified by the (m,n) pair. The lines joining the data points were drawn for convenience. (Online version in colour.)
Table 2.
Lifetimes: τ values derived from the exponential fit 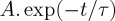 of the temporal decay of the PAH radical cation abundance.
of the temporal decay of the PAH radical cation abundance.
| species | E (eV) | τ (ns) | ntraj. | duration of 1 traj. (ps) |
|---|---|---|---|---|
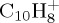 |
14 | 4.4 | 100 | 5000 |
| 18 | 0.03 | 1000 | 100 | |
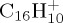 |
22 | 3.5 | 1000 | 500 |
| 30 | 0.01 | 1000 | 100 | |
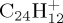 |
30 | 4.3 | 1000 | 500 |
| 40 | 0.04 | 1000 | 200 |
Based on this preliminary study of 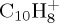 , we explored the statistical electronic ground state dissociation dynamics of larger PAHs, namely pyrene
, we explored the statistical electronic ground state dissociation dynamics of larger PAHs, namely pyrene 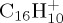 and coronene
and coronene 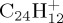 , without applying the charge constraint approach.
, without applying the charge constraint approach.
(b). C16H10+
The dissociation of the pyrene radical cation 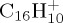 was studied with 30 and 22 eV of internal energy. The temporal evolution of the parent and fragment ions are reported in figure 7a–d for 30 and 22 eV of internal energy, respectively. The fragments were considered as cations if their partial charges were larger than 0.5, and this led us to consider down to
was studied with 30 and 22 eV of internal energy. The temporal evolution of the parent and fragment ions are reported in figure 7a–d for 30 and 22 eV of internal energy, respectively. The fragments were considered as cations if their partial charges were larger than 0.5, and this led us to consider down to 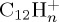 fragments.
fragments.
Figure 7.

Dissociation of 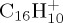 with 30 eV (a,b) and 22 eV (c,d) of internal energy. Time evolution of the parent and fragment
with 30 eV (a,b) and 22 eV (c,d) of internal energy. Time evolution of the parent and fragment 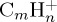 ions for a given number m of C atoms (a), for a selection of the most abundant fragments (b), for all present ions (c). (d) is a zoom of (c) on the fragment ions. In (b–d), fragments are identified by the (m,n) pair. The lines joining the data points were drawn for convenience. (Online version in colour.)
ions for a given number m of C atoms (a), for a selection of the most abundant fragments (b), for all present ions (c). (d) is a zoom of (c) on the fragment ions. In (b–d), fragments are identified by the (m,n) pair. The lines joining the data points were drawn for convenience. (Online version in colour.)
At 30 eV, the parent ion is totally dissociated after 100 ps (τ=10 ps), whereas only 13% are dissociated after 500 ps at 22 eV, leading to a τ value estimated at 3.5 ns. At 30 eV, the dominant fragment 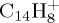 corresponds to the loss of C2H2 that was identified as the ethyne molecule. As can be seen in figure 7b, it reaches a maximum ratio of 26% at 25 ps, and continues fragmenting. The next most abundant fragment is
corresponds to the loss of C2H2 that was identified as the ethyne molecule. As can be seen in figure 7b, it reaches a maximum ratio of 26% at 25 ps, and continues fragmenting. The next most abundant fragment is 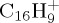 and corresponds to the loss of H (maximum ratio of 11% at 25 ps too). The loss of molecular H2 remains minor (steady ratio of 3% for t>50 ps). The loss of odd-numbered carbon chains is non-negligible when all dehydrogenation states are considered, but becomes individually small with respect to the losses of C2H2 and H with a maximum ratio of 6% for
and corresponds to the loss of H (maximum ratio of 11% at 25 ps too). The loss of molecular H2 remains minor (steady ratio of 3% for t>50 ps). The loss of odd-numbered carbon chains is non-negligible when all dehydrogenation states are considered, but becomes individually small with respect to the losses of C2H2 and H with a maximum ratio of 6% for 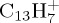 . Interestingly, at 22 eV, the loss of H becomes the major path with a maximum ratio of 5% at 500 ps, whereas the path corresponding to the loss of C2H2 evolves parallely as a function of time with a maximum ratio of 4% at 500 ps (figure 7d). The loss of H2 remains a minor path (ratio less than 1%), so as the loss of C3H and C4H2. The loss of H proceeds either through a direct mechanism or after H-migration at the surface of the PAH or on C atoms. From the observation of several trajectories, the loss of H2 may arise either from two H from a sp3 C atom after H-shift, which is known as the lowest energy path, of from the concerted release of two H atoms attached to two adjacent sp2 C atoms. Two types of mechanisms have been identified for the loss of C2H2: either direct or preceded by external ring openings and H-shifts. Some examples are shown in figure 8 for cationic pyrene and coronene, that undergo similar mechanisms (figure 8a–c: from open rings, figure 8d,e: direct).
. Interestingly, at 22 eV, the loss of H becomes the major path with a maximum ratio of 5% at 500 ps, whereas the path corresponding to the loss of C2H2 evolves parallely as a function of time with a maximum ratio of 4% at 500 ps (figure 7d). The loss of H2 remains a minor path (ratio less than 1%), so as the loss of C3H and C4H2. The loss of H proceeds either through a direct mechanism or after H-migration at the surface of the PAH or on C atoms. From the observation of several trajectories, the loss of H2 may arise either from two H from a sp3 C atom after H-shift, which is known as the lowest energy path, of from the concerted release of two H atoms attached to two adjacent sp2 C atoms. Two types of mechanisms have been identified for the loss of C2H2: either direct or preceded by external ring openings and H-shifts. Some examples are shown in figure 8 for cationic pyrene and coronene, that undergo similar mechanisms (figure 8a–c: from open rings, figure 8d,e: direct).
Figure 8.

Snapshots of BOMD simulations during dissociation (loss of C2H2) for 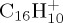 (a–c, 22 eV of internal energy, 500 fs between each snapshot),
(a–c, 22 eV of internal energy, 500 fs between each snapshot), 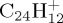 (d,e, 32 eV of internal energy, 250 fs between the two snapshots). (Online version in colour.)
(d,e, 32 eV of internal energy, 250 fs between the two snapshots). (Online version in colour.)
(c). C24H12+
The dissociation of the coronene radical cation 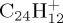 was studied with 40 and 30 eV of internal energy, with an intermediate study at 32 eV (for which only 400 trajectories have been performed). The temporal evolution of the parent and fragment ions is reported in figure 9a,b and figure 9c,d for 40 and 30 eV of internal energy, respectively. At 40 eV, there was no surviving parent ion after 200 ps (τ= 40 ps), whereas only 11% ions were dissociated after 500 ps at 30 eV, leading to τ value estimated at 4.3 ns. At 40 eV, at short times (t<50 ps), the dominant processes are the formation of
was studied with 40 and 30 eV of internal energy, with an intermediate study at 32 eV (for which only 400 trajectories have been performed). The temporal evolution of the parent and fragment ions is reported in figure 9a,b and figure 9c,d for 40 and 30 eV of internal energy, respectively. At 40 eV, there was no surviving parent ion after 200 ps (τ= 40 ps), whereas only 11% ions were dissociated after 500 ps at 30 eV, leading to τ value estimated at 4.3 ns. At 40 eV, at short times (t<50 ps), the dominant processes are the formation of 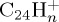 fragment ions, corresponding to losses of H and H2. After 50 ps, the formation of
fragment ions, corresponding to losses of H and H2. After 50 ps, the formation of 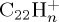 , with similar ratio as
, with similar ratio as 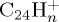 at 50 ps, keeps increasing although that of
at 50 ps, keeps increasing although that of 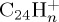 starts decreasing (figure 9a). Fragment ions with an odd number of carbons are minor, probably resulting from a higher energy process. Regarding the individual fragments of astrophysical interest (figure 9b), we note that the loss of one H atom is major at short times (20% at 50 ps), in line with a direct process, whereas the loss of H2 and sequential losses of H increase slowly, leading to a maximum ratio for
starts decreasing (figure 9a). Fragment ions with an odd number of carbons are minor, probably resulting from a higher energy process. Regarding the individual fragments of astrophysical interest (figure 9b), we note that the loss of one H atom is major at short times (20% at 50 ps), in line with a direct process, whereas the loss of H2 and sequential losses of H increase slowly, leading to a maximum ratio for 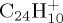 of 10% at 200 ps. Interestingly, loss of H2 and sequential loss of two H are found to occur. The loss of C2H2 appears as the major path between 50 and 200 ps. As for naphthalene and pyrene, various processes lead to the H2 and C2H2 molecules, the latter being the ethyne molecule. At 40 eV, as at 30 eV for pyrene and 18 eV for naphthalene, complex fragmentation patterns are observed (figure 9a,b). The amount of energy indeed allows for sequential fragmentation, in particular, the formation of various hydrogenation states for a given number of carbon atoms. When decreasing the energy, a limited number of fragments is observed. This is, in particular, the case when the internal energy of coronene is decreased down to 30 eV. As can be seen in figure 9d, only the loss of H (with a maximum ratio of 9%), and to less extent H2 and C2H2, are observed. We achieved test calculations at the intermediate energy of 32 eV, where we derived an intermediate τ value of 92 ps with a parallel evolution of the H and C2H2 loss pathways, the loss of H being predominant.
of 10% at 200 ps. Interestingly, loss of H2 and sequential loss of two H are found to occur. The loss of C2H2 appears as the major path between 50 and 200 ps. As for naphthalene and pyrene, various processes lead to the H2 and C2H2 molecules, the latter being the ethyne molecule. At 40 eV, as at 30 eV for pyrene and 18 eV for naphthalene, complex fragmentation patterns are observed (figure 9a,b). The amount of energy indeed allows for sequential fragmentation, in particular, the formation of various hydrogenation states for a given number of carbon atoms. When decreasing the energy, a limited number of fragments is observed. This is, in particular, the case when the internal energy of coronene is decreased down to 30 eV. As can be seen in figure 9d, only the loss of H (with a maximum ratio of 9%), and to less extent H2 and C2H2, are observed. We achieved test calculations at the intermediate energy of 32 eV, where we derived an intermediate τ value of 92 ps with a parallel evolution of the H and C2H2 loss pathways, the loss of H being predominant.
Figure 9.

Dissociation of 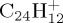 with 40 (a,b) and 30 eV (c,d) of internal energy. Time evolution of the parent and fragment
with 40 (a,b) and 30 eV (c,d) of internal energy. Time evolution of the parent and fragment 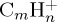 ions for a given number m of C atoms (a), for a selection of the most abundant fragments (b), for all present ions (c). (d) is a zoom of (c) on the fragment ions. In (b–d), fragments are identified by the (m,n) pair. The lines joining the data points were drawn for convenience. (Online version in colour.)
ions for a given number m of C atoms (a), for a selection of the most abundant fragments (b), for all present ions (c). (d) is a zoom of (c) on the fragment ions. In (b–d), fragments are identified by the (m,n) pair. The lines joining the data points were drawn for convenience. (Online version in colour.)
Reflected by the two top curves in figures 7d and 9d, the trends in losses of H versus C2H2 in pyrene and coronene cations appear to be in line with DFT–PES explorations. Indeed, while the asymptote with respect to H loss is at about the same height in both cations (around 5.6 eV; table 1), the paths with respect to C2H2 elimination are different in both cases. Not given in table 1 and detailed elsewhere [49], the asymptotes with respect to C2H2 eliminations are calculated as follows. For 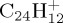 , the dissociation limit is calculated at 7.4 eV, with a barrier of 8.1 eV to be overcome. For
, the dissociation limit is calculated at 7.4 eV, with a barrier of 8.1 eV to be overcome. For 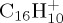 , two paths have been explored. The most direct one, similar to that of coronene, corresponds to an asymptote of 7.1 eV, with a barrier of 7.6 eV to be overcome. A second one, less direct, seems much more favourable, associated with an asymptote lying at only 6.4 eV, with a corresponding barrier estimated at less than 7 eV.
, two paths have been explored. The most direct one, similar to that of coronene, corresponds to an asymptote of 7.1 eV, with a barrier of 7.6 eV to be overcome. A second one, less direct, seems much more favourable, associated with an asymptote lying at only 6.4 eV, with a corresponding barrier estimated at less than 7 eV.
(d). Discussion
The presented MD simulations do not completely cover all the processes occurring in photodissociation or collision experiments. Besides not describing possible ionization, one could, for instance, mention the neglect of the excited states’ contribution to the process. This is probably reasonable assuming the general statement that internal conversion is very effective and that the excited electronic states reached by excitation either by collision or by photon absorption rapidly decay non-adiabatically to the ground state. The non-radiative lifetime for PAH radical cations in excited electronic states is indeed expected to be very short (less than several tens of fs [50,51]) because of numerous conical intersections, where non-adiabatic decay prevails [52,53]. However, the presence of conical intersections may lead to overexcitation in specific modes, which could orient further fragmentation. Nevertheless, in the following, we compare theoretical and experimental branching ratios, and show some interesting common features regarding dependence on PAH size and energy. Our approach also allows us to derive kinetic data hardly accessible from experiments.
In the case of naphthalene dissociation simulations, we recognize carbonated fragment ions that have been identified in photodissociation experiments [11], 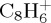 and
and 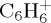 , the former being more abundant than the latter in both experiment and theory. We note that experimentally [11], the photons both ionize and fragment the subsequent ion. So loss of H and C2H2 is observed with similar branching ratios for photon energy between roughly 16 and 19 eV, with a photoionization energy of 8.12 eV [7]. This means that 8–11 eV should be available for fragmentation, which is below the energy domain studied here. As expected, we observe some discrepancies with photodissociation experiments: the loss of H remains a minor path, although it was observed experimentally to be of similar branching ratio as the loss of C2H2 in a reasonably wide range of energy. Besides, the loss of C3H3 is an important channel in our simulations, and is not observed in photodissociation experiments. The discrepancy for the H/C2H2 ratio cannot be accounted for by the DFTB energetics, the respective energy difference between the loss of H and C2H2 even being underestimated with DFTB with respect to DFT (table 1). In addition to the possible role of excited states, one may also invoke simply and most probably the most significant role of the internal energy of the system: when decreasing the energy, one might be able to favour some direct H loss rather than C2H2 and C3H3 that are expected to require structural rearrangement with subsequent barriers. Motivated by such an idea, the dissociation of
, the former being more abundant than the latter in both experiment and theory. We note that experimentally [11], the photons both ionize and fragment the subsequent ion. So loss of H and C2H2 is observed with similar branching ratios for photon energy between roughly 16 and 19 eV, with a photoionization energy of 8.12 eV [7]. This means that 8–11 eV should be available for fragmentation, which is below the energy domain studied here. As expected, we observe some discrepancies with photodissociation experiments: the loss of H remains a minor path, although it was observed experimentally to be of similar branching ratio as the loss of C2H2 in a reasonably wide range of energy. Besides, the loss of C3H3 is an important channel in our simulations, and is not observed in photodissociation experiments. The discrepancy for the H/C2H2 ratio cannot be accounted for by the DFTB energetics, the respective energy difference between the loss of H and C2H2 even being underestimated with DFTB with respect to DFT (table 1). In addition to the possible role of excited states, one may also invoke simply and most probably the most significant role of the internal energy of the system: when decreasing the energy, one might be able to favour some direct H loss rather than C2H2 and C3H3 that are expected to require structural rearrangement with subsequent barriers. Motivated by such an idea, the dissociation of 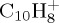 with 14 eV internal energy was tried. One hundred simulations of 5 ns were run. The dissociation rate was found to be much slower (τ ∼ 4.4 ns), the loss of C3H3 becomes minor (now in line with experiment) but the H loss remains minor. The internal energy should probably be decreased, and it would necessitate longer dynamics (up to 5 or 10 ns). Performing a sufficient number of such simulations to reach ergodicity would then become computationally expensive. We may note that if the energy becomes too low, i.e. of the order of magnitude of the zero-point energy (4.0 eV for
with 14 eV internal energy was tried. One hundred simulations of 5 ns were run. The dissociation rate was found to be much slower (τ ∼ 4.4 ns), the loss of C3H3 becomes minor (now in line with experiment) but the H loss remains minor. The internal energy should probably be decreased, and it would necessitate longer dynamics (up to 5 or 10 ns). Performing a sufficient number of such simulations to reach ergodicity would then become computationally expensive. We may note that if the energy becomes too low, i.e. of the order of magnitude of the zero-point energy (4.0 eV for 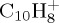 , 5.6 eV for
, 5.6 eV for 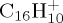 and 7.7 eV for
and 7.7 eV for 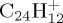 ), the classical description of the nuclei may become questionable and nuclear quantum effects should be taken into account. Of course, full quantum dynamics for such systems are out of reach. One could think about path integral MD [54] but achieving exhaustive simulations would be currently too computationally demanding. An interesting alternative could be to use a semiclassical preparation scheme such as in reference [55] consisting of a quantum preparation of the system distributing the initial energy into the harmonic oscillators before propagating the classical equations of motion.
), the classical description of the nuclei may become questionable and nuclear quantum effects should be taken into account. Of course, full quantum dynamics for such systems are out of reach. One could think about path integral MD [54] but achieving exhaustive simulations would be currently too computationally demanding. An interesting alternative could be to use a semiclassical preparation scheme such as in reference [55] consisting of a quantum preparation of the system distributing the initial energy into the harmonic oscillators before propagating the classical equations of motion.
Although for the highest internal energies studied here, the fragmentation patterns are very complex and the observed CmHn fragments posses various hydrogenation states (figures 6, 7 and 9a), we observe a limited number of channels at lower energy (figures 6, 7 and 9c,d). In the latter conditions, we are able to distinguish fast processes such as the loss of H that occurs either directly or after H-migration, which is well-known, low-energy process in the case of PAHs [22,28], and slower processes such as the loss of C2H2, that requires structural rearrangements leading to external ring opening. The H/C2H2 ratio was found to increase when decreasing the energy in the cases of cationic pyrene and coronene. In experiments on pyrene [12], the loss of H appears as the dominant channel for an incident energy in the range (19–21) eV used to ionize and fragment the pyrene cation. Knowing that the photo-ion appearance of 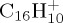 is 7.45 eV [7], between 11 and 13 eV should be available for dissociation. From our MD simulations, we may infer that either less than 22 eV would lead to the H-loss as the only major dissociation channel or, conical intersections may play a crucial role, favouring the H-loss path. In the particular case of
is 7.45 eV [7], between 11 and 13 eV should be available for dissociation. From our MD simulations, we may infer that either less than 22 eV would lead to the H-loss as the only major dissociation channel or, conical intersections may play a crucial role, favouring the H-loss path. In the particular case of 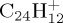 , we found conditions (less than 30 eV of internal energy) where the loss of H would become the unique dissociation channel, which is in line with experimental studies such as photodissociation ion trap experiments (there is sequential loss of H atoms following the sequential absorption of several photons) [56]. With the perspective of the study of larger compact PAHs, we expect to encounter the loss of H as the unique dissociation path at the minimum energy to observe dissociation (in our simulations). The few determined τ characteristic times show the drastic influence of internal energy on the dissociation kinetics. One may note that the orders of magnitude (10–44 ns) obtained at the energies chosen in this work are much below the typical IR radiative lifetime of PAHs (10−2 s).
, we found conditions (less than 30 eV of internal energy) where the loss of H would become the unique dissociation channel, which is in line with experimental studies such as photodissociation ion trap experiments (there is sequential loss of H atoms following the sequential absorption of several photons) [56]. With the perspective of the study of larger compact PAHs, we expect to encounter the loss of H as the unique dissociation path at the minimum energy to observe dissociation (in our simulations). The few determined τ characteristic times show the drastic influence of internal energy on the dissociation kinetics. One may note that the orders of magnitude (10–44 ns) obtained at the energies chosen in this work are much below the typical IR radiative lifetime of PAHs (10−2 s).
To summarize, we found with our approach based on extensive MD simulations in the ground state that, for a chosen set of energies, PAH radical cations major dissociation pathways are the losses of H and ethylene C2H2. These are known to be the dominant dissociation channels for PAHs following photo-absorption processes or low-energy collision-induced dissociation experiments. We have also shown that the H/C2H2 ratio depends on the PAH, on energy and on time and that it increases with the size of the PAH, which seems in line with experimental data and modelling [8,7].
5. Conclusion
In this work, we first show that the SCC–DFTB ground states PES of PAH+ is similar enough to the DFT one, so that it can be used for MD simulations. By studying the statistical dissociation of cationic naphthalene, pyrene and coronene with high internal energies, interesting trends could be derived. We have studied the evolution of the branching ratios and kinetics as a function of energy for a given PAH, and as a function of the PAH. The timescale as revealed by the typical lifetime disappearance of the initial parent is very sensitive to the total internal energy deposited in the PAH, the higher energy, the shorter the lifetime. In fair agreement with other experimental results and modelling, the major fragments are C2H2, under its ethylene form, and H. The H loss has been analysed as a fast and direct process being favoured at low energy and with increasing PAH size. On the opposite, the loss of C2H2 under its ethylene form is major for small PAHs, and is the dominant path at long times at high energy, which is a signature of an indirect process requiring structural reorganization with significant barriers. The loss of H2 has a smaller branching ratio. The loss of CHn fragment was found to be very minor, in line with a probably higher energy process. Our simulations also show that, prior to dissociation, many structural rearrangements occur, involving H migration, vinylidene type complexes and external ring openings.
Such a semiquantitative study is interesting for astrochemistry as important trends on branching ratios, kinetics, structures and hints on the formation mechanism of the ejected neutral fragments can be derived. Although the electronic excited states are not taken into account, this is probably reasonable assuming fast internal conversion, thanks to the presence of conical intersections. However, their presence may affect kinetics and branching ratios, and that will be the object of future studies. The direct comparison with experimental data is delicate owing to the uncertainties in the processes’ timescales and in the amount of internal energy transferred to the system. A perspective could be to distinguish isomers from the difference in their dissociation paths and compare with experimental results.
Acknowledgements
The authors acknowledge the computing facility CALMIP for allocation of computer resources. This work is part of the SWEET project (Stellar Wind and Electron interactions on astrophysical molecules. Experiment and Theory), integrated in the NEXT collaborative project. It was conducted in collaboration with the LCAR laboratory (CNRS-UMR5589, IIM team), and the LPT laboratory (CNRS-UMR5152, Cluster team). We thank Drs Martine Sence, Jean-Philippe Champeaux, Patrick Moretto-Capelle and Phuong Mai Dinh for productive discussions.
Authors' contributions
A.S. achieved the DFTB and dynamic calculations, the data analysis and drafted the manuscript. M.R. modified the DFTB code to achieve this work and contributed to the writing of the manuscript. G.R. is a student and contributed to the calculations. G.T. achieved the DFT calculations. F.X.G. designed the project. All authors contributed to the scientific discussions, read and approved the manuscript.
Competing interests
We declare we have no competing interest.
Funding
This study is supported through the grant NEXT no. ANR-10-LABX-0037 in the framework of the ‘Programme des Investissements d’Avenir’.
References
- 1.Léger A, Puget JL. 1984. Identification of the ‘unidentified’ IR emission features of interstellar dust? Astron. Astrophys. 137, L5–L8. [Google Scholar]
- 2.Allamandola LJ, Tielens AGGM, Barker JR. 1985. Polycyclic aromatic-hydrocarbons and the unidentified infrared-emission bands - auto exhaust along the milky-way. Astrophys. J. 290, L25–L28. ( 10.1086/184435) [DOI] [Google Scholar]
- 3.Herbig GH. 1995. The diffuse interstellar bands. Annu. Rev. Astron. Astrophys. 33, 19–73. ( 10.1146/annurev.aa.33.090195.000315) [DOI] [Google Scholar]
- 4.Boschman L, Cazaux S, Spaans M, Hoekstra R, Schlatholter T. 2015. H-2 formation on PAHs in photodissociation regions: a high-temperature pathway to molecular hydrogen. Astron. Astrophys. 579, A72 ( 10.1051/0004-6361/201323165) [DOI] [Google Scholar]
- 5.Joblin C, Tielens AGGM (eds). 2011. PAHs and the Universe: a Symp. to Celebrate the 25th Anniversary of the PAH Hypothesis, Toulouse, France, 31 May–4 June 2010, vol. 46 of EAS Publications Series. Paris, France: EDP Sciences. [Google Scholar]
- 6.Allain T, Sedlmayr E, Leach S. 1995. Formation and photodestruction of PAHs. Astrophys. Space Sci. 224, 417–418. ( 10.1007/BF00667884) [DOI] [Google Scholar]
- 7.Jochims HW, Ruhl E, Baumgartel H, Tobita S, Leach S. 1994. Size effects on dissociation rates of polycyclic aromatic hydrocarbons cation - laboratory studies and astrophysical implications. Astrophys. J. 420, 307–317. ( 10.1086/173560) [DOI] [Google Scholar]
- 8.Allain T, Leach S, Sedlmayr E. 1996. Photodestruction of PAHs in the interstellar medium.1. Photodissociation rates for the loss of an acetylenic group. Astron. Astrophys. 305, 602–615. [Google Scholar]
- 9.Allain T, Leach S, Sedlmayr E. 1996. Photodestruction of PAHs in the interstellar medium. 2. Influence of the states of ionization and hydrogenation. Astron. Astrophys. 305, 616–630. [Google Scholar]
- 10.Boissel P, deParseval P, Marty P, Lefevre G. 1997. Fragmentation of isolated ions by multiple photon absorption: a quantitative study. J. Chem. Phys. 106, 4973–4984. ( 10.1063/1.473545) [DOI] [Google Scholar]
- 11.West B, Joblin C, Blanchet V, Bodi A, Sztaray B, Mayer PM. 2012. On the dissociation of the naphthalene radical cation: new iPEPICO and tandem mass spectrometry results. J. Phys. Chem. A 116, 10 999–11 007. ( 10.1021/jp3091705) [DOI] [PubMed] [Google Scholar]
-
12.West B, Useli-Bacchitta F, Sabbah H, Blanchet V, Bodi A, Mayer PM, Joblin C.
2014.
Photodissociation of pyrene cations: structure and energetics from
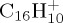 to
to 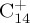 and almost everything in between. J. Phys. Chem. A
118, 7824–7831. ( 10.1021/jp506420u) [DOI] [PubMed] [Google Scholar]
and almost everything in between. J. Phys. Chem. A
118, 7824–7831. ( 10.1021/jp506420u) [DOI] [PubMed] [Google Scholar] - 13.West B, Sit A, Mohamed S, Joblin C, Blanchet V, Bodi A, Mayer PM. 2014. Dissociation of the anthracene radical cation: a comparative look at iPEPICO and collision-induced dissociation mass spectrometry results. J. Phys. Chem. A 118, 9870–9878. ( 10.1021/jp505438f) [DOI] [PubMed] [Google Scholar]
- 14.Zhen J, Castellanos R, Paardekooper DM, Ligterink N, Linnartz H, Nahon L, Joblin C, Tielens AGGM. 2015. Laboratory photo-chemistry of PAHs: ionization versus fragmentation. Astrophys. J. Lett. 804, L7 ( 10.1088/2041-8205/804/1/L7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ekern SP, Marshall AG, Szczepanski J, Vala M. 1998. Photodissociation of gas-phase polycylic aromatic hydrocarbon cations. J. Phys. Chem. A 102, 3498–3504. ( 10.1021/jp980488e) [DOI] [Google Scholar]
- 16.Bouwman J, de Haas AJ, Oomens J. 2016. Spectroscopic evidence for the formation of pentalene+ in the dissociative ionization of naphthalene. Chem. Commun. 52, 2636–2638. ( 10.1039/c5cc10090a) [DOI] [PubMed] [Google Scholar]
- 17.Micelotta ER, Jones AP, Tielens AGGM. 2010. Polycyclic aromatic hydrocarbon processing in a hot gas. Astron. Astrophys. 510, A37 ( 10.1051/0004-6361/200911683) [DOI] [Google Scholar]
- 18.Micelotta ER, Jones AP, Tielens AGGM. 2010. Polycyclic aromatic hydrocarbon processing in interstellar shocks. Astron. Astrophys. 510, A36 ( 10.1051/0004-6361/200911682) [DOI] [Google Scholar]
- 19.Micelotta ER, Jones A, Tielens AGGM. 2011. Polycyclic aromatic hydrocarbon processing by cosmic rays. Astron. Astrophys. 526, A52 ( 10.1051/0004-6361/201015741) [DOI] [Google Scholar]
- 20.Lawicki A. et al 2011. Multiple ionization and fragmentation of isolated pyrene and coronene molecules in collision with ions. Phys. Rev. A 83, 022704 ( 10.1103/PhysRevA.83.022704) [DOI] [Google Scholar]
- 21.Martin S, Chen L, Bredy R, Montagne G, Ortega C, Schlatholter T, Reitsma G, Bernard J. 2012. Statistical fragmentation of doubly charged anthracene induced by fluorine-beam impact at 3 keV. Phys. Rev. A 85, 052715 ( 10.1103/PhysRevA.85.052715) [DOI] [Google Scholar]
- 22.Chen T. et al 2015. Formation of H2 from internally heated polycyclic aromatic hydrocarbons: excitation energy dependence. J. Chem. Phys. 142, 144305 ( 10.1063/1.4917021) [DOI] [PubMed] [Google Scholar]
- 23.Chen T. et al 2014. Absolute fragmentation cross sections in atom-molecule collisions: scaling laws for non-statistical fragmentation of polycyclic aromatic hydrocarbon molecules. J. Chem. Phys. 140, 224306 ( 10.1063/1.4881603) [DOI] [PubMed] [Google Scholar]
- 24.Stockett MH. et al 2014. Nonstatistical fragmentation of large molecules. Phys. Rev. A 89, 032701 ( 10.1103/PhysRevA.89.032701) [DOI] [Google Scholar]
- 25.Stockett MH. et al 2015. Isomer effects in fragmentation of polycyclic aromatic hydrocarbon. Int. J. Mass Spectrom. 392, 58–62. ( 10.1016/j.ijms.2015.09.005) [DOI] [Google Scholar]
- 26.Champeaux JP, Moretto-Capelle P, Cafarelli P, Deville C, Sence M, Casta R. 2014. Is the dissociation of coronene in stellar winds a source of molecular hydrogen? application to the HD 44179 nebula. Month. Not. Roy. Astron. Soc. 441, 1479–1487. ( 10.1093/mnras/stu665) [DOI] [Google Scholar]
- 27.Solano EA, Mayer PM. 2015. A complete map of the ion chemistry of the naphthalene radical cation? DFT and RRKM modeling of a complex potential energy surface. J. Chem. Phys. 143, 104305 ( 10.1063/1.4930000) [DOI] [PubMed] [Google Scholar]
- 28.Jolibois F, Klotz A, Gadea FX, Joblin C. 2005. Hydrogen dissociation of naphthalene cations: a theoretical study. Astron. Astrophys. 444, 629–634. ( 10.1051/0004-6361:20053508) [DOI] [Google Scholar]
- 29.Paris C, Alcami M, Martin F, Diaz-Tendero S. 2014. Multiple ionization and hydrogen loss from neutral and positively-charged coronene. J. Chem. Phys. 140, 204307 ( 10.1063/1.4875805) [DOI] [PubMed] [Google Scholar]
- 30.Postma J, Hoekstra R, Tielens AGGM, Schlatholter T. 2014. A molecular dynamics study on slow ion interactions with the polycyclic aromatic hydrocarbon molecular anthracene. Astrophys. J 783, 61 ( 10.1088/0004-637X/783/1/61) [DOI] [Google Scholar]
- 31.Porezag D, Frauenheim T, Köhler T, Seifert G, Kaschner R. 1995. Construction of tight-binding-like potentials on the basis of density functional theory - application to carbon. Phys. Rev. B 51, 12 947–12 957. ( 10.1103/PhysRevB.51.12947) [DOI] [PubMed] [Google Scholar]
- 32.Seifert G, Porezag D, Frauenheim T. 1996. Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA scheme. Int. J. Quant. Chem. 58, 185–192. ( 10.1002/(SICI)1097-461X(1996)58:2%3C185::AID-QUA7%3E3.0.CO;2-U) [DOI] [Google Scholar]
- 33.Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G. 1998. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 58, 7260–7268. ( 10.1103/PhysRevB.58.7260) [DOI] [Google Scholar]
- 34.Frauenheim T, Seifert G, Elsterner M, Hajnal Z, Jungnickel G, Porezag D, Suhai S, Scholz R. 2000. A self-consistent charge density-functional based tight-binding method for predictive materials simulations in physics, chemistry and biology. Phys. Stat. Solidi (B) 217, 41–62. ( 10.1002/(SICI)1521-3951(200001)217:1%3C41::AID-PSSB41%3E3.0.CO;2-V) [DOI] [Google Scholar]
- 35.Frauenheim T. et al 2002. Atomistic simulations of complex materials: ground-state and excited-state properties. J. Phys. Condensed Matter 14, 3015 ( 10.1088/0953-8984/14/11/313) [DOI] [Google Scholar]
- 36.Oliveira A, Seifert G, Heine T, Duarte H. 2009. Density functional based tight binding : an approximate DFT method. J. Braz. Chem. Soc. 20, 1193–1205. ( 10.1590/S0103-50532009000700002) [DOI] [Google Scholar]
- 37.Krüger T, Elstner M, Schiffels P, Frauenheim T. 2005. Validation of the density-functional based tight-binding approximation method for the calculation of reaction energies and other data. J. Chem. Phys. 122, 114110 ( 10.1063/1.1871913) [DOI] [PubMed] [Google Scholar]
- 38.Rapacioli M, Spiegelman F, Talbi D, Mineva T, Goursot A, Heine T, Seifert G. 2009. Correction for dispersion and coulombic interactions in molecular clusters with density functional derived methods: Application to polycyclic aromatic hydrocarbon clusters. J. Chem. Phys. 130, 244304–10. ( 10.1063/1.3152882) [DOI] [PubMed] [Google Scholar]
- 39.Kukk E. et al 2015. Internal energy dependence in X-ray-induced molecular fragmentation: an experimental and theoretical study of thiophene. Phys. Rev. A 91, 043417 ( 10.1103/PhysRevA.91.043417) [DOI] [Google Scholar]
- 40.Gräfenstein J, Cremer D. 2009. The self-interaction error and the description of non-dynamic electron correlation in density functional theory. Theor. Chem. Acc.: Theor. Comp. Model. 123, 171–182. ( 10.1007/s00214-009-0545-9) [DOI] [Google Scholar]
- 41.Ruzsinszky A, Perdew JP, Csonka GI, Vydrov OA, Scuseria GE. 2006. Spurious fractional charge on dissociated atoms: pervasive and resilient self-interaction error of common density functionals. J. Chem. Phys. 125, 194112 ( 10.1063/1.2387954) [DOI] [PubMed] [Google Scholar]
- 42.Gräfenstein J, Kraka E, Cremer D. 2004. Effect of the self-interaction error for three-electron bonds: on the development of new exchange-correlation functionals. Phys. Chem. Chem. Phys. 6, 1096–1112. ( 10.1039/B311840A) [DOI] [Google Scholar]
- 43.Kaduk B, Kowalczyk T, Van Voorhis T. 2011. Constrained density functional theory. Chem. Rev. 112, 321–370. ( 10.1021/cr200148b) [DOI] [PubMed] [Google Scholar]
- 44.Rapacioli F, Spiegelman M. 2009. Modelling singly ionized coronene clusters. Eur. Phys. J. D 52, 55–58. ( 10.1140/epjd/e2008-00280-2) [DOI] [Google Scholar]
- 45.Janeček I, Cintavá S, Hrivňák D, Kalus R, Fárník M. 2009. Postionization fragmentation of rare-gas trimers revisited with new theoretical approaches. J. Chem. Phys. 131, 114306 ( 10.1063/1.3224855) [DOI] [PubMed] [Google Scholar]
- 46.Heine T. et al 2009. deMonNano, http://demon-nano.ups-tlse.fr/ .
- 47.Frisch MJ. et al 2009. Gaussian revision D.01. Wallingford, CT: Gaussian Inc. [Google Scholar]
- 48.Gatchell M. et al 2014. Non-statistical fragmentation of PAHs and fullerenes in collisions with atoms. Int. J. Mass Spectrom. 365, 260–265. ( 10.1016/j.ijms.2013.12.013) [DOI] [Google Scholar]
- 49.Trinquier G, Simon A, Rapacioli M, Gadéa FX. In press PAH chemistry at eV internal energies. 2. Ring alteration and dissociation. Mol. Astrophys. ( 10.1016/j.molap.2017.02.002) [DOI] [Google Scholar]
- 50.Reddy SN, Mahapatra S. 2013. Theoretical study on molecules of interstellar interest. I. Radical cation of noncompact polycyclic aromatic hydrocarbons. J. Phys. Chem. A 117, 8737–8749. ( 10.1021/jp4033645) [DOI] [PubMed] [Google Scholar]
- 51.Reddy SN, Mahapatra S. 2015. Theoretical Study on molecules of interstellar interest. II. Radical cation of compact polycyclic aromatic hydrocarbons. J. Phys. Chem. B 119, 11 391–11 402. ( 10.1021/acs.jpcb.5b03614) [DOI] [PubMed] [Google Scholar]
- 52.Hall KF, Boggio-Pasqua M, Bearpark MJ, Robb MA. 2006. Photostability via sloped conical intersections: computational study of the excited states of the naphthalene radical cation. J. Phys. Chem. A 110, 13 591–13 599. ( 10.1021/jp064711g) [DOI] [PubMed] [Google Scholar]
- 53.Tokmachev AM, Boggio-Pasqua M, Bearpark MJ, Robb MA. 2008. Photostability via sloped conical intersections: a computational study of the pyrene radical cation. J. Phys. Chem. A 112, 10 881–10 886. ( 10.1021/jp8044109) [DOI] [PubMed] [Google Scholar]
- 54.Habershon S, Manolopoulos DE, Markland TE, Miller TF III. 2013. Ring-polymer molecular dynamics: quantum effects in chemical dynamics from classical trajectories in an extended phase space. Annu. Rev. Phys. Chem. 64, 387–413. ( 10.1146/annurev-physchem-040412-110122) [DOI] [PubMed] [Google Scholar]
- 55.Van-Oanh NT, Falvo C, Calvo F, Lauvergnat D, Basire M, Gaigeot MP, Parneix P. 2012. Improving anharmonic infrared spectra using semiclassically prepared molecular dynamics simulations. Phys. Chem. Chem. Phys. 14, 2381–2390. ( 10.1039/c2cp23101h) [DOI] [PubMed] [Google Scholar]
- 56.Useli-Bacchitta F, Bonnamy A, Mulas G, Malloci G, Toublanc D, Joblin C. 2010. Visible photodissociation spectroscopy of PAH cations and derivatives in the PIRENEA experiment. Chem. Phys. 371, 16–23. ( 10.1016/j.chemphys.2010.03.012) [DOI] [Google Scholar]