

Abstract
This article surveys the empirical information which originated both by laboratory experiments and by computational simulations, and expands previous understanding of the rates of chemical processes in the low-temperature range, where deviations from linearity of Arrhenius plots were revealed. The phenomenological two-parameter Arrhenius equation requires improvement for applications where interpolation or extrapolations are demanded in various areas of modern science. Based on Tolman's theorem, the dependence of the reciprocal of the apparent activation energy as a function of reciprocal absolute temperature permits the introduction of a deviation parameter d covering uniformly a variety of rate processes, from those where quantum mechanical tunnelling is significant and d < 0, to those where d > 0, corresponding to the Pareto–Tsallis statistical weights: these generalize the Boltzmann–Gibbs weight, which is recovered for d = 0. It is shown here how the weights arise, relaxing the thermodynamic equilibrium limit, either for a binomial distribution if d > 0 or for a negative binomial distribution if d < 0, formally corresponding to Fermion-like or Boson-like statistics, respectively. The current status of the phenomenology is illustrated emphasizing case studies; specifically (i) the super-Arrhenius kinetics, where transport phenomena accelerate processes as the temperature increases; (ii) the sub-Arrhenius kinetics, where quantum mechanical tunnelling propitiates low-temperature reactivity; (iii) the anti-Arrhenius kinetics, where processes with no energetic obstacles are rate-limited by molecular reorientation requirements. Particular attention is given for case (i) to the treatment of diffusion and viscosity, for case (ii) to formulation of a transition rate theory for chemical kinetics including quantum mechanical tunnelling, and for case (iii) to the stereodirectional specificity of the dynamics of reactions strongly hindered by the increase of temperature.
This article is part of the themed issue ‘Theoretical and computational studies of non-equilibrium and non-statistical dynamics in the gas phase, in the condensed phase and at interfaces’.
Keywords: sub-Arrhenius, super-Arrhenius, anti-Arrhenius, deformed Arrhenius, d-transition state theory
1. Introduction
For understanding, monitoring and controlling the physical chemistry of materials and the biophysics of environments, information is needed on the kinetics of the involved elementary processes, specifically on their rates—often in a wide range of conditions and notably as a function of temperature. In the late nineteenth century, the increasing phenomenology started to be compacted, principally within the Arrhenius equation [1,2]; in the mid-1930s, the heuristic formulations of the transition-state theory (TST) were introduced ([3–6]; see also [7–14]); experimental progress in the age of nanotechnologies has been mainly extended in the low-temperature range: advances were propitiated to assist the new sciences of astrochemistry and astrobiology, demanding alternative paradigms. Computational progress has occurred by a variety of approaches, benchmarks being those aimed at exploring quantum mechanical advances (here the exemplary story is that of the F + H2 reaction and its variants; see §4b): crucial progress came from time-independent methods, accompanied by those employing carefully checked time-dependent, classical trajectories or semi-classical techniques. The applications of the TST type of formulations benefit quantum mechanics ‘equilibrium’ calculations, where most degrees of freedom are frozen (or allowed to adiabatically adjust) except for a few, mostly one only, and specifically that corresponding motion along a reaction coordinate running through saddles in potential energy surfaces (PESs). Knowledge of PESs is thus crucial, and demands the enormous corpus of theories and computer information coming from quantum chemical calculations that make use of advanced electronic structure codes.
An early remarkable attempt at application to chemical kinetics of the then newly born science of statistical mechanics was initiated by Tolman in 1920 [15]: he concludes his later 1927 book [16, p. 323] saying
The problem of reaction velocities is probably nearer to the heart of most chemists than anything else in their whole range of activity. Rates of reaction are the factors that determine yields, and costs, and possibilities, and their theory must eventually succumb to scientific treatment
However, his optimism was soon frustrated. He subsequently abandoned the field and did not dedicate any attention to it in his monumental treatise [17], which from 1938 was the standard reference on the foundations of statistical mechanics. According to Laidler & King [18], both the use of old quantum theory and some assumptions on the role of radiation in activating chemical processes became obsolete; however, the relevance of what is now often referred to as the ‘Tolman theorem’ (see §2a) was definitely reformulated from a quantum-mechanical perspective by Fowler & Guggenheim [19]. They start a long chapter dedicated to chemical kinetics in their 1939 book on statistical thermodynamics by warning that
The subject of chemical kinetics strictly lies outside the province of this book. Equilibrium theory alone gives no information as to how equilibrium is attained. Nevertheless, equilibrium theory can be a useful tool for dealing with this problem, if combined with certain assumptions concerning the mechanisms involved. We therefore include a survey of such use of equilibrium theory, and shall lay particular stress on the assumptions involved, as these are too frequently ignored in discussions of reaction kinetics [19, p. 489].
We will do the same in this paper, where in order to illustrate the new results we will move directly to them from early motivations, paying only a little attention to some important intermediate developments.
The next section accounts for the basic theory, introducing the apparent activation energy (§2a) and its inverse, the transitivity, admitting an expansion in inverse absolute temperature (§2b), which leads directly to the deformed Arrhenius formula (§2c). Section 3 provides links to the rigorous statistical mechanics foundations, discussing the deviation from the thermodynamic limit (§3a) and introducing the non-equilibrium statistical distributions (§3b). A report on case studies, their treatment and an account of the state of the phenomenological progress is given in §4, which includes a presentation of the recently developed d-TST theory [20]. Final comments are given in the concluding §5.
2. The basic theory
(a). The apparent activation energy
The so-called Tolman's theorem was introduced in a 1920 paper [15], which provides a statistical-mechanics foundation to chemical kinetics, developing a formulation based on the kinetic theory of gases. Its interpretative and predictive power is controversial, but the attention paid by Tolman to the following function is remarkable:
| 2.1 |
which, as is well known, is now to be taken as the definition of the (apparent) activation energy Ea.
This function has been recommended by the International Union for Pure and Applied Chemistry (IUPAC) [21] as directly linked to the key experimental quantity, the reaction rate constant k(T) and obtainable by the familiar Arrhenius plots according to the practice of chemical kinetics. In (2.1), k is the Boltzmann constant and T is the absolute temperature. Formula (2.1) simplifies introducing, as is customary in statistical mechanics, the Lagrange parameter
| 2.2 |
to yield
| 2.3 |
The Tolman expression for Ea, as the difference between the average energy of chemically successful collisions and the total kinetic energy of the gas where the reaction occurs, attributes to this quantity the meaning of an energetic requirement for the reaction to occur. However, its often discussed identification with the Arrhenius activation energy turns out to be transparent only in particular cases [22–25], specifically in temperature ranges where it is a constant or varies slowly.
(b). The reciprocal temperature dependence of the reciprocal activation energy
In general, Ea is to be considered as temperature-dependent. In her comprehensive review ‘On the temperature dependence of Ea’, Berta Perlmutter–Hayman in 1976 [26] has examined in detail two power-series expansions for Ea, one directly in the temperature T and another in terms of its reciprocal (essentially our β, equation (2.2)). We have found it interesting to endorse as the basic expansion the reciprocal-activation–reciprocal-temperature relationship [27], for which we can provide a formal mathematical justification by extending the so-called Tolman theorem [15], following the quantum mechanical formulation in [19]. Tolman's function Ea, when written as the logarithmic derivative of the rate constants with respect to β (equation (2.3)), is indeed akin to the concept of an activation energy, in the sense that it represents a measure of an energetic obstacle to the progress of the reaction; therefore, its reciprocal can be interpreted as a measure of the propensity for the reaction to proceed and defined as the specific transitivity [20] of the process:
| 2.4 |
Our notation emphasizes the fact that in general the transitivity may not be constant but may take a gamma of values: it is assumed to be a smooth function of β in a sufficiently ample range of temperature, not including abrupt changes, e.g. in the mechanism or in the phases of reactants: in this range it will admit a Laurent expansion in a neighbourhood around a reference value denoted as β0:
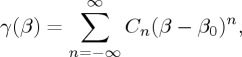 |
2.5 |
where the coefficients Cn will contain n-order derivatives of γ(β) with respect to β and taken at β0. The theory of chemical reaction kinetics is focused on the task of providing a set of such coefficients to connect to the experiment on k(T) via equations ((2.1)–(2.4)).
Since we require a description of the deviations of rates from their high-temperature behaviour, we now consider β0 = 0 and look for a theory with a minimal number of parameters (Occam's razor), limited to only two terms of the Taylor–McLaurin series
| 2.6 |
where E is constant and represents an energetic obstacle at high temperatures and O(β2) indicates neglect of terms of order higher than one. In (2.6), d is the first-order coefficient (C1) and is defined as the deformation parameter. The relationship was first given in [28,29] (see also [20]).
The expansion (2.6) in (2.3), as detailed previously [27,28], leads to a linear first-order differential equation for k(β) of the Bernoulli type that can be integrated by quadrature. It is immediate, but reassuring, to recover the Arrhenius equation within the validity of the first term, i.e. assuming Ea a constant. And it is simple, but remarkable, that inclusion of the second term leads directly to the deformed expression for the rate constant, which is to be considered next.
(c). The deformed Arrhenius rate formula
Further insight into the relationship (2.6) is gained by inserting it in the first equality in equation (2.3), considered as a differential equation for k(β). Equation (2.6) is of first order in the variable β and is easily integrated, specifying the lower limit of the integration range, β0, as shown below:
 |
2.7 |
yielding the deformed Arrhenius equation (d-Arrhenius)
| 2.8 |
when β0, as remarked, can be taken to be zero at high temperature and the symbol A is introduced for the last term in (2.7) in view of its correspondence to the Arrhenius pre-exponential factor: indeed, it can be immediately realized that in the limit d → 0, the term (1 − dEβ)1/d can be identified with the Arrhenius exponential law
| 2.9 |
where the apparent activation energy Ea becomes a constant identified with E, appearing as the first term in equation (2.6).
3. A statistical mechanics interlude
The key step of the previous section, which connects equations (2.8) and (2.9), is a well-known mathematical limit, which is due to Euler and is further under focus in the following developments (§3b).
For d < 0 in the equation (2.8) (sub-Arrhenius behaviour), the identification of d with features of the potential barrier permits entering the deformed exponential formula for describing quantum tunnelling [20,27]. For d > 0 (super-Arrhenius behaviour), one obtains a uniform generalization to classical statistical mechanics, where the Boltzmann–Gibbs distribution is deformed into that of the Pareto–Tsallis statistics (Here, the identification d = 1 − q must be made; see [30–33]). It is not difficult to show that the deformed distribution can be interpreted following lines of thought initiated by Maxwell and developed much later: it is obtained without taking the thermodynamic limit assumption of an infinite number of particles, a step clearly seen e.g. in [34–36] corresponding to an incomplete equipartition over available degrees of freedom (§3b).
(a). Deviation from the thermodynamic limit
Let's attempt to give a preliminary and perhaps more insightful schematic approach within the present context. In general, one can describe the deviation from the thermodynamic limit as due to the interruption of a discrete temporal sequence of events, which is equivalent to avoiding taking the assumption of a continuous time variable. We are inspired by Hinshelwood [37], who gave an illuminating elementary illustration of how exponential expressions come in throughout physical chemistry, and in particular determine the Arrhenius dependence of reaction rate constants upon temperature. The probability that a molecule possesses energy, E, in excess of the average is generally taken as proportional to e−Eβ according to a Boltzmann distribution. One can understand this considering that to accumulate such an energy a molecule needs a fortunate sequence of favourable collisions and the exponential expression arises directly from the limit on probabilities of lucky runs, obtained by Euler in the eighteenth century and a mathematical milestone. That is the limit that we exploited in the previous section. We use it as in §3b.
Notoriously, Euler obtained the limit solving a problem posed by Jakob Bernoulli more than 100 years before: the problem is the important one on how to establish a connection between discrete and continuous computations of interest rates. (Incidentally, Eyring and co-workers [6] pointed out that a theory of reaction rates ‘is not merely a theory of the kinetics of chemical reactions; it is one that can, in principle, be applied to any rate process’). As remarked before, it has been known for a long time [26] that if one wants to keep the exponential form of the Arrhenius equation, there is an interdependence between the energy of activation and the pre-exponential factor, so that if Ea is temperature-dependent, the same must be true for A. The way out that is coherent with the IUPAC definition of Ea, given in equation (2.1), is to follow our procedure and to consider it as a differential equation for k, involving abandoning the exponential form in favour of its d-deformation, equation (2.8).
(b). Non-equilibrium statistical distributions
Let's elaborate further on the centrality of the road to the deformed exponential rate equation (2.8) via the remarkable discovery by Euler that the exponential function can be considered as the limit of succession, alternative to the previously known power-series:
| 3.1 |
The utilization of this expression, where for us the d parameter is the reciprocal of the continuous generalization of the integer N, marks a bifurcation between equilibrium and non-equilibrium treatments, leading to non-equilibrium statistical distribution when the number of microscopic ‘entities’ (particles, or in general sequences of events) is not considered to tend to infinity. When the assumption of the existence of the thermodynamic limit is relaxed, a deformed exponential distribution arises naturally to describe the probability distribution for fluctuations in many-particle systems. Invariably, all treatments (e.g. [38]) proceed directly to the limit.
The deformed exponential distribution arises naturally to account for probabilities attributed to the occurrence of long series of events generated randomly. Considering two mutually excluding events (A and B). For N trials (or for N particles in a microscopic description), let there be a pn chance that the event A occurs and (1 − p)N−n be the chance for event B. In more detail, n is the number of times which the trial experiment yields event A (for example, n can be the number of given particles on a specific configuration A in the microscopic description of the distribution). Then, the number of different ways to yield event A is given by the binomial coefficient
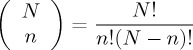 |
3.2 |
and since the trials are independent, the Bernoulli probability distribution for n is the well-known binomial distribution
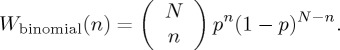 |
3.3 |
Considering the mean of events 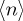 = pN, equation (3.3) can be conveniently rewritten as
= pN, equation (3.3) can be conveniently rewritten as
 |
3.4 |
If N → ∞, p → 0 in the binomial distribution (this step would amount to take the thermodynamic limit), the Poisson distribution is obtained
| 3.5 |
It can be shown that a modification of (3.4) accounts for the probability distribution of a specific event involving distinguishable (or Fermion) particles [39–43]:
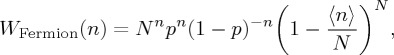 |
3.6 |
which is the distribution that applies prior to the N → ∞ limit, leading to the Fermi–Dirac distribution.
In microscopic systems where quantum effects are operative and the indistinguishability of particles has to be taken into account separately, another physically significant case applies, wherein no limitation occurs on the number of particles per state (Boson particles):
 |
3.7 |
recognized as a negative binomial distribution [40,44].
Proceeding as previously, the large N limit now leads to the Bose–Einstein distribution. In summary, the first case is the Fermi–Dirac distribution and the second case is the Bose–Einstein distribution [40,41,44] and appears that ‘tertium non datur’ [45]. The most probable distributions resulting for equations (3.4), (3.6) and (3.7) lead to Boltzmann, Fermi–Dirac and Bose–Einstein statistics [39,46], respectively, when the thermodynamic limit is taken. However, the presentation emphasizes that an intermediate step in the derivation of all the three statistical distributions in equations (3.4), (3.6) and (3.7) includes what we refer to as the Euler deformed exponential function, (1 − (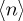 /N))N. This step is crucial here as well as to discuss the role of the Tsallis distribution in non-equilibrium statistics for finite heat baths: when finite size effects are negligible, we can establish the connection between the thermodynamic and the microscopic parameters: n = Eβ and d = 1/N [44,47–49]. See also important previous work on statistical distributions in the kinetics of rate processes [50–52].
/N))N. This step is crucial here as well as to discuss the role of the Tsallis distribution in non-equilibrium statistics for finite heat baths: when finite size effects are negligible, we can establish the connection between the thermodynamic and the microscopic parameters: n = Eβ and d = 1/N [44,47–49]. See also important previous work on statistical distributions in the kinetics of rate processes [50–52].
4. Low-temperature behaviour in chemical kinetics and materials science
Modern experimental techniques and theoretical approaches are providing an ample phenomenology of deviations from the well-known Arrhenius behaviour, especially occurring at low temperatures. As a classification for the phenomenology of these processes, we illustrate the use of the deformed Arrhenius equation (equation (2.8)), permitting one to evaluate prototypical systems where the temperature dependence of the rate constant according to equation (2.8) is described by E > 0 and d > 0 or d < 0, corresponding to convex (super-Arrhenius), concave (sub-Arrhenius) in the semi-log plots against reciprocal temperature. A case for E < 0 will also be documented and indicated as anti-Arrhenius.
(a). Super-Arrhenius
The super-Arrhenius behaviour is one that deserves particular attention, and its occurrence is varied and demanding (figure 1). It often manifests because of collective phenomena, such as those amenable to treatment by the non-extensive thermodynamics of Tsallis, and covers an ample set of phenomena: rates of enzymatic catalysis-promoted processes [53–55], food preservation processes [56,57] and basic features of the dynamics of complex or glass-forming liquids and solids [58–63].
Figure 1.
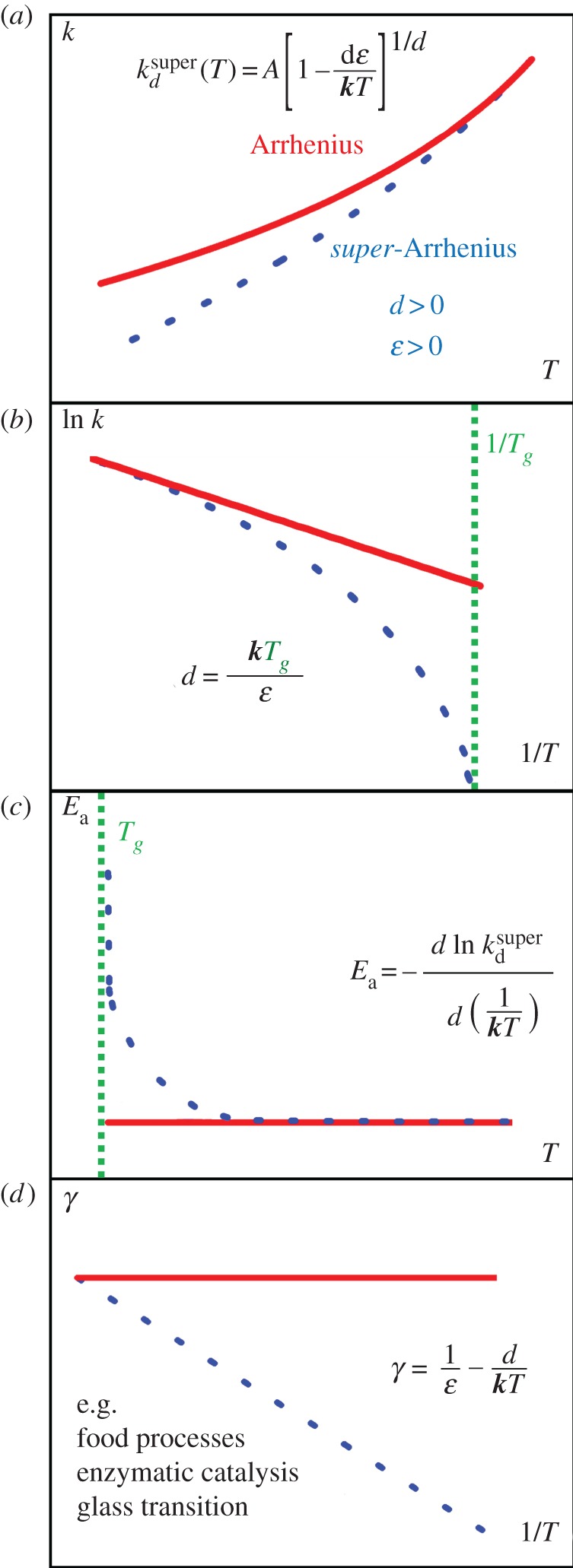
(a) The exponential dependence of reaction rates k(T) upon absolute temperature T. Deviations from linearity at low temperatures can be observed in the plot as showing a ‘convex’ curvature, i.e. lower than expected rates as temperature decreases. In (b), the super-Arrhenius behaviour is accentuaded in an Arrhenius plot view, where ln k(T) is reported against 1/T. In supercooled systems, an approximate relationship between the deformation parameter d and the glass transition temperature Tg illustrates the trend of this complicated phenomenon (see §4a). (c) Deviations from constancy of the apparent activation energy, expected from the Arrhenius Law, manifests for a super-Arrhenius behaviour the apparent activation energy increase with decreasing temperature. Panel (d) exhibits the linear relationship of the transitivity (equation (2.4)) with inverse temperature, basic to our derivation of the d-Arrhenius formula. Super-Arrhenius behaviour, often arising for collective phenomena, is amenable to a classical mechanics interpretation for the examples mentioned and discussed in §4a. (Online version in colour.)
A significant number of studies in the temperature dependence of rates of enzymatic catalysis reactions has inspired several formulations for the description of the mechanism involved in these processes [64–67]. Results in the kinetics of catalytic reactions of the dehydrogenase and oxidase enzymes have shown an undoubted super-Arrhenius behaviour [25,53,54,68].
The theory has interesting applications. A similar behaviour is found in the temperature dependence of food processes. We propose in [56] the use of equation (2.8) to describe the non-Arrhenius behaviour in these processes, showing that the d-Arrhenius formula is suitable for describing the effect of temperature on non-enzymatic browning of onion and on the rate of growth of several species of bacteria. The d-Arrhenius rate law provides a means to account for convex curvature. Such factors include particle diffusion and constraints on the proposed microscopic model, in particular requiring that any successful approach to super-Arrhenius processes should be consistent with the microcanonical rate constant (see §3a, [25,56]).
A most fundamental case of super-Arrhenius temperature dependence occurs for the diffusion in supercooled systems near the glass transition temperature Tg. The following discussion is a simplification for a didactic purpose of a very complicated and still controversial issue. The mechanism involved in convex curvature of Arrhenius plots in supercooled liquids is one of the less understood unsolved processes in condensed matter science [58,69,70]. An interesting example is the diffusion of krypton in methanol and ethanol mixtures at low temperature near their glass transition temperatures [71–73]. Since the diffusion coefficient D and viscosity η are connected (see e.g. the Stokes–Einstein formulation, D = kT/6πrη, where r is the range parameter [6,74]), a manifestation occurs for a super-Arrhenius behaviour of this property. A variety of liquids with different composition exhibit variations of the temperature-dependent viscosity [75]. Around Tg temperature, the Arrhenius-like formulation,
| 4.1 |
of course breaks down. In (4.1), η0 is the viscosity when the temperature tends to infinity (β → 0) and the energy parameter E has to be substantiated by a proper microscopic model. It is no surprise that a variety of proposed non-Arrhenius equations provide options to tackle this problem: we mention those by William–Landel–Ferry [76], Bässler [77], Vogel–Tammann–Fulcher [78–80] and other variants [81,82]. The treatment leading us to equation (2.8) can inspire an option to describe the temperature-dependent viscosity, exploiting the flexibility and continuity of the deformed exponential function introduced owing again to the Euler limit, equation (3.1).
Let the deformed temperature rate law for viscosity be written as follows:
| 4.2 |
where the transitivity (see equation (2.6)) goes to zero, and Tg is identified as the temperature where the glass activation energy tends to infinity, so it is possible to propose a relationship with the deformation parameter d:
| 4.3 |
From the Kauzmann–Eyring [6,83] pre-exponential factor, where Na is the Avogadro number and 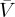 is the molar volume and
is the molar volume and 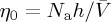 , one can write, for the deformed viscosity rate law,
, one can write, for the deformed viscosity rate law,
| 4.4 |
Several previous formulations described the dependence of Tg and E parameters from the intrinsic properties of the molecular system, and encourage us to give a heuristic interpretation for equation (4.4): from the Flory–Kauzmann hypothesis [83–85], Tg is proportional to the molecular mass (M) and the molecular dimensionality (L), Tg = M1/2/L. According to Eyring and colleagues, the E parameter is connected to the energy of vapourization, ΔEvap [6,86]: the work required to make a hole of molecular size is intuitively identified with the ΔEvap, and so E may be estimated to correspond to a certain fraction of the ΔEvap. This treatment of course does not provide further insight into these amply investigated issues, but points at a simple and perhaps useful parametrization of a long-standing intriguing rate phenomenon.
(b). Sub-Arrhenius cases
There are uncountable cases of concave deviation on the temperature rate constants for the elementary chemical reactions that can be classified as exhibiting a sub-Arrhenius behaviour (figure 2). In systems with no apparent changes in the chemical mechanism, this behaviour can be attributed in most cases to quantum mechanical tunnelling [28,29]. A number of investigations have provided examples of several chemical reactions within this regime. A series of experimental kinetic data and of benchmark quantum mechanical calculations [87–94] has established that, for example, the reaction of a fluorine atom with molecular hydrogen represents a prototypical case of sub-Arrhenius behaviour [95,96] has been experimentally confirmed [97].
Figure 2.
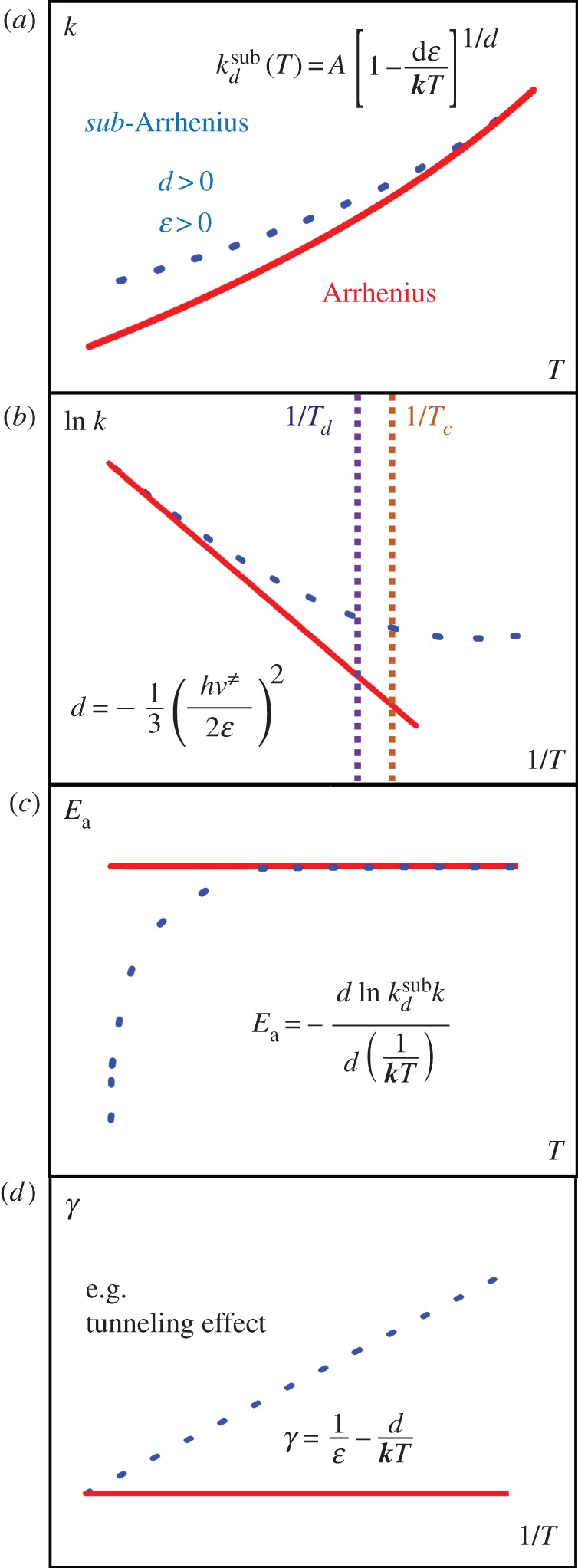
(a) The exponential dependence of reaction rates 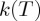 upon absolute temperature
upon absolute temperature 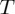 . Deviations from linearity at low temperatures can be observed in the plot as showing a ‘concave’ curvature, i.e. higher than expected rates as temperature decreases. (b) The sub-Arrhenius behaviour is accentuaded in an Arrhenius plot view, where ln
. Deviations from linearity at low temperatures can be observed in the plot as showing a ‘concave’ curvature, i.e. higher than expected rates as temperature decreases. (b) The sub-Arrhenius behaviour is accentuaded in an Arrhenius plot view, where ln 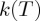 is reported against
is reported against 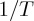 . In elementary reactions at temperatures where quantum mechanical tunnelling is operative, there is a relation between the deformation parameter and features of (height and width) of the barrier (see 4b(i)). Panel (c) shows that deviations from constancy of the apparent activation energy, expected from the Arrhenius Law, for a sub-Arrhenius behaviour manifests as a decrease with decreasing temperature. Panel (d) exhibits the linear relationship of the transitivity (equation (2.4)) with inverse temperature, basic to our derivation of the
. In elementary reactions at temperatures where quantum mechanical tunnelling is operative, there is a relation between the deformation parameter and features of (height and width) of the barrier (see 4b(i)). Panel (c) shows that deviations from constancy of the apparent activation energy, expected from the Arrhenius Law, for a sub-Arrhenius behaviour manifests as a decrease with decreasing temperature. Panel (d) exhibits the linear relationship of the transitivity (equation (2.4)) with inverse temperature, basic to our derivation of the 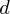 -Arrhenius formula. Sub-Arrhenius behaviour, discussed as a manifestation of quantum mechanical tunnelling through a potential energy barrier as exemplified in the panel. (Online version in colour.)
-Arrhenius formula. Sub-Arrhenius behaviour, discussed as a manifestation of quantum mechanical tunnelling through a potential energy barrier as exemplified in the panel. (Online version in colour.)
Concerning atmospheric and industrial reactions, the direct H-atom abstraction from organic compounds by the hydroxyl radical often presents an evident concave curvature in the Arrhenius plot [98–101]. Within an entirely different chemical environment, experimental rate constants related to multiple proton transfer reactions in condensed matter also show strong sub-Arrhenius behaviour [102–104]. Also, sub-Arrhenius behaviour has been revealed for rates of processes promoted by enzymatic catalysis [55,68,105]. In the next sections, we address the TST and deform it to include the tunnelling correction as a simple tool to describe sub-Arrhenius behaviour.
(i). Transition state theory
The Eyring formulation [4,6] of chemical reaction rates provided chemists with the basic ingredients for understanding and even predicting both parameters A and E of the Arrhenius equation (equation (2.9)) [1] and is assumed as a paradigm for describing the temperature dependence of the rate coefficient of chemical reactions:
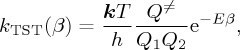 |
4.5 |
(for simplicity, we consider a bimolecular process), where h is the Planck constant, Q1 and Q2 are the (translational, vibrational and rotational) partition functions of the two reactants, and Q≠ is the partition function of the activated complex, from which the translational contribution along the reaction coordinate is excluded. A recent paper [20] exploits the fact that the parameters of equation (4.5) can be explicitly obtained from features of barriers on PESs, as generated from current codes for quantum chemistry calculations. The factor (kT/h) Q≠/Q1Q2 has sometimes been called the collision number, but a more satisfactory term used is frequency factor. The quantity E is the energy of activation of the reaction, encountered before as representing the energy that the molecule in the initial state of the process must acquire before it can take part in the reaction.
Early concerns on possible deviations from Arrhenius behaviour because of quantum mechanical tunnelling (sub-Arrhenius behaviour) were circumvented ad hoc, e.g. by a multiplicative transmission coefficient κ evaluated according to recipes, such as those due to Eckart [106], Pelzer & Wigner [3] and Bell [107]. The tunnelling correction κ is introduced in the TST rate constant as a multiplicative factor, k = κ × kTST and Henriksen & Hansen [7,108] have shown that indeed it is a natural extension of the original formulation. Recent attention has been focused on the role of non-equilibrium or deviation from assumed Maxwell–Boltzmann distributions in the reaction rate theory [109–111] and, in the next section, we show a proposal remarkably distinct from others (see [20]).
(ii). Deformed transition-state theory
What we call the sub-Arrhenius behaviour would be accounted for traditionally by introducing a tunnelling parameter κ, as discussed above. In the deformed transition-state theory (d-TST) formulation, we replace the κ × e−Eβ factor in the TST rate constant, equation (4.5), by the deformed exponential function, yielding
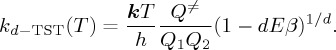 |
4.6 |
The symbols are the same as in (4.5), to which (4.6) tends as d tends to zero, according to (3.1). In [27], the significance of the d parameter and an explicit procedure for its calculation were proposed comparing expansions for the apparent activation energies, Ea. It is shown that d is inversely proportional to the square of the barrier height (E) and directly proportional to the square of the frequency for crossing the barrier (ν≠) at a maximum in the PES:
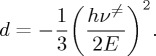 |
4.7 |
The application of d-TST to several hydrogen transfer reactions has been showing promising results [20]. Temperature ranges for the validity of the approach are assessed with respect to features of the potential energy barrier to reaction [20,112], permitting comparison with experiments and tests against alternative formulations. Elementary reactions, widely investigated both experimentally and theoretically, which have been described successfully, are F + H2 [28,96], F + HD [113], CH4 + OH [20], CH3Cl + OH [20], H2 + CN [20], and also abstraction and dissociation in the nitrogen trifluoride channels [114], and proton rearrangement in curcumin [115] and methylhydroxycarbene [116]. The deformed formulation has also been found useful to describe other reactions not involving tunnelling but showing non-negligible sub-Arrhenius behaviour, such as the C + CH+ reaction [117], which is of relevance in cold interstellar clouds.
As discussed in [107,118], the degree of concavity in the Arrhenius plot can characterize the degree of tunnelling in chemical reactions. The crossover temperature, Tc = hν≠/k, is the parameter that delimits the degree of tunnelling regimes. The ranges of tunnelling regimes are important to quantify how the tunnelling affects the rate constant in particular cases.
From a mathematical viewpoint, the d-TST formulation has clear limitations in the description of the deep tunnelling regime (Wigner limit) [119,120], since the Euler limit deformation of the exponential function fails to quantify the distributions of reactive particles with energy less than the height of the barrier. However, the flexibility of the distribution formula permits one to cover the sub-Arrhenius behaviour, specifically typical of quantum tunnelling, smoothly extending into the Boltzmann distribution [27] of the classical regime.
A definition of a validity temperature that delimits the applicability within the negligible or moderate tunnelling ranges can be obtained from the limit of the Bell equation [107,113,121]. In the case of the Wigner limit, when T tends to zero, k(T) tends to a constant. Assuming the Bell limit, 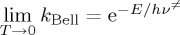 in contrast with the d-TST limit,
in contrast with the d-TST limit, 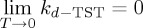 , the temperature that marks the change of validity ranges can be found at the intersection of the two limiting behaviours:
, the temperature that marks the change of validity ranges can be found at the intersection of the two limiting behaviours:
| 4.8 |
where Td is therefore the validity temperature of the d-TST formalism: at temperatures below Td under deep tunnelling conditions, there is no guarantee of the accuracy of the d-TST description.
Although several other papers [109,110,122–125] have proposed ways to insert the Pareto–Tsallis distribution in the description of rate constants in chemical reaction theory, none gave a physical meaning and an explicitly tractable expression for the deformation parameter d as provided by (4.7).
(c). An anti-Arrhenius case
The rates of some processes increase as the temperature decreases according to an apparently negative activation energy. These processes can be classified as anti-Arrhenius. In a gas-phase reaction the anti-Arrhenius behaviour is frequently found in molecule–radical reactions [126–131]. Among these processes, the OH + HBr → H2O + Br reaction is one of the most studied experimentally. Recently, salient features of the PES have been characterized and most kinetic aspects can be considered as satisfactorily reproduced by classical trajectory simulations [132,133]. An insightful illustration of the origin of this behaviour is concerned with the stereodirectional effect assessed by first-principles Born–Oppenheimer ‘canonical’ molecular dynamics [134,135]. The anti-Arrhenius behaviour is documented as being due to the adjustment of the reactants' mutual orientation in order to encounter the entrance into the ‘cone of acceptance’ for reactivity. The aperture angle of this cone is dictated by a range of directions of approach compatible with the formation of the specific HOH angle of the product water molecule; and consistently the adjustment is progressively less effective the higher the kinetic energy. Qualitatively, this emerging picture corroborates experiments on this reaction [136,137], involving collisions of aligned and oriented molecular beams, and covering a range of energies higher than the thermal ones. The rate constant from this molecular dynamic approach was estimated larger than expected and a calibration process was necessary for accounting of the limited dynamical sampling of the involved phase space.
Currently, there is ample activity investigating whether advances in molecular dynamics simulations can provide quantitative values for rate constants [138–140]. However, the methods have difficulty in estimating the rate constants, generally leading to overestimates, and the discrepancy with experimental data is larger for high temperatures, as documented for the OH + HBr reaction [134,135]. These uncertainties are often associated with inherent difficulties of possible direct evaluations from molecular dynamics simulation, and are ascribed to the statistical validity of samplings of the system phase space and the accurate characterization of transition state features [141–143]. These crucial issues in the applications of TST-type approaches to calculations of rates constitute hard problems in the extraction of rate constants from first-principles molecular dynamics experiments, preventing them from representing an at least semi-quantitative alternative to direct exact or approximate quantum mechanical methods, often prohibitive to be implemented.
As a final example on how the study of the anti-Arrhenius behaviour offers opportunities for fundamental research and can guide scientific progress in different areas, we note a recent paper where the stereodirectionality effect contributed to understanding the negative activation energy in addition reactions of arylchlorocarbenes to alkenes [144].
5. Concluding remarks
These notes were written to appear in a collection of papers presented on the occasion of a workshop on ‘Theoretical and Computational Studies of Non-Equilibrium and Non-Statistical Dynamics in Gas-Phase, Condensed-Phase, and Interfacial Reactions’ and the effort of organizers and the stimulus of the participants are acknowledged. The venue was the Institut Henri Poincaré in Paris in April 2016. To Poincaré we owe the observation that ‘mathematics is the art of giving the same name to different things'. Accordingly, we can interpret as a task for scientists that of providing samples of phenomena to be compacted within a common frame. In this presentation of progress on the kinetics of rate processes, the focus of our interest has been to emphasize the unifying role of the treatment of a different type of rate, mentioning the relationship with those occurring in the calculation of interest in bank accounts. The description of the rates of chemical changes is a subject where the main issue is to understand how systems depart from equilibrium states and how they approach new ones: the rates are typically exponentially influenced by the reciprocal of the temperature (Arrhenius behaviour). The description of deviations at low temperatures exploits Euler's solution of the problem posed by Jakob Bernoulli on the computation of compound rates. The key is the Euler formula, equation (3.1), established as a continuous limit of a discrete succession of events: in essence, our deformation formulas are based on the same now elementary mathematical tool.
Acknowledgements
Valter Henrique Carvalho-Silva thanks PrP/UEG for research funding through the PROBIP programme and FAPEG. Nayara Dantas Coutinho thanks FAPDF and CAPES. Vincenzo Aquilanti thanks CAPES for the appointment as Special Visiting Professor (Professor Visitante Especial) at the Institute of Physics, Federal University of Bahia, Salvador (Brazil). For the work reported here, we have enjoyed collaborations with T. Kasai, K. C. Mundim, S. Cavalli, H. C. B. de Oliveira, A. J. Camargo, D. De Fazio, E. Munusami and S. Kleijn.
Authors' contributions
Equal contributions by all authors.
Competing interests
We declare that we have no competing interests.
Funding
Financial support for the Italian Ministry for Education, University and Research, MIUR. Grant no. SIR2014(RBSI14U3VF).
References
- 1.Arrhenius S. 1889. On the reaction rate of the inversion of non-refined sugar upon souring. Z. Phys. Chem. 4, 226–248. [Google Scholar]
- 2.Stiller W. 1989. Arrhenius equation and non-equilibrium kinetics: 100 years Arrhenius equation. Leipzig, Germany: BSB B.G. Teubner. [Google Scholar]
- 3.Pelzer H, Wigner E. 1932. Über das Überschreiten von Potentialschwellen bei chemischen Reaktionen. Z. Phys. Chem. Abt. B, 19, 203–216. [Google Scholar]
- 4.Eyring H. 1935. The activated complex in chemical reactions. J. Chem. Phys. 3, 107–115. ( 10.1063/1.1749604) [DOI] [Google Scholar]
- 5.Polanyi M, Wigner E. 1928. Über die Interferenz von Eigenschwingungen als Ursache von Energieschwankungen und chemischer Umsetzungen. Z. Phys. Chem. Abt. A 139, 439–452. ( 10.1515/zpch-1928-0130) [DOI] [Google Scholar]
- 6.Glasstone S, Laidler KJ, Eyring H. 1941. The theory of rate processes: the kinetics of chemical reactions, viscosity, diffusion and electrochemical phenomena. New York, NY: McGraw-Hill. [Google Scholar]
- 7.Henriksen NE, Hansen FY. 2008. Theories of molecular reaction dynamics: the microscopic foundation of chemical kinetics. Oxford, UK: Oxford University Press. [Google Scholar]
- 8.Keck JC. 1960. Variational theory of chemical reaction rates applied to three-body recombinations. J. Chem. Phys. 32, 1035–1050. ( 10.1063/1.1730846) [DOI] [Google Scholar]
- 9.Truhlar DG, Garrett BC. 1984. Variational transition state theory. Annu. Rev. Phys. Chem. 35, 159–189. ( 10.1146/annurev.pc.35.100184.001111) [DOI] [Google Scholar]
- 10.Keck JC. 2007. Variational theory of reaction rates. In Advances in chemical physics, pp. 85–121. New York, NY: John Wiley & Sons, Inc. [Google Scholar]
- 11.Kramers HA. 1940. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica 7, 284–304. ( 10.1016/S0031-8914(40)90098-2) [DOI] [Google Scholar]
- 12.Marcus RA. 1993. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 65, 599–610. ( 10.1103/RevModPhys.65.599) [DOI] [Google Scholar]
- 13.Callan CG, Coleman S. 1977. Fate of the false vacuum. II. First quantum corrections. Phys. Rev. D 16, 1762–1768. ( 10.1103/PhysRevD.16.1762) [DOI] [Google Scholar]
- 14.Coleman S. 1977. Fate of the false vacuum: semiclassical theory. Phys. Rev. D 15, 2929–2936. ( 10.1103/PhysRevD.15.2929) [DOI] [Google Scholar]
- 15.Tolman RC. 1920. Statistical mechanics applied to chemical kinetics. J. Am. Chem. Soc. 42, 2506–2528. ( 10.1021/ja01457a008) [DOI] [Google Scholar]
- 16.Tolman RC. 1927. Statistical mechanics with applications to physics and chemistry, p 323. (Reprint 2013 in India by Isha Books).
- 17.Tolman RC. 1938. The principles of statistical mechanics. London, UK: Oxford University. [Google Scholar]
- 18.Laidler KJ, King MC. 1983. Development of transition-state theory. J. Phys. Chem. 87, 2657–2664. ( 10.1021/j100238a002) [DOI] [Google Scholar]
- 19.Fowler R, Guggenheim EA. 1939. Statistical thermodynamics. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 20.Carvalho-Silva VH, Aquilanti V, de Oliveira HCB, Mundim KC. 2017. Deformed transition-state theory: deviation from arrhenius behavior and application to bimolecular hydrogen transfer reaction rates in the tunneling regime. J. Comput. Chem. 38, 178–188. ( 10.1002/jcc.24529) [DOI] [PubMed] [Google Scholar]
- 21.Laidler KJ. 1996. A glossary of terms used in chemical kinetics, including reaction dynamics. Pure Appl. Chem. 68, 149–192. ( 10.1351/pac199668010149) [DOI] [Google Scholar]
- 22.Menzinger M, Wolfgang R. 1969. The meaning and use of the Arrhenius activation energy. Angew. Chem. Int. Ed. 9, 438–444. ( 10.1002/anie.196904381) [DOI] [Google Scholar]
- 23.Craven GT, Nitzan A. 2016. Electron transfer across a thermal gradient. Proc. Natl Acad. Sci. USA 13, 9421–9429. ( 10.1073/pnas.1609141113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matyushov DV. 2016. Activated kinetics in a nonequilibrium thermal bath. Proc. Natl Acad. Sci. USA 113, 9401–9403. ( 10.1073/pnas.1610542113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truhlar DG, Kohen A. 2001. Convex Arrhenius plots and their interpretation. Proc. Natl Acad. Sci. USA 98, 848–851. ( 10.1073/pnas.98.3.848) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perlmutter-Hayman B. 1976. The temperature dependence of Ea. In Progress in inorganic chemistry, vol. 20 (ed. Lippard SJ.), pp. 229–297. New York, NY: John Wiley & Sons, Inc. [Google Scholar]
- 27.Silva VHC, Aquilanti V, de Oliveira HC, Mundim KC. 2013. Uniform description of non-Arrhenius temperature dependence of reaction rates, and a heuristic criterion for quantum tunneling vs classical non-extensive distribution. Chem. Phys. Lett. 590, 201–207. ( 10.1016/j.cplett.2013.10.051) [DOI] [Google Scholar]
- 28.Aquilanti V, Mundim KC, Elango M, Kleijn S, Kasai T. 2010. Temperature dependence of chemical and biophysical rate processes: Phenomenological approach to deviations from Arrhenius law. Chem. Phys. Lett. 498, 209–213. ( 10.1016/j.cplett.2010.08.035) [DOI] [Google Scholar]
- 29.De Fazio D, Aquilanti V, Cavalli S, Aguilar A, Lucas JM. 2006. Exact quantum calculations of the kinetic isotope effect: cross sections and rate constants for the F+HD reaction and role of tunneling. J. Chem. Phys. 125, 133109 ( 10.1063/1.2221695) [DOI] [PubMed] [Google Scholar]
- 30.Arnold BC. 2008. Pareto and generalized Pareto distributions. In Modeling income distributions and Lorenz curves, pp. 119–145. New York, NY: Springer [Google Scholar]
- 31.Pareto V, Bousquet GH, Busino G. 1964. Cours d’économie politique. Paris, France: Librairie Droz. [Google Scholar]
- 32.Borges EP.2012. Nonextensive local composition models in theories of solutions. (http://arxiv.org/abs/1206.0501. )
- 33.Tsallis C. 1988. Possible generalization of Boltzmann-Gibbs statistics. J. Stat. Phys. 52, 479–487. ( 10.1007/BF01016429) [DOI] [Google Scholar]
- 34.Jeans J. 1913. The dynamical theory of gases. New York, NY: Dover Publications Incorporated. [Google Scholar]
- 35.Condon EU. 1938. A simple derivation of the Maxwell-Boltzmann Law. Phys. Rev. 54, 937–940. ( 10.1103/PhysRev.54.937) [DOI] [Google Scholar]
- 36.Kennard EH. 1938. Kinetic theory of gases: with an introduction to statistical mechanics. New York, NY: McGraw-Hill. [Google Scholar]
- 37.Hinshelwood CN. 1940. The kinetics of chemical change. Oxford, UK: The Clarendon press. [Google Scholar]
- 38.Landau LD, Lifshitz EM. 1958. Statistical Physics, 5th edn Oxford, UK: Pergamon Press. [Google Scholar]
- 39.Laurendeau NM. 2005. Statistical thermodynamics: fundamentals and applications. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 40.Lavenda BH. 2016. Statistical physics: a probabilistic approach. New York, NY: Dover Publications. [Google Scholar]
- 41.Lavenda BH. 1988. Derivation of quantum statistics from Gauss's principle and the second law. Int. J. Theor. Phys. 27, 1371–1381. ( 10.1007/BF00671315) [DOI] [Google Scholar]
- 42.Walter JE, Eyring H, Kimball GE. 1944. Quantum chemistry. New York, NY: Wiley & Sons. [Google Scholar]
- 43.Van Vliet CM. 2008. Equilibrium and non-equilibrium statistical mechanics. Singapore: World Scientific Pub. [Google Scholar]
- 44.Biró T, Ván P, Barnaföldi G, Ürmössy K. 2014. Statistical power law due to reservoir fluctuations and the universal thermostat independence principle. Entropy 16, 6497–6514. ( 10.3390/e16126497) [DOI] [Google Scholar]
- 45.Lavenda BH, Dunning-Davies J. 1989. The case against intermediate statistics. J. Math. Phys. 30, 1117 ( 10.1063/1.528330) [DOI] [Google Scholar]
- 46.Eyring H, Walter J, Kimball GE. 1961. Quantum chemistry. New York, NY: Wiley. [Google Scholar]
- 47.Almeida MP. 2001. Generalized entropies from first principles. Phys. A 300, 424–432. ( 10.1016/S0378-4371(01)00353-3) [DOI] [Google Scholar]
- 48.Adib AB, Moreira AA, Andrade JS Jr, Almeida MP. 2003. Tsallis thermostatistics for finite systems: a Hamiltonian approach. Phys. A Stat. Mech. Appl. 322, 276–284. ( 10.1016/S0378-4371(02)01601-1) [DOI] [Google Scholar]
- 49.Wada T. 2003. Model-free derivations of the Tsallis factor: constant heat capacity derivation. Phys. Lett. A 318, 491–498. ( 10.1016/j.physleta.2003.09.056) [DOI] [Google Scholar]
- 50.Schulten K, Schulten Z, Szabo A. 1980. Reactions governed by a binomial redistribution process—the Ehrenfest Urn problem. Phys. A Stat. Mech. Appl. 100, 599–614. ( 10.1016/0378-4371(80)90170-3) [DOI] [Google Scholar]
- 51.Hoare MR, Rahman M. 1983. Cumulative Bernoulli trials and Krawtchouk processes. Stoch. Process. Appl. 16, 113–139. ( 10.1016/0304-4149(84)90014-0) [DOI] [Google Scholar]
- 52.Hoare MR, Rahman M. 1988. Cumulative hypergeometric processes: a statistical role for the nFn-1 functions. J. Math. Anal. Appl. 135, 615–626. ( 10.1016/0022-247X(88)90176-X) [DOI] [Google Scholar]
- 53.Kohen A, Cannio R, Bartolucci S, Klinman JP. 1999. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature 399, 496–499. ( 10.1038/20981) [DOI] [PubMed] [Google Scholar]
- 54.Nagel ZD, Dong M, Bahnson BJ, Klinman JP. 2011. Impaired protein conformational landscapes as revealed in anomalous Arrhenius prefactors. Proc. Natl Acad. Sci. USA 108, 10 520–10 525. ( 10.1073/pnas.1104989108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer MP, Tomchick DR, Klinman JP. 2008. Enzyme structure and dynamics affect hydrogen tunneling: the impact of a remote side chain (I553) in soybean lipoxygenase-1. Proc. Natl Acad. Sci. USA 105, 1146–1151. ( 10.1073/pnas.0710643105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coutinho ND, Silva VHC, Mundim KC, de Oliveira HCB. 2015. Description of the effect of temperature on food systems using the deformed Arrhenius rate law: deviations from linearity in logarithmic plots vs. inverse temperature. Rend. Lincei 26, 141–149. ( 10.1007/s12210-015-0407-4) [DOI] [Google Scholar]
- 57.Peleg M, Normand MD, Corradini MG. 2012. The Arrhenius equation revisited. Crit. Rev. Food Sci. Nutr. 52, 830–851. ( 10.1080/10408398.2012.667460) [DOI] [PubMed] [Google Scholar]
- 58.Debenedetti PG, Stillinger FH. 2001. Supercooled liquids and the glass transition. Nature 410, 259–267. ( 10.1038/35065704) [DOI] [PubMed] [Google Scholar]
- 59.Smith RS, Kay BD. 2012. Breaking through the glass ceiling: recent experimental approaches to probe the properties of supercooled liquids near the glass transition. J. Phys. Chem. Lett. 3, 725–730. ( 10.1021/jz201710z) [DOI] [PubMed] [Google Scholar]
- 60.Luiggi Agreda NJ. 2016. Aquilanti–Mundim deformed Arrhenius model in solid-state reactions. J. Thermal Anal. Calorim. 126, 1175–1184. ( 10.1007/s10973-016-5566-8) [DOI] [Google Scholar]
- 61.Hecksher T, Nielsen AI, Olsen NB, Dyre JC. 2008. Little evidence for dynamic divergences in ultraviscous molecular liquids. Nat. Phys. 4, 737–741. ( 10.1038/nphys1033) [DOI] [Google Scholar]
- 62.Vyazovkin S, et al. 2016. A time to search: finding the meaning of variable activation energy. Phys. Chem. Chem. Phys. 18, 18 643–18 656. ( 10.1039/C6CP02491B) [DOI] [PubMed] [Google Scholar]
- 63.de Souza VK, Wales DJ. 2008. Energy landscapes for diffusion: analysis of cage-breaking processes. J. Chem. Phys. 129, 164507 ( 10.1063/1.2992128) [DOI] [PubMed] [Google Scholar]
- 64.Warshel A, Bora RP. 2016. Perspective: defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 144, 180901 ( 10.1063/1.4947037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Truhlar DG. 2015. Transition state theory for enzyme kinetics. Arch. Biochem. Biophys. 582, 10–17. ( 10.1016/j.abb.2015.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh P, Islam Z, Kohen A. 2016. Examinations of the chemical step in enzyme catalysis. Methods Enzymol. 577, 287–318. ( 10.1016/bs.mie.2016.05.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klinman JP, Kohen A. 2014. Evolutionary aspects of enzyme dynamics. J. Biol. Chem. 289, 30 205–30 212. ( 10.1074/jbc.R114.565515) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klinman JP, Kohen A. 2013. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 82, 471–496. ( 10.1146/annurev-biochem-051710-133623) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kivelson SA, Tarjus G. 2008. In search of a theory of supercooled liquids. Nat. Mater. 7, 831–833. ( 10.1038/nmat2304) [DOI] [PubMed] [Google Scholar]
- 70.Stillinger FH, Debenedetti PG. 2013. Glass transition thermodynamics and kinetics. Annu. Rev. 4, 263–285. ( 10.1146/annurev-conmatphys-030212-184329) [DOI] [Google Scholar]
- 71.Matthiesen J, Smith RS, Kay BD. 2011. Mixing it up: measuring diffusion in supercooled liquid solutions of methanol and ethanol at temperatures near the glass transition. J. Phys. Chem. Lett. 2, 557–561. ( 10.1021/jz2000744) [DOI] [Google Scholar]
- 72.Matthiesen J, Smith RS, Kay BD. 2009. Using rare gas permeation to probe methanol diffusion near the glass transition temperature. Phys. Rev. Lett. 245902, 1–4. ( 10.1103/PhysRevLett.103.245902) [DOI] [PubMed] [Google Scholar]
- 73.Rosa ACP, Vaveliuk P, Mundim KC, Moret MA. 2016. A model for diffusive systems: beyond the Arrhenius mechanism. Phys. A Stat. Mech. Appl. 450, 317–322. ( 10.1016/j.physa.2015.12.122) [DOI] [Google Scholar]
- 74.Debenedetti PG, Truskett TM, Lewis CP, Stillinger FH. 2001. Theory of supercooled liquids and glasses: energy landscape and statistical geometry perspectives. Adv. Chem. Eng. 28, 21–79. ( 10.1016/S0065-2377(01)28003-X) [DOI] [Google Scholar]
- 75.Angell CA. 1995. Formation of glasses from liquids and biopolymers. Science 267, 1924–1935. ( 10.1126/science.267.5206.1924) [DOI] [PubMed] [Google Scholar]
- 76.Williams ML, Landel RF, Ferry JD. 1955. The temperature dependence of relaxation mechanisms in amorphous polymers and other glass-forming liquids. J. Am. Chem. Soc. 77, 3701–3707. ( 10.1021/ja01619a008) [DOI] [Google Scholar]
- 77.Bässler H. 1987. Viscous flow in supercooled liquids analyzed in terms of transport theory for random media with energetic disorder. Phys. Rev. Lett. 58, 767–770. ( 10.1103/PhysRevLett.58.767) [DOI] [PubMed] [Google Scholar]
- 78.Vogel H. 1921. Das temperature-abhangigketsgesetz der viskositat von flussigkeiten. Phys. Z 22, 645–646. [Google Scholar]
- 79.Tammann G, Hesse W. 1926. Die Abhängigkeit der Viscosität von der Temperatur bie unterkühlten Flüssigkeiten. Z. Anorg. Allg. Chem. 156, 245–257. ( 10.1002/zaac.19261560121) [DOI] [Google Scholar]
- 80.Fulcher GS. 1925. Analysis of recent measurements of the viscosity of glasses. J. Am. Ceram. Soc. 8, 339–355. ( 10.1111/j.1151-2916.1925.tb16731.x) [DOI] [Google Scholar]
- 81.Angell CA, Ngai KL, McKenna GB, McMillan PF, Martin SW. 2000. Relaxation in glass forming liquids and amorphous solids. J. Appl. Phys. 88, 3113 ( 10.1063/1.1286035) [DOI] [Google Scholar]
- 82.Hou F, Martin JD, Dill ED, Folmer JCW, Josey AA. 2015. Transition zone theory of crystal growth and viscosity. Chem. Mater. 27, 3526–3532. ( 10.1021/acs.chemmater.5b00956) [DOI] [Google Scholar]
- 83.Kauzmann W, Eyring H. 1940. The viscous flow of large molecules. J. Am. Chem. Soc. 62, 3113–3125. ( 10.1021/ja01868a059) [DOI] [Google Scholar]
- 84.Flory PJ. 1940. Viscosities of linear polyesters. An exact relationship between viscosity and chain length. J. Am. Chem. Soc. 62, 1057–1070. ( 10.1021/ja01862a020) [DOI] [Google Scholar]
- 85.Novikov VN, Rössler EA. 2013. Correlation between glass transition temperature and molecular mass in non-polymeric and polymer glass formers. Polymer (Guildf). 54, 6987–6991. ( 10.1016/j.polymer.2013.11.002) [DOI] [Google Scholar]
- 86.Powell RE, Roseveare WE, Eyring H. 1941. Diffusion, thermal conductivity, and viscous flow of liquids. Ind. Eng. Chem. 33, 430–435. ( 10.1021/ie50376a003) [DOI] [Google Scholar]
- 87.Rayez J-C, Bonnet L, Larrégaray P, Perrier A. 2009. Transition state theory: a reaction dynamics tool applied to gas-surface reactions. Mol. Sci. 3, A0029. ( 10.3175/molsci.3.A0029) [DOI] [Google Scholar]
- 88.Bonnet L, Rayez J-C. 2010. Dynamical derivation of Eyring equation and the second-order kinetic law. Int. J. Quantum Chem. 110, 2355–2359. ( 10.1002/qua.22545) [DOI] [Google Scholar]
- 89.Bonnet L. 2004. On the dynamical foundations of transition state theory: a semiclassical analysis. Ann. Phys. NY 314, 99–118. ( 10.1016/j.aop.2004.05.007) [DOI] [Google Scholar]
- 90.Klippenstein SJ, Pande VS, Truhlar DG. 2014. Chemical kinetics and mechanisms of complex systems: a perspective on recent theoretical advances. J. Am. Chem. Soc. 136, 528–546. ( 10.1021/ja408723a) [DOI] [PubMed] [Google Scholar]
- 91.Bao JL, Sripa P, Truhlar DG. 2015. Path-dependent variational effects and multidimensional tunneling in multi-path variational transition state theory: rate constants calculated for the reactions of HO2 with tert-butanol by including all 46 paths for abstraction at C and all six paths for. Phys. Chem. Chem. Phys. 18, 1032–1041. ( 10.1039/c5cp05780a) [DOI] [PubMed] [Google Scholar]
- 92.Chapman S, Garrett BC, Miller WH. 1975. Semiclassical transition state theory for nonseparable systems: application to the collinear H+H2 reaction. J. Chem. Phys. 63, 2710–2716. ( 10.1063/1.431620) [DOI] [Google Scholar]
- 93.Hänggi P, Borkovec M. 1990. Reaction-rate theory: fifty years after Kramers. Rev. Mod. Phys. 62, 251–341. ( 10.1103/RevModPhys.62.251) [DOI] [Google Scholar]
- 94.Suleimanov YV, Aoiz FJ, Guo H. 2016. Chemical reaction rate coefficients from ring polymer molecular dynamics: theory and practical applications. J. Phys. Chem. A 120, 8488–8502. ( 10.1021/acs.jpca.6b07140) [DOI] [PubMed] [Google Scholar]
- 95.Aquilanti V, Cavalli S, De Fazio D, Volpi A, Aguilar A, Lucas JM. 2005. Benchmark rate constants by the hyperquantization algorithm. The F+H2 reaction for various potential energy surfaces: features of the entrance channel and of the transition state, and low temperatur e reactivity. Chem. Phys. 308, 237–253. ( 10.1016/j.chemphys.2004.03.027) [DOI] [Google Scholar]
- 96.Aquilanti V, Mundim KC, Cavalli S, De Fazio D, Aguilar A, Lucas JM. 2012. Exact activation energies and phenomenological description of quantum tunneling for model potential energy surfaces. The F+H2 reaction at low temperature. Chem. Phys. 398, 186–191. ( 10.1016/j.chemphys.2011.05.016) [DOI] [Google Scholar]
- 97.Tizniti M, Le Picard SD, Lique F, Berteloite C, Canosa A, Alexander MH, Sims IR. 2014. The rate of the F + H2 reaction at very low temperatures. Nat. Chem. 6, 141–145. ( 10.1038/nchem.1835) [DOI] [PubMed] [Google Scholar]
- 98.Atkinson R. 1986. Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions. Chem. Rev. 86, 69–201. ( 10.1021/cr00071a004) [DOI] [Google Scholar]
- 99.Meana-Paneda R, Truhlar DG, Fernandez-Ramos A. 2011. High-level direct-dynamics variational transition state theory calculations including multidimensional tunneling of the thermal rate constants, branching ratios, and kinetic isotope effects of the hydrogen abstraction reactions from methanol by atomic hyd. J. Chem. Phys. 134, 94302. ( 10.1063/1.3555763) [DOI] [PubMed] [Google Scholar]
- 100.Shannon RJ, Blitz MA, Goddard A, Heard DE. 2013. Accelerated chemistry in the reaction between the hydroxyl radical and methanol at interstellar temperatures facilitated by tunnelling. Nat. Chem. 5, 745–749. ( 10.1038/nchem.1692) [DOI] [PubMed] [Google Scholar]
- 101.Sims IR. 2013. Low-temperature reactions: tunnelling in space. Nat. Chem. 5, 734–736. ( 10.1038/nchem.1736) [DOI] [PubMed] [Google Scholar]
- 102.Limbach H-H, Miguel Lopez J, Kohen A. 2006. Arrhenius curves of hydrogen transfers: tunnel effects, isotope effects and effects of pre-equilibria. Phil. Trans. R. Soc. B 361, 1399–1415. ( 10.1098/rstb.2006.1872) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Limbach H-H, Schowen KB, Schowen RL. 2010. Heavy atom motions and tunneling in hydrogen transfer reactions: the importance of the pre-tunneling state. J. Phys. Org. Chem. 23, 586–605. ( 10.1002/poc.1663) [DOI] [Google Scholar]
- 104.Torres V, Lopez J-M, Langer U, Buntkowsky G, Vieth H-M, Elguero J, Limbach H-H. 2012. Kinetics of coupled double proton and deuteron transfer in hydrogen-bonded ribbons of crystalline pyrazole-4-carboxylic acid. Z. Phys. Chem. 226, 1125–1148. ( 10.1524/zpch.2012.0305) [DOI] [Google Scholar]
- 105.Liang Z-X, Lee T, Resing KA, Ahn NG, Klinman JP. 2004. Thermal-activated protein mobility and its correlation with catalysis in thermophilic alcohol dehydrogenase. Proc. Natl Acad. Sci. USA 101, 9556–9561. ( 10.1073/pnas.0403337101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eckart C. 1930. The penetration of a potential barrier by electrons. Phys. Rev. 35, 1303–1309. ( 10.1103/PhysRev.35.1303) [DOI] [Google Scholar]
- 107.Bell RP. 1980. The tunnel effect in chemistry. London, UK: Champman and Hall. [Google Scholar]
- 108.Henriksen NE, Hansen FY. 2002. Transition-state theory and dynamical corrections. Phys. Chem. Chem. Phys. 4, 5995–6000. ( 10.1039/b207021a) [DOI] [Google Scholar]
- 109.Quapp W, Zech A. 2009. Transition state theory with Tsallis statistics. J. Comput. Chem. 31, 573–585. ( 10.1002/jcc.21349) [DOI] [PubMed] [Google Scholar]
- 110.Yin C, Du J. 2014. The power-law reaction rate coefficient for an elementary bimolecular reaction. Phys. A Stat. Mech. Appl. 395, 416–424. ( 10.1016/j.physa.2013.10.038) [DOI] [Google Scholar]
- 111.Pollak E, Talkner P. 2005. Reaction rate theory: what it was, where is it today, and where is it going? Chaos Interdiscip. J. Nonlinear Sci. 15, 26116. ( 10.1063/1.1858782) [DOI] [PubMed] [Google Scholar]
- 112.Carvalho VH, Aquilanti V, de Oliveira HCB, Mundim KC. 2015. Deformed transition state theory: inclusion of the tunneling effect by Euler exponential, limit of validity and description of bimolecular reactions. Rev. Process. Químicos 9, 226–228. [Google Scholar]
- 113.Cavalli S, Aquilanti V, Mundim KC, De Fazio D. 2014. Theoretical reaction kinetics astride the transition between moderate and deep tunneling regimes: the F + HD case. J. Phys. Chem. A 118, 6632–6641. ( 10.1021/jp503463w) [DOI] [PubMed] [Google Scholar]
- 114.Claudino D, Gargano R, Carvalho-Silva VH, e Silva GM, da Cunha WF. 2016. Investigation of the abstraction and dissociation mechanism in the nitrogen trifluoride channels: combined post-Hartree–Fock and transition state theory approaches. J. Phys. Chem. A 120, 5464–5473. ( 10.1021/acs.jpca.6b04947) [DOI] [PubMed] [Google Scholar]
- 115.Santin LG, Toledo EM, Carvalho-Silva VH, Camargo AJ, Gargano R, Oliveira SS. 2016. Methanol solvation effect on the proton rearrangement of curcumin's enol forms: an ab initio molecular dynamics and electronic structure viewpoint. J. Phys. Chem. C 120, 19 923–19 931. ( 10.1021/acs.jpcc.6b02393) [DOI] [Google Scholar]
- 116.de A Morais SF, Mundim KC, Ferreira DAC. 2015. An alternative interpretation of the ultracold methylhydroxycarbene rearrangement mechanism: cooperative effects. Phys. Chem. Chem. Phys. 17, 7443–7448. ( 10.1039/c4cp05842a) [DOI] [PubMed] [Google Scholar]
- 117.Rampino S, Pastore M, Garcia E, Pacifici L, Laganà A. 2016. On the temperature dependence of the rate coefficient of formation of C2+ from C + CH+. Mon. Notes R. Astron. Soc. 460, 2368–2375. ( 10.1093/mnras/stw1116) [DOI] [Google Scholar]
- 118.Christov SG. 1997. The characteristic (crossover) temperature in the theory of thermally activated tunneling processes. Mol. Eng. 7, 109–147. ( 10.1023/A:1008274213168) [DOI] [Google Scholar]
- 119.Wigner EP. 1948. On the behavior of cross sections near thresholds. Phys. Rev. 73, 1002–1009. ( 10.1103/PhysRev.73.1002) [DOI] [Google Scholar]
- 120.Takayanagi T, Masaki N, Nakamura K, Okamoto M, Sato S, Schatz GC. 1987. The rate constants for the H+H2 reaction and its isotopic analogs at low temperatures: Wigner threshold law behavior. J. Chem. Phys. 86, 6133 ( 10.1063/1.452453) [DOI] [Google Scholar]
- 121.Bell RP. 1935. Quantum mechanical effects in reactions involving hydrogen. Proc. R. Soc. Lond. A 148, 241–250. ( 10.1098/rspa.1935.0016) [DOI] [Google Scholar]
- 122.Zhao J-L, Bao J-D. 2005. Transition state theory rate in nonlinear environment: the under-damping case. Commun. Theor. Phys. (Beijing, China) 44, 752–756. ( 10.1088/6102/44/4/752) [DOI] [Google Scholar]
- 123.Jiulin D. 2012. Transition state theory: a generalization to nonequilibrium systems with power-law distributions. Phys. A 391, 1718–1728. ( 10.1016/j.physa.2012.01.010) [DOI] [Google Scholar]
- 124.Lenzi EK, Anteneodo C, Borland L. 2001. Escape time in anomalous diffusive media. Phys. Rev. E 63, 51 105–51 109. ( 10.1103/PhysRevE.63.051109) [DOI] [PubMed] [Google Scholar]
- 125.Yin C, Zhou Y, Du J. 2014. The power-law TST reaction rate coefficient with tunneling correction. Phys. A Stat. Mech. Appl. 413, 294–300. ( 10.1016/j.physa.2014.06.051) [DOI] [Google Scholar]
- 126.Timonen RS, Seetula JA, Gutman D. 1990. Kinetics of the reactions of alkyl radicals (CH, C2H5,i-C3H7,and t-C4H9) with molecular bromine. J. Chem. Inf. Model. 94, 3005–3008. ( 10.1017/CBO9781107415324.004) [DOI] [Google Scholar]
- 127.Seetula JA. 1998. Kinetics and thermochemistry of the R+HBr reversible arrow RH+Br (R=C2H5 or β-C2H4Cl) equilibrium—an ab initio study of the bond energies in partly chlorinated ethanes and propanes. J. Chem. Soc. Trans. 94, 891–898. ( 10.1039/a706440c) [DOI] [Google Scholar]
- 128.Jaramillo VI, Gougeon S, Le Picard SD, Canosa A, Smith MA, Rowe BR. 2002. A consensus view of the temperature dependence of the gas phase reaction: OH + HBr → H2O + Br. Int. J. Chem. Kinet. 34, 339–344. ( 10.1002/kin.10056) [DOI] [Google Scholar]
- 129.Stone D, Rowley DM. 2005. Kinetics of the gas phase HO2 self-reaction: effects of temperature, pressure, water and methanol vapours. Phys. Chem. Chem. Phys. 7, 2156–2163. ( 10.1039/b502673c) [DOI] [PubMed] [Google Scholar]
- 130.Atkinson R, Perry R, Pitts JN Jr. 1978. Rate constants for the reactions of the OH radical with (CH) NH,(CH) N, and CHNH over the temperature range 298–426°K. J. Chem. Phys. 68, 1850–1853. ( 10.1063/1.435906) [DOI] [Google Scholar]
- 131.Campuzano-Jost P, Crowley JN. 1999. Kinetics of the reaction of OH with HI between 246 and 353 K. J. Phys. Chem. A 103, 2712–2719. ( 10.1021/jp984321x) [DOI] [Google Scholar]
- 132.de Oliveira-Filho AGS, Ornellas FR, Bowman JM. 2014. Quasiclassical trajectory calculations of the rate constant of the OH + HBr → Br + H2O reaction using a full-dimensional ab initio potential energy surface over the temperature range 5 to 500 K. J. Phys. Chem. Lett. 5, 706–712. ( 10.1021/jz5000325) [DOI] [PubMed] [Google Scholar]
- 133.de Oliveira-Filho AGS, Ornellas FR, Bowman JM. 2014. Energy disposal and thermal rate constants for the OH + HBr and OH + DBr reactions: quasiclassical trajectory calculations on an accurate potential energy surface. J. Phys. Chem. A 118, 12 080–12 088. ( 10.1021/jp509430p) [DOI] [PubMed] [Google Scholar]
- 134.Coutinho ND, Silva VHC, de Oliveira HCB, Camargo AJ, Mundim KC, Aquilanti V. 2015. Stereodynamical origin of anti-Arrhenius kinetics: negative activation energy and roaming for a four-atom reaction. J. Phys. Chem. Lett. 6, 1553–1558. ( 10.1021/acs.jpclett.5b00384) [DOI] [PubMed] [Google Scholar]
- 135.Coutinho ND, Aquilanti V, Silva VHC, Camargo AJ, Mundim KC, de Oliveira HCB. 2016. Stereodirectional origin of anti-Arrhenius kinetics for a tetraatomic hydrogen exchange reaction: Born-Oppenheimer molecular dynamics for OH + HBr. J. Phys. Chem. A 120, 5408–5417. ( 10.1021/acs.jpca.6b03958) [DOI] [PubMed] [Google Scholar]
- 136.Che D-C, Matsuo T, Yano Y, Bonnet L, Kasai T. 2008. Negative collision energy dependence of Br formation in the OH + HBr reaction. Phys. Chem. Chem. Phys. 10, 1419–1423. ( 10.1039/b713322g) [DOI] [PubMed] [Google Scholar]
- 137.Che D-C, Doi A, Yamamoto Y, Okuno Y, Kasai T. 2009. Collision energy dependence for the Br formation in the reaction of OD + HBr. Phys. Scr. 80, 48110 ( 10.1088/0031-8949/80/04/048110) [DOI] [Google Scholar]
- 138.Döntgen M, Przybylski-Freund M-D, Kröger LC, Kopp WA, Ismail AE, Leonhard K. 2015. Automated discovery of reaction pathways, rate constants, and transition states using reactive molecular dynamics simulations. J. Chem. Theory Comput. 11, 2517–2524. ( 10.1021/acs.jctc.5b00201) [DOI] [PubMed] [Google Scholar]
- 139.Fleming KL, Tiwary P, Pfaendtner J. 2016. New approach for investigating reaction dynamics and rates with ab initio calculations. J. Phys. Chem. A 120, 299–305. ( 10.1021/acs.jpca.5b10667) [DOI] [PubMed] [Google Scholar]
- 140.Imandi V, Chatterjee A. 2016. Estimating Arrhenius parameters using temperature programmed molecular dynamics. J. Chem. Phys. 145, 34104 ( 10.1063/1.4958834) [DOI] [PubMed] [Google Scholar]
- 141.Xie J, Hase WL. 2016. Rethinking the SN2 reaction. Science. 352, 32–33. ( 10.1126/science.aaf5172) [DOI] [PubMed] [Google Scholar]
- 142.Manikandan P, Zhang J, Hase WL. 2012. Chemical dynamics simulations of X– + CH3Y → XCH3 + Y– gas-phase SN2 nucleophilic substitution reactions. Nonstatistical dynamics and nontraditional reaction mechanisms. J. Phys. Chem. A 116, 3061–3080. ( 10.1021/jp211387c) [DOI] [PubMed] [Google Scholar]
- 143.de Souza MAF, Correra TC, Riveros JM, Longo RL. 2012. Selectivity and mechanisms driven by reaction dynamics: the case of the gas-phase OH- + CH3ONO2 reaction. J. Am. Chem. Soc. 134, 19 004–19 010. ( 10.1021/ja3057166) [DOI] [PubMed] [Google Scholar]
- 144.Wang L, Krogh-Jespersen K, Moss RA. 2015. Activation parameters for additions to alkenes of arylchlorocarbenes with enhanced electrophilicity. J. Org. Chem. 80, 7590–7593. ( 10.1021/acs.joc.5b01210) [DOI] [PubMed] [Google Scholar]