

Abstract
Ants have long been renowned for their intimate mutualisms with trophobionts and plants and more recently appreciated for their widespread and diverse interactions with microbes. An open question in symbiosis research is the extent to which environmental influence, including the exchange of microbes between interacting macroorganisms, affects the composition and function of symbiotic microbial communities. Here we approached this question by investigating symbiosis within symbiosis. Ant–plant–hemipteran symbioses are hallmarks of tropical ecosystems that produce persistent close contact among the macroorganism partners, which then have substantial opportunity to exchange symbiotic microbes. We used metabarcoding and quantitative PCR to examine community structure of both bacteria and fungi in a Neotropical ant–plant–scale-insect symbiosis. Both phloem-feeding scale insects and honeydew-feeding ants make use of microbial symbionts to subsist on phloem-derived diets of suboptimal nutritional quality. Among the insects examined here, Cephalotes ants and pseudococcid scale insects had the most specialized bacterial symbionts, whereas Azteca ants appeared to consume or associate with more fungi than bacteria, and coccid scale insects were associated with unusually diverse bacterial communities. Despite these differences, we also identified apparent sharing of microbes among the macro-partners. How microbial exchanges affect the consumer-resource interactions that shape the evolution of ant–plant–hemipteran symbioses is an exciting question that awaits further research.
Keywords: Cordia alliodora, bacteria, fungi, metabarcoding, myrmecophyte, scale insects
1. Introduction
Mutualistic symbioses, i.e. mutually beneficial interactions where the partners live in prolonged physical contact, have been a major driver of the evolution of life on Earth, from the origin of eukaryotes to coral-reef diversity hotspots. Mutualistic symbioses are currently receiving unprecedented attention across biological subdisciplines as the use of high-throughput sequencing begins to reveal the ubiquitous and dynamic associations between microbes and macroorganisms. These associations provide vital functions for both partners, including increased metabolic capacity, nutrition and protection, but have so far been examined mostly as two-way host–microbe interactions. A central question in mutualism biology is how mutualistic interactions shape and are shaped by their surrounding ecological communities [1].
Symbioses can be classified as ‘open’ or ‘closed’, where open symbioses have symbionts that can be gained or lost from the environment and are thus typically subject to greater influence from the outside community than are symbionts in closed systems [2]. Closed symbioses, by contrast, are usually transmitted vertically from parent to offspring and tend to show higher partner fidelity, which may lead more easily to coevolution between partners [2]. Although the distinction between open and closed symbioses provides a bird's-eye view of mutualistic symbiosis in its ecological context, the complete framework will probably require ‘open’ and ‘closed’ at two ends of a continuum of possible environmental interactions. What constitutes the space between requires investigation into how organisms' evolutionary history and present ecology simultaneously influence the extent of interaction between a given mutualistic symbiosis and its environment.
Both coevolution and the key role of the ecological community in shaping a mutualistic symbiosis were first demonstrated in a single pioneering experiment: Janzen [3] separated tropical acacia plants that provide housing and food for ant symbionts from those ant symbionts and showed that herbivore damage to plants increased, strongly reducing plant fitness. Ant–plant protective mutualisms, including acacia-like ‘ant-plants’ (myrmecophytes) with hollow cavities (domatia) that host symbiotic ant defenders, have been central to developing our understanding of mutualism ever since [4]. The diversity of ant–plant interactions, as well as the sheer abundance of ants that is maintained by them, may have been driven primarily by the evolution of protective-nutritional mutualism between ants and honeydew-producing hemipteran insects (e.g. scale insects and aphids) [5,6]. Hemipterans feed on plant sap and metabolize it into carbohydrate-rich honeydew, which allows honeydew-feeding ants to take on primarily arboreal lifestyles as ‘cryptic herbivores’ [6]. Indeed, the vast majority of myrmecophytes feed ants in part via the honeydew of hemipteran scale insects [7].
Although both ants and hemipterans can obtain their required energy from plant phloem sap, several key nutrients, and essential amino acids in particular, are in much shorter supply [8]. Hemipterans were early and enduring models for studying how animals can obtain such key nutrients from microbes [9], but the broad relevance of bacteria and fungi as potential sources of nutrition for ants, which are typically omnivorous, was recognized only recently [10,11]. Most phloem-feeding hemipterans are associated with just one or a few highly specialized intracellular bacteria that are vertically transmitted, i.e. ‘closed’ symbiosis, whereas ants are more typically associated with gut bacterial symbionts, i.e. ‘open’ symbiosis. Nevertheless, evidence to date suggests that these associations may provide good examples of the continuum of ecological interaction with symbiosis: some ants exhibit core microbiota that, though subject to dietary influence, vary little through evolutionary time [12–14], and hemipteran honeydew can contain proteins derived from their bacterial symbionts as well as even some of the bacteria themselves [15,16]. Microbial symbionts have in fact recently been suggested to mediate ant–hemipteran mutualisms [17]. Because there is a potentially strong feedback loop among plant chemistry, hemipteran honeydew, and the quantity and quality of ant defensive behaviour [18], which may be modified by the insect-associated microbes, microbial symbionts may also play key roles in the eco-evolutionary outcomes of ant–plant protective mutualisms.
To investigate the extent of environmental influence on patterns of microbial communities in an ant–plant–hemipteran symbiosis, we investigated the abundance and composition of bacteria and fungi in the myrmecophytic tree Cordia alliodora in Costa Rica. Cordia alliodora is a widespread Neotropical tree that forms hollow domatia at stem nodes, where ant symbionts nest and tend several species of scale insects. In Costa Rica, Co. alliodora trees are commonly inhabited by both Azteca spp. (Dolichoderinae) and Cephalotes setulifer Emery (Myrmicinae) ants, either in separate trees or in different domatia on the same tree [19]. Whereas Azteca ants appear to be omnivorous and to host very few specialized gut symbionts, Cephalotes is the quintessential cryptic herbivore, and this habit may be facilitated by a core gut bacterial microbiome that is maintained throughout the genus [10,12–14]. In Co. alliodora, however, both Azteca and Cephalotes share a very similar environment and diet: individual tree domatia can transition between ant occupants over time, and both ants tend large numbers of honeydew-producing scale insects in two subfamilies: Pseudococcidae : Pseudococcinae and Coccidae : Myzolecaniinae (E. G. Pringle 2007, personal observation). The vertically transmitted intracellular symbionts of a major clade of Pseudococcinae species have been well studied and are unusual in that the metabolic pathways are shared between two nested bacteria—a Gammaproteobacteria within the cytoplasm of the Betaproteobacteria Tremblaya—and the host insect genome [20,21]. To our knowledge, the microbial symbionts of Myzolecanniinae remain virtually unstudied. Relatives in Coccidae : Coccinae were reported to have abundant fungal symbionts [9].
Using high-throughput sequencing of 16S rRNA from bacteria and internal transcribed spacer (ITS) region rDNA from fungi, we tested the hypothesis that the persistent physical contact created by the symbiosis among the tree, ants and scale insects would influence the composition of the insects' microbial associates and symbionts. For bacteria, we predicted that environmental influence would be strongest in Azteca, weaker in Cephalotes and weakest for the coccoid scale insects, based on the expected differences in localization, transmission and partner fidelity of the associated bacteria. For fungi, we predicted that Azteca ants would consistently associate with domatia-derived fungi: these associations have been previously reported from other Azteca-plant symbioses [22], and we have observed black, fungal patches within the ant domatia. We had few predictions for the fungal associations of the rest of the insect taxa, which have been very little studied.
2. Material and methods
(a). Sample collection
Samples were collected in June 2012 in the Area de Conservación Guanacaste, Sector Santa Rosa, Costa Rica (10°50′ N, 85°36′ W). Two species of Azteca commonly occupy Co. alliodora trees at the site (Azteca pittieri and Azteca beltii; [23]), and they are difficult to distinguish in the field. We therefore chose study trees based on which ant genera were present and subsequently identified Azteca to species level with molecular barcoding (electronic supplementary material, text S1). We collected samples from five trees (approx. 9 cm average diameter at breast height) (electronic supplementary material, table S1). From each tree, we collected ants (Dolichoderinae: Azteca spp. and/or Myrmicinae: Ce. setulifer), hereafter Azteca and Ce. setulifer, scale insects (Coccidae: Myzolecaniinae: Cryptostigma spp. and/or Pseudococcidae: Pseudococcinae: Paraputo cf. larai), hereafter Cryptostigma and Paraputo, and environmental samples from the ant domatia and tree leaves. Domatia samples consisted of 2 mm2 scrapings of an interior domatium wall occupied by each ant species per tree; leaf samples consisted of approximately 4 cm2 per tree from each of two leaves from separate whorls. Stable-isotope samples were transferred to a freezer within 1 h of collection and frozen for less than 12 h before they were dried for ≥48 h at 60°C. Microbial-survey samples were stored in 100% ethanol until processing.
(b). Stable isotopes
To test whether there was a relationship between the trophic position of the insects and the abundance of their internal microbes, we measured ∂15N in the ants and scale insects. Sets of approximately 20 ant worker bodies (head and thorax) (n = 6 Azteca and n = 6 Ce. setulifer) and entire scale insects (n = 3 Cryptostigma and n = 3 Paraputo) were ground with a mortar and pestle. Four larval leaf-chewing herbivores, three spiders, and nine leaves were analysed for comparison. Analyses were performed at the UC Davis Stable Isotope Facility. We tested for differences in ∂15N among sample types using a generalized linear model because of the unbalanced sampling design and conducted Tukey post hoc comparisons in the multcomp package in R v. 3.3.1 [24,25].
(c). DNA extraction
Insects were surface sterilized prior to DNA extraction. Ant gasters and whole scale insects were dipped in 95% ethanol, soaked for 1 min in 5% bleach and rinsed in sterile water. We clipped gasters (i.e. posterior abdomens, which contain the entire digestive tract apart from the mouth and oesophagus) from ant bodies and pooled 1–3 gasters for each ant sample (n = 15 Azteca samples, n = 15 Ce. setulifer samples). We dissected the midgut from one additional Ce. setulifer ant and extracted it separately. Ethanol was evaporated from the domatia (n = 10) and leaf (n = 6) samples prior to extraction. DNA was extracted using a modified version of the PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA) (see [26]).
(d). Quantitative PCR to determine microbe copy numbers
To estimate the abundance of bacteria and fungi in our samples, we performed real-time quantitative PCR (qPCR) of the bacterial 16S rRNA gene and fungal ITS rDNA gene (see also the electronic supplementary material, text S1). For 16S rRNA, we used the universal bacterial primers 515F and 806R [27]. For ITS rDNA, we used ITS1-F and 5.8S primers [28,29]. All qPCRs were performed on a CFX Connect Real-Time System (Bio-Rad, Hercules, CA, USA) using SsoAdvanced 2X SYBR green supermix (Biorad) and 2 µl of DNA extract. For bacterial 16S rRNA, standard curves were generated from the Escherichia coli 16S rRNA gene. For fungal rDNA, standard curves were generated from the ITS rDNA gene of Pleurotus sp., obtained from store-bought oyster mushrooms (electronic supplementary material, text S1). To determine the number of gene copies per microgram of DNA, we measured the DNA concentration of each sample on a Qubit (Life Technologies, Grand Island, NY, USA) (electronic supplementary material, text S1).
We tested for differences among sample types in the abundance of 16S rRNA and ITS rDNA separately using the nparcomp package in R v. 3.3.1 [25,30] because the sampling was unbalanced and the data exhibited a strong right skew. We used two-sided tests on a multivariate t-distribution with a Satterthwaite approximation. We tested for a relationship between the abundance of bacteria in our ant and scale-insect samples and their ∂15N signatures using a general linear model in R.
(e). Sequencing of bacterial and fungal communities
We first sequenced both 16S bacterial rRNA and ITS fungal rDNA amplicons using tag-encoded 454 FLX-titanium amplicon pyrosequencing (Research and Testing Laboratory, Lubbock, TX, USA). 16S rRNA from bacteria was amplified in the V1–3 region using primers 28F and 519R [31]. ITS rDNA from fungi was amplified using fungal-specific primers ITS1-F and ITS4 [28]. Two blank negative controls from the PowerSoil DNA extraction kit produced no sequences. The 454 sequences were extracted and processed in mothur v. 1.36.0 [32], following the standard operating procedure (accessed 2 February 2015; [33]) with some modifications.
For ITS sequences, after running sff.multiple with minflows = 360, we trimmed all sequences at both ends to 250 total bases, discarding shorter sequences. This trimming scheme produced a good match to a mock community that we sequenced for quality control (electronic supplementary material, text S1). We detected chimeras using the uchime algorithm implemented in mothur. Operational taxonomic units (OTUs) were then determined by calculating pairwise distances between sequences and clustering using the average neighbour method and a 0.03 distance. Although the 97% OTU cut-off may approximate species imprecisely in some taxa [34], it is currently the most common threshold for community analysis of both bacteria and fungi and allowed us to compare within and between these hyperdiverse kingdoms. Representative OTUs and phylotypes were classified using a recent UNITE database (UNITEv6_sh_dynamic) [35]. We included singletons in the analyses presented here; analyses excluding singletons were run in parallel (see the electronic supplementary material).
Because our preliminary analyses of the 454 16S rRNA sequences indicated unusually high diversity among the bacteria associated with our two Cryptostigma (Coccidae: Myzolecaniinae) samples, we resequenced all of our samples for bacterial 16S rRNA, including five additional Cryptostigma spp. samples (electronic supplementary material, table S1) and 13 additional Myzolecaniinae (electronic supplementary material, text S4), on an Illumina MiSeq platform (Argonne National Laboratory, Lemont, IL, USA), amplifying the V4 region using primers 515F and 806R [27]. Because these MiSeq 16S rRNA sequences produced very similar results and more total reads after filtering (241 077 from MiSeq versus 201 246 from 454), here we focus on the MiSeq results for 16S rRNA. MiSeq sequences were processed in mothur v. 1.37.0 [32], following the standard operating procedure (accessed 1 April 2016; [36]). We again detected and removed chimeras using the uchime algorithm. Sequences were classified using the SILVA database (www.arb-silva.de). We removed sequences classified as Archaea, chloroplasts, or mitochondria, and clustered sequences into OTUs using the dist.seqs and cluster commands. OTUs were subsequently classified at 97% (see above). Sequences from four blank negative controls from the PowerSoil DNA extraction kit were analysed in parallel to test for possible reagent- or platform-derived contamination.
(f). Analysis of bacterial and fungal communities
To investigate the alpha diversity of our samples, we compared composition based on phylotypes, conducted rarefaction analysis and calculated diversity. We calculated phylotypes from the SILVA and UNITE database for bacteria and fungi, respectively. Rarefaction curves were calculated from data binned at the OTU level with the rarefaction.single command in mothur. To compare diversity, we calculated an inverse Simpson index for all replicates subsampled at 1100 sequences for bacteria and 902 sequences for fungi (see below) using the summary.single command in mothur. Because the distributions of diversity estimates were right-skewed, we assessed differences in diversity among samples using the nparcomp package in R.
Before beta-diversity analyses, the mothur sub.sample command was used to subsample the data. Bacteria were subsampled at 1100 sequences, which excluded nine samples. Fungi were subsampled at 902 sequences, which excluded three samples. Community composition was compared in two ways. First, we used nonmetric multidimensional scaling (NMDS) to visualize differences in community composition among all of our sample types. The vegan package in R v. 3.3.1 was used to calculate Bray–Curtis distances (vegdist function), and stable solutions to the first two NMDS axes were calculated using the metaMDS function [25,37]. Second, we visualized the extent to which each ant shared bacterial and fungal OTUs with their scale insects and domatia environment using Venn diagrams. We searched for overlap among the Azteca, Azteca domatia and Azteca-tended scale insects and among the Ce. setulifer, Ce. setulifer domatia and Ce. setulifer-tended scale insects (electronic supplementary material, figure S1). The merge.groups and venn commands in mothur were used to visualize OTU overlap.
(g). Functional inference from inferred hemipteran metagenomes
To examine whether the two genera of hemipteran scale insects (Cryptostigma coccids and Paraputo pseudococcids) harboured bacterial endosymbionts with similar predicted functional activity, we used the online Galaxy version of Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt, v. 1.0.0) to predict metagenome function from 16S rRNA sequences [38] (electronic supplementary material, text S1). To compare the relative abundance of predicted gene families from PICRUSt, we calculated the relative abundance of each gene family per sample and tested for differences between Cryptostigma and Paraputo using t-tests with unequal variances and a Bonferroni correction for multiple comparisons.
3. Results
(a). Trophic position and microbial abundance
Among our insects, there was no relationship between bacterial abundance and ∂15N natural abundance (figure 1). Unexpectedly, the Paraputo pseudococcids, which feed on tree phloem, exhibited ∂15N levels significantly higher than tree leaves and not significantly different than the Azteca ants or the spiders (electronic supplementary material, figure S2a). By contrast, the Cryptostigma coccids, which also feed on tree phloem, exhibited ∂15N levels similar to leaf-chewing herbivores and not significantly different than tree leaves. Cryptostigma coccids and Paraputo pseudococcids contained similar abundances of bacteria, and both contained low abundances of fungi (electronic supplementary material, figure S2b). Cephalotes setulifer ants exhibited much lower ∂15N than Azteca ants (figure 1). The posterior abdomens of the Ce. setulifer ants contained the highest bacterial abundance of any of our samples (electronic supplementary material, figure S2b), consistent with the indication that these bacteria play a role in Cephalotes nutrition [10]. By contrast, the posterior abdomens of Azteca ants contained very few bacteria but a higher abundance of fungi, suggesting that perhaps fungi are an important source of nutrition for these ants, as has been shown in other symbiotic ant–plant mutualisms [11].
Figure 1.
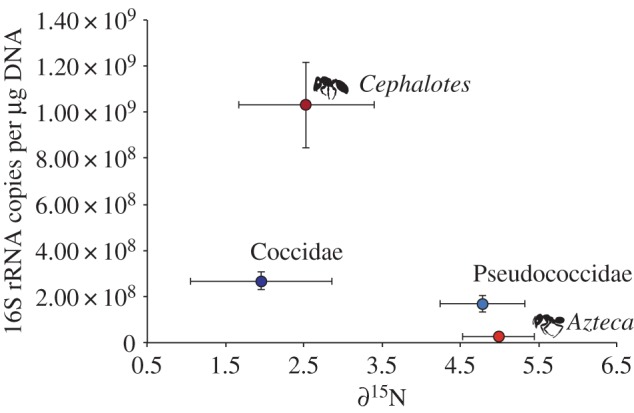
Relationship between the abundance of bacteria in ants (posterior abdomens; red dots) and whole scale insects (blue dots) and their ∂15N signatures. Error bars indicate s.e. Sample sizes are as follows: Azteca n = 4; Cephalotes n = 5; Coccidae n = 3; Pseudococcidae n = 3. (Online version in colour.)
(b). Alpha diversity of communities
(i). Bacteria
There were 20 identifiable bacterial phyla among all of our sample types, including ants, scale insects, domatia and leaves, with Proteobacteria, Actinobacteria, Bacteroidetes and Verrucomicrobia representing nearly 92% of all 241 077 high-quality sequences in the MiSeq dataset. Nearly 7% of sequences were not classified at the phylum level. Classification by phylotype produced 50 classes, 90 orders and 203 families. A class-level barplot indicated 15 abundant classes and particular abundance of Alphaproteobacteria and Gammaproteobacteria in all sample types (figure 2a). Surprisingly, at least 11 classes were present in high abundance in the Cryptostigma coccids, compared to only two classes in the Paraputo pseudococcids (see §3d, below).
Figure 2.

Taxonomic composition of (a) bacterial and (b) fungal communities for all sample types. Each bar corresponds to the pooled sequence counts for all replicates within a sample type. Coccidae (Myzolecaniinae: Cryptostigma) and Pseudococcidae (Pseudococcinae: Paraputo) are set apart in (a) for easier comparison of their distinct alpha diversity (see also the electronic supplementary material, figure S5). Note that nearly all of the sequences in the insects and approximately 50% of the sequences in the domatia that are classified by UNITE as ‘Ascomycota_unclassified’ provide good BLASTn hits to Chaetothyriales.
The 16S rRNA sequences were binned at 97% similarity into 5069 OTUs, including 1555 non-singletons. 0.6% of these OTUs clustered with sequences from the blank negative controls and represented potential reagent- or platform-derived contaminants (electronic supplementary material, table S2). Rarefaction analysis indicated that our sequence coverage was exhaustive in the case of non-singletons (electronic supplementary material, figure S3b)—except in the case of leaves and Cephalotes domatia, probably owing to low bacterial sequence counts and/or abundance, respectively (electronic supplementary material, figure S2a)—and the relative richness of the different sample types was very similar when singletons were included (electronic supplementary material, figure S3a; see also the electronic supplementary material, figure S4a). Bacteria found in Azteca ants and Cryptostigma coccids exhibited higher OTU richness that was less stable across individual replicates than the bacteria in Ce. setulifer ants or Paraputo pseudococcids (electronic supplementary material, figure S5). Indeed, Paraputo pseudococcids exhibited the lowest bacterial diversity among our sample types, whereas Azteca domatia exhibited particularly high diversity (electronic supplementary material, figure S6a). The core microbiome of Ce. setulifer ants strongly mirrored previous reports of core bacterial species common to the genus (electronic supplementary material, table S3; [10,12,13]).
(ii). Fungi
There were 17 identifiable fungal classes among all of our samples, with Dothideomycetes, Sordariomycetes, Agaricomycetes and Eurotiomycetes representing approximately 50% of all 139 646 high-quality sequences. Unclassified Ascomycota represented another approximately 28% of sequences, and nearly 18% of sequences were unclassified at the phylum level. Phylotypes at the order and family level produced 56 and 112 operational units, respectively. An order-level barplot indicated 18 abundant orders and particularly high abundance of Pleosporales and Chaetothyriales in all sample types, and in ants and ant domatia especially (figure 2b).
The fungal ITS rDNA sequences were binned at 97% similarity into 1602 OTUs, which included 1398 non-singletons. Rarefaction analysis indicated that our sequence coverage was exhaustive for all sample types, including for coccids, which had the fewest total sequences (electronic supplementary material, figure S3c,d). Fungal OTUs were more variable than bacteria among replicates within sample types (electronic supplementary material, figure S7). Leaves exhibited particularly high fungal diversity compared to the rest of the samples (electronic supplementary material, figure S6b).
Chaetothyriales comprised three of the five fungal OTUs with the highest relative abundance. These three OTUs produced BLAST hits at approximately 97% sequence identity to the so-called ‘domatia symbiont clade’ [39] (E-values: e-97). They were present in the domatia and the gasters of both Azteca and Ce. setulifer. The ninth most abundant fungal OTU was also Chaetothyriales but produced a BLAST hit at 100% sequence identity to a previously described domatia-carton-associated OTU from an ant-plant in Thailand [39]. This OTU was virtually absent from Azteca and Ce. setulifer gasters.
(c). Beta diversity of communities
Beta-diversity analyses were conducted on 2026 bacterial OTUs and 1092 fungal OTUs after subsampling at 1100 and 902 sequences per replicate, respectively. The NMDS ordinations revealed that the different sample types grouped much more tightly by their communities of bacteria (stress = 0.12) than by their communities of fungi (stress = 0.19) (figure 3; electronic supplementary material, figures S4b and S8). In fact, in the bacterial communities, the only two samples that exhibited any considerable overlap were the Cryptostigma coccids and the Azteca domatia. The Cephalotes ants and the Paraputo pseudococcids grouped the farthest apart from the rest of the samples, consistent with a role for highly specialized bacterial endosymbionts in these taxa. By contrast, there was substantial NMDS overlap among all samples in their fungal communities, with the sole exception of leaves. The leaves were composed of a very distinct community of endophytes and leaf pathogens (electronic supplementary material, table S4).
Figure 3.
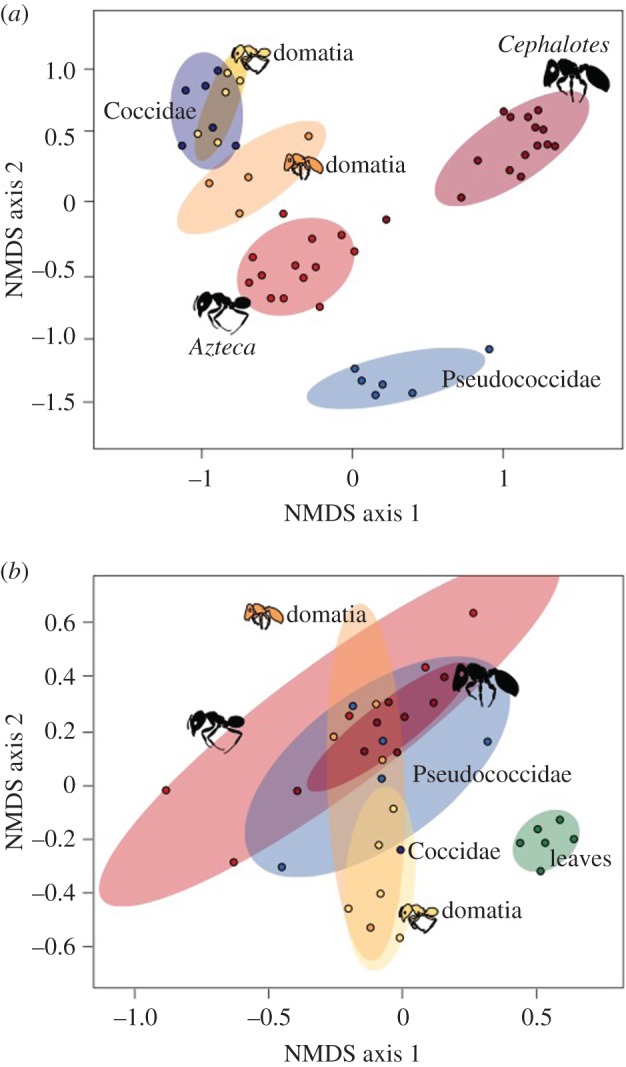
Nonmetric multidimensional scaling for (a) bacteria (stress = 0.12) and (b) fungi (stress = 0.19). Ellipses indicate 80% confidence intervals (CI). Leaves are not included in (a) because no leaf samples included ≥1100 bacterial sequences after chloroplast sequences were discarded. Note that the only Cephalotes bacterial sample that falls outside of its 80% CI (halfway towards Azteca) was the sample where DNA was extracted from only the midgut.
The Venn diagrams of OTU overlap between a given ant taxon and its environment suggested that, despite the overall separation in bacterial communities indicated in the NMDS analysis, there is leakage of individual bacterial OTUs among ants, their tended scale insects, and their domatia (figure 4a; electronic supplementary material, table S5 and figure S4c,d). The fungal Venn diagrams, by contrast, showed that despite extensive leakage between sample types of the most common fungal OTUs, which was also suggested by the NMDS, these shared OTUs comprised a relatively small proportion of the overall OTU richness: only 22% of all fungal OTUs were shared among samples for Azteca and 18% for Ce. setulifer (figure 4b; electronic supplementary material, table S6). Many fungal OTUs were unique to ants or their domatia, as well as, somewhat surprisingly, to the Paraputo pseudococcids tended by Ce. setulifer ants.
Figure 4.
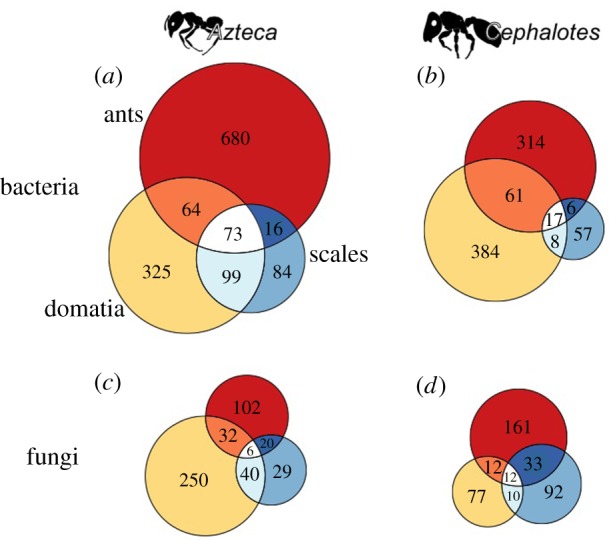
Venn diagrams of OTU overlap calculated separately for (a,c) Azteca and (b,d) Cephalotes microbial communities (see also the electronic supplementary material, tables S5 and S6). Ants are the top circles (red), domatia are the lefthand circles (yellow) and scale insects (Cryptostigma coccids and Paraputo pseudococcids) are the righthand circles (blue). For both bacteria (a,b) and fungi (c,d), the size of the circle is proportional to the number of OTUs present in the sample type. (Online version in colour.)
(d). Bacterial diversity in Cryptostigma coccids
The bacterial diversity in the Cryptostigma spp. coccids was unusually high for an insect in the Sternorrhyncha suborder of Hemiptera (figure 2a; electronic supplementary material, figure S5c). To probe this unexpected result further, we explored the diversity and function of these bacteria. In our dataset, the Paraputo pseudococcids exhibited much more typical diversity for Sternorrhyncha—93.5% of all sequences came from only two OTUs (figure 2a; electronic supplementary material, figure S5d). A search of the NCBI database revealed good BLAST hits to known pseudococcid endosymbionts, Tremblaya princeps (E-value e-121; Betaproteobacteria in figure 2a) and a Gammaproteobacteria from Dysmicoccus neobrevipes (E-value e-128; Gammaproteobacteria in figure 2a) [20,40] that presumably comes from the Sodalis-allied bacteria that have repeatedly replaced the symbiont within Tremblaya [21]. A third OTU (OTU00031) that made up 3.2% of sequences from our Paraputo pseudococcids (Proteobacteria_unclassified in figure 2a) did not produce BLAST hits to pseudococcid symbionts but instead to Sodalis-like uncultured symbionts from stinkbugs and beetles. In contrast to this low bacterial diversity in the Paraputo pseudococcids, our six samples of Cryptostigma spp. coccids contained 1175 bacterial OTUs, of which 192 were present in 50% or more of our samples (89% of sequences) and 41 were present in all six of our samples (31% of sequences) (electronic supplementary material, table S7).
Despite the striking differences in the composition of the Cryptostigma coccid and Paraputo pseudococcid bacterial communities, the functional characteristics of the bacterial metagenomes predicted to Level 2 KEGG orthologs by PICRUSt were remarkably similar (electronic supplementary material, figure S9). NSTI values ranged from 0.03 to 0.12. Well represented gene categories included those of presumed symbiotic importance, with prominent roles for amino acid and carbohydrate metabolism. Genes involved in the metabolism of secondary plant compounds were significantly more abundant in the Cryptostigma coccids, whereas genes involved in cellular and environmental processes were significantly more abundant in the Paraputo pseudococcids (electronic supplementary material, figure S9 and table S8).
4. Discussion
In this study, we examined how the overlap in environment and natural history of the insects in an ant–plant–hemipteran symbiosis affected the community composition of their associated symbiotic microbes. Consistent with our predictions, Ce. setulifer ants and Paraputo pseudococcids exhibited distinct, apparently specialized bacterial communities that were comparatively closed to environmental influence. In addition, based on the qPCR results, Azteca ants consumed (or associated with) significantly more fungus than Ce. setulifer ants, which may provide the Azteca ants with their required nitrogen in the absence of a specialized bacterial microbiome. Counter to our predictions, the Cryptostigma coccids exhibited an unexpectedly diverse bacterial community, and the Ce. setulifer ants were associated with many of the same fungi as the Azteca ants.
The bacterial communities of Azteca and Ce. setulifer posterior abdomens were very different despite the ants' similar ecology (sharing the same individual trees and subsisting at least in part on similar honeydew diets) and convergent adaptation to the Co. alliodora host tree (both A. pittieri and Ce. setulifer are Co. alliodora specialists [41,42]). Substantial evidence now suggests that Cephalotes ants have coevolved with their gut microbiome [10,12,13,43], and our Ce. setulifer ants reflected these previously reported patterns in gut-bacteria alpha-diversity (electronic supplementary material, table S3). The variance in bacterial diversity among Azteca replicates was also higher (electronic supplementary material, figures S5a,b and S6a) and the overall abundance of bacteria was lower (electronic supplementary material, figure S2b) than in Ce. setulifer, consistent with the hypothesis that the Azteca gut bacteria represent a more transient and less functionally important community than in Cephalotes [12].
What the Co. alliodora-associated Azteca lack in a stable bacterial gut community, however, may be compensated for by their consumption of (or association with) fungi, as indicated by the high abundance of fungal ITS sequences in Azteca posterior abdomens (electronic supplementary material, figure S2b). Recent studies have found intimate co-feeding relationships between Chaetothyriales fungi and plant-ants, in which the ants fertilize the fungi and also consume it [11,44]. In other ant–plant symbioses, there has been some evidence for distinct fungal communities associated with different ant species (e.g. [39]), although a recent study found evidence against codiversification between Azteca and their associated Chaetothyriales [22]. Here we found that Ce. setulifer and both Azteca species were associated with what appeared to be the same strains of Chaetothyriales at the 97% OTU level, showing a potentially strong effect of the ants' shared environment (host tree) on the identity of their fungal associations (electronic supplementary material, text S2). Consistent with the suggestion that the thinner cell walls of domatia-associated Chaetothyriales make them easier to digest than carton-associated taxa [39,45], our ant posterior abdomens contained OTUs from the ‘domatia-symbiont clade’ [39] but not from a carton-associated OTU. In all of our sample types, we also found abundant Pleosporales fungi, which, like Chaetothyriales, is thought to be saprotrophic in other contexts; the potential role of Pleosporales in ant–plant symbioses remains to be explored.
Despite the distinct compositions of the insects' symbiotic bacterial communities, several bacterial OTUs appeared to be shared among the ants, their tended scales and/or their domatia (figure 4a,b; electronic supplementary material, table S5). Although the levels of sharing of diverse OTUs supports this result overall (electronic supplementary material, tables S5 and S6), individual cases of shared OTUs need to be verified because of the possibility for cross-talk among multiplexed samples [46]. Two potential ecological pathways for microbes to be shared between ants and scale insects are: (i) if the ants are ‘farming’ the scale insects for meat, or (ii) if these bacteria are passed to the ants in low abundance via scale-insect honeydew, as has been shown for some gut-associated bacteria in aphids [15]. This kind of trophic microbial sharing could have nutritional or other effects on the consumers in ant–plant–hemipteran interactions [17].
Some surprising results emerged from our investigation of the Co. alliodora scale insects. First, the Paraputo pseudococcids exhibited unusually high ∂15N for herbivores (electronic supplementary material, text S3). Another unexpected result was that our Paraputo pseudococcids contained non-negligible quantities of fungi (electronic supplementary material, figure S2b) and a surprisingly high diversity of fungal OTUs (figure 4d; electronic supplementary material, figure S7d). It is not clear how the pseudococcids acquire these fungi or whether they play a functional role. Unlike the Paraputo pseudococcids, our Cryptostigma coccids contained few fungi, suggesting that these insects have very different microbial symbionts than their relatives in the Coccinae subfamily, which contain dense aggregations of lymph-associated and intracellular fungi [9]. Even more surprisingly, our Cryptostigma insects had very diverse bacterial associates, which, if accurate (electronic supplementary material, text S4 and figure S10), has not been documented for any other insect in the Sternorrhyncha, the suborder of Hemiptera whose members include scale insects, whiteflies, psyllids and aphids [47]. Two caveats, however, are: (i) that we used adult female coccids in all cases, and it can be difficult to know whether these insects are still alive without careful dissection (P. J. Gullan 2016, personal communication); and (ii) we do not know where these symbionts are located within the insect except that they were present after surface sterilization.
Although the metagenome prediction analysis conducted with PiCRUST is a coarse tool that should be interpreted with some caution, it too suggested possible symbiotic function of the Cryptostigma coccid bacteria. It is tempting to speculate that if the Myzolecaniinae are associated with diverse bacterial symbionts, this unusual symbiosis could be related to the insects' long, immobile period as adults. Immobility creates little if any chance to feed on different plant phloem sieve-tubes over time, and this stationary lifestyle may require the Myzolecaniinae to process many unusual and possibly toxic metabolites from the plant in its lifetime [48]. Indulging in this speculation, we note that some of the few gene functions that were more abundant in the Cryptostigma coccids compared to in the Paraputo pseudococcids were those related to the metabolism of plant secondary metabolites (electronic supplementary material, figure S9).
This study represents a first step towards a holistic understanding of how microbial symbionts are integrated with ecological interactions among macroorganisms. The enormous diversity of microbes in this ant–plant–hemipteran symbiosis (like, presumably, in most interactions among macroorganisms) presents a challenge for determining the interactions of functional relevance, but such complexity may be of profound importance to organismal ecology and evolution. Ants are emerging models for microbial study that also have wide-ranging effects on the functions of entire ecosystems, many of which are threatened by global change. Elucidating how ants interact in a microbial world is thus a necessary challenge.
Supplementary Material
Acknowledgements
We thank Brian Wray for his invaluable assistance in the laboratory and Alexandra Westrich for the ant drawings. We are grateful to Aileen Berastegui, Paul Dunlap, Penny Gullan, Timothy James, Takumasa Kondo, Manuela Ramalho and Benjamin Rubin for their analytical insights. The participants of both the 2015 Animal-Microbe Symbioses GRC and the 2016 Ants Symposium in Munich provided helpful feedback. The molecular portion of this research was completed in the Pritzker Laboratory for Molecular Systematics and Evolution at the Field Museum of Natural History, Chicago, Illinois.
Data accessibility
DNA sequences are available in GenBank (accessions: SRP095765). Code, OTU tables, taxonomy and representative sequences of 97% OTUs are deposited in Dryad (accession URL: http://dx.doi.org/10.5061/dryad.16830) [49].
Authors' contributions
E.G.P. and C.S.M. conceived the study; E.G.P. and C.S.M. designed the experiments; E.G.P. performed the experiments and analysed the data. E.G.P. wrote the initial manuscript, and C.S.M. contributed to revising the manuscript.
Competing interests
We declare we have no competing interests.
Funding
E.G.P. was supported by the Michigan Society of Fellows and start-up funds from the University of Nevada, Reno. C.S.M. was supported by the National Science Foundation (DEB-1442316 and IOS-1354193) and an anonymous donor.
References
- 1.Bronstein JL. 2015. Mutualism, 297 p. Oxford, UK: Oxford University Press. [Google Scholar]
- 2.Douglas AE. 2015. The special case of symbioses: mutualisms with persistent contact. In Mutualism (ed. Bronstein JL.), pp. 20–34. Oxford, UK: Oxford University Press. [Google Scholar]
- 3.Janzen DH. 1966. Coevolution of mutualism between ants and Acacias in Central America. Evolution 20, 249–275. ( 10.2307/2406628) [DOI] [PubMed] [Google Scholar]
- 4.Bronstein JL. 1998. The contribution of ant-plant protection studies to our understanding of mutualism. Biotropica 30, 150–161. ( 10.1111/j.1744-7429.1998.tb00050.x) [DOI] [Google Scholar]
- 5.Wilson EO, Hölldobler B. 2005. The rise of the ants: a phylogenetic and ecological explanation. Proc. Natl Acad. Sci. USA 102, 7411–7414. ( 10.1073/pnas.0502264102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davidson DW, Cook SC, Snelling RR, Chua TH. 2003. Explaining the abundance of ants in lowland tropical rainforest canopies. Science 300, 969–972. ( 10.1126/science.1082074) [DOI] [PubMed] [Google Scholar]
- 7.Davidson DW, McKey D. 1993. The evolutionary ecology of symbiotic ant-plant relationships. J. Hymenopt. Res. 2, 13–83. [Google Scholar]
- 8.Douglas AE. 2006. Phloem-sap feeding by animals: problems and solutions. J. Exp. Bot. 57, 747–754. ( 10.1093/jxb/erj067) [DOI] [PubMed] [Google Scholar]
- 9.Buchner P. 1965. Endosymbiosis of animals with plant microorganisms, 909 p. New York, NY: John Wiley & Sons, Inc. [Google Scholar]
- 10.Russell JA, Moreau CS, Goldman-Huertas B, Fujiwara M, Lohman DJ, Pierce NE. 2009. Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Natl Acad. Sci. USA 106, 21 236–21 241. ( 10.1073/pnas.0907926106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blatrix R, Djieto-Lordon C, Mondolot L, La Fisca P, Voglmayr H, McKey D. 2012. Plant-ants use symbiotic fungi as a food source: new insight into the nutritional ecology of ant-plant interactions. Proc. R. Soc. B 279, 3940–3947. ( 10.1098/rspb.2012.1403) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanders JG, Powell S, Kronauer DJC, Vasconcelos HL, Frederickson ME, Pierce NE. 2014. Stability and phylogenetic correlation in gut microbiota: lessons from ants and apes. Mol. Ecol. 23, 1268–1283. ( 10.1111/mec.12611) [DOI] [PubMed] [Google Scholar]
- 13.Hu Y, Lukasik P, Moreau CS, Russell JA. 2014. Correlates of gut community composition across an ant species (Cephalotes varians) elucidate causes and consequences of symbiotic variability. Mol. Ecol. 23, 1284–1300. ( 10.1111/mec.12607) [DOI] [PubMed] [Google Scholar]
- 14.Lanan MC, Rodrigues PAP, Agellon A, Jansma P, Wheeler DE. 2016. A bacterial filter protects and structures the gut microbiome of an insect. ISME J. 10, 1866–1876. ( 10.1038/ismej.2015.264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leroy PD et al. 2011. Microorganisms from aphid honeydew attract and enhance the efficacy of natural enemies. Nat. Commun. 2, 348 ( 10.1038/ncomms1347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabri A, Vandermoten S, Leroy PD, Haubruge E, Hance T, Thonart P, De Pauw E, Francis F. 2013. Proteomic investigation of aphid honeydew reveals an unexpected diversity of proteins. PLoS ONE 8, e74656 ( 10.1371/journal.pone.0074656) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henry LM, Maiden MCJ, Ferrari J, Godfray HCJ. 2015. Insect life history and the evolution of bacterial mutualism. Ecol. Lett. 18, 516–525. ( 10.1111/ele.12425) [DOI] [PubMed] [Google Scholar]
- 18.Pringle EG, Novo A, Ableson I, Barbehenn RV, Vannette RL. 2014. Plant-derived differences in the composition of aphid honeydew and their effects on colonies of aphid-tending ants. Ecol. Evol. 4, 4065–4079. ( 10.1002/ece3.1277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tillberg CV. 2004. Friend or foe? A behavioral and stable isotopic investigation of an ant-plant symbiosis. Oecologia 140, 506–515. ( 10.1007/s00442-004-1601-8) [DOI] [PubMed] [Google Scholar]
- 20.Husnik F, et al. 2013. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153, 1567–1578. ( 10.1016/j.cell.2013.05.040) [DOI] [PubMed] [Google Scholar]
- 21.Husnik F, McCutcheon JP. 2016. Repeated replacement of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. Proc. Natl Acad. Sci. USA 113, E5416–E5424. ( 10.1073/pnas.1603910113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nepel M, Voglmayr H, Blatrix R, Longino JT, Fiedler K, Schönenberger J, Mayer VE. 2016. Ant-cultivated Chaetothyriales in hollow stems of myrmecophytic Cecropia sp. trees - diversity and patterns. Fungal Ecol. 23, 131–140. ( 10.1016/j.funeco.2016.07.007) [DOI] [Google Scholar]
- 23.Pringle EG, Ramírez SR, Bonebrake TC, Gordon DM, Dirzo R. 2012. Diversification and phylogeographic structure in widespread Azteca plant-ants from the northern Neotropics. Mol. Ecol. 21, 3576–3592. ( 10.1111/j.1365-294X.2012.05618.x) [DOI] [PubMed] [Google Scholar]
- 24.Hothorn T, Bretz F, Westfall P. 2008. Simultaneous inference in general parametric models. Biom. J. 50, 346–363. ( 10.1002/bimj.200810425) [DOI] [PubMed] [Google Scholar]
- 25.R Core Team. 2016. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See https://www.R-project.org/. [Google Scholar]
- 26.Rubin BER, Sanders JG, Hampton-Marcell J, Owens SM, Gilbert JA, Moreau CS. 2014. DNA extraction protocols cause differences in 16S rRNA amplicon sequencing efficiency but not in community profile composition or structure. MicrobiologyOpen 3, 910–921. ( 10.1002/mbo3.216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caporaso JG, et al. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. ( 10.1038/ismej.2012.8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardes M, Bruns TD. 1993. ITS primers with enhanced specificity for Basidiomycetes—application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. ( 10.1111/j.1365-294X.1993.tb00005.x) [DOI] [PubMed] [Google Scholar]
- 29.Vilgalys R, Hester M. 1990. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 172, 4238–4246. ( 10.1128/jb.172.8.4238-4246.1990) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konietschke F, Placzek M, Schaarschmidt F. 2015. nparcomp: an R software package for nonparametric multiple comparisons and simultaneous confidence intervals. J. Stat. Softw. 64, 1–17. ( 10.18637/jss.v064.i09) [DOI] [Google Scholar]
- 31.Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS. 2008. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 8 125 ( 10.1186/1471-2180-8-125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schloss PD, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. ( 10.1128/aem.01541-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schloss PD, Gevers D, Westcott SL. 2011. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6, e27310 ( 10.1371/journal.pone.0027310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen NP, Warnow T, Pop M, White B. 2016. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2, 16004 ( 10.1038/npjbiofilms.2016.4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koljalg U, et al. 2013. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. ( 10.1111/mec.12481) [DOI] [PubMed] [Google Scholar]
- 36.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. ( 10.1128/aem.01043-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oksanen J, et al. 2016. vegan: community ecology package. R package version 24-0. https://CRAN.R-project.org/package=vegan .
- 38.Langille MGI, et al. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. ( 10.1038/nbt.2676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voglmayr H, Mayer V, Maschwitz U, Moog J, Djieto-Lordon C, Blatrix R. 2011. The diversity of ant-associated black yeasts: insights into a newly discovered world of symbiotic interactions. Fungal Biol. 115, 1077–1091. ( 10.1016/j.funbio.2010.11.006) [DOI] [PubMed] [Google Scholar]
- 40.Thao ML, Gullan PJ, Baumann P. 2002. Secondary (gamma-Proteobacteria) endosymbionts infect the primary (beta-Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Appl. Environ. Microbiol. 68, 3190–3197. ( 10.1128/aem.68.7.3190-3197.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longino JT. 1996. Taxonomic characterization of some live-stem inhabiting Azteca (Hymenoptera: Formicidae) in Costa Rica, with special reference to the ants of Cordia (Boraginaceae) and Triplaris (Polygonaceae). J. Hymenopt. Res. 5, 131–156. [Google Scholar]
- 42.Carroll CR. 1983. Azteca (hormiga azteca, Azteca ants, Cecropia ants). In Costa Rican natural history (ed. Janzen DH.), pp. 691–693. Chicago, IL: University of Chicago Press. [Google Scholar]
- 43.Anderson KE et al. 2012. Highly similar microbial communities are shared among related and trophically similar ant species. Mol. Ecol. 21, 2282–2296. ( 10.1111/j.1365-294X.2011.05464.x) [DOI] [PubMed] [Google Scholar]
- 44.Defossez E, Djieto-Lordon C, McKey D, Selosse MA, Blatrix R. 2011. Plant-ants feed their host plant, but above all a fungal symbiont to recycle nitrogen. Proc. R. Soc. B 278, 1419–1426. ( 10.1098/rspb.2010.1884) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayer VE, Voglmayr H. 2009. Mycelial carton galleries of Azteca brevis (Formicidae) as a multi-species network. Proc. R. Soc. B 276, 3265–3273. ( 10.1098/rspb.2009.0768) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nelson MC, Morrison HG, Benjamino J, Grim SL, Graf J. 2014. Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS ONE 9, e94249 ( 10.1371/journal.pone.0094249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jing XF, Wong ACN, Chaston JM, Colvin J, McKenzie CL, Douglas AE. 2014. The bacterial communities in plant phloem-sap-feeding insects. Mol. Ecol. 23, 1433–1444. ( 10.1111/mec.12637) [DOI] [PubMed] [Google Scholar]
- 48.Douglas AE. 2009. The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47. ( 10.1111/j.1365-2435.2008.01442.x) [DOI] [Google Scholar]
- 49.Pringle EG, Moreau CS. 2017. Data from: Community analysis of microbial sharing and specialization in a Costa Rican ant–plant–hemipteran symbiosis. Dryad Digital Repository. ( 10.5061/dryad.16830) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Pringle EG, Moreau CS. 2017. Data from: Community analysis of microbial sharing and specialization in a Costa Rican ant–plant–hemipteran symbiosis. Dryad Digital Repository. ( 10.5061/dryad.16830) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
DNA sequences are available in GenBank (accessions: SRP095765). Code, OTU tables, taxonomy and representative sequences of 97% OTUs are deposited in Dryad (accession URL: http://dx.doi.org/10.5061/dryad.16830) [49].