

Abstract
Aortic medial amyloid is the most prevalent amyloid found to date, but remarkably little is known about it. It is characterised by aberrant deposition of a 5.4 kDa protein called medin within the medial layer of large arteries. Here we employ a combined approach of ab initio protein modelling and 13C-direct detection NMR to generate a model for soluble monomeric medin comprising a stable core of three β-strands and shorter more labile strands at the termini. Molecular dynamics simulations suggested that detachment of the short, C-terminal β-strand from the soluble fold exposes key amyloidogenic regions as a potential site of nucleation enabling dimerisation and subsequent fibril formation. This mechanism resembles models proposed for several other amyloidogenic proteins suggesting that despite variations in sequence and protomer structure these proteins may share a common pathway for amyloid nucleation and subsequent protofibril and fibril formation.
The most common form of localised amyloid occurs in the aortic media (aortic medial amyloid; AMA) and is estimated to occur in 97% of Caucasian people above the age of 501. The pathological impact of AMA is unknown, but it is believed that amyloid accumulation contributes to age-related diminished elasticity of the vessels and that prefibrillar intermediates of medin may underlie the pathogenesis of sporadic thoracic aortic aneurysm2. The main constituent of AMA is a 50 amino acid polypeptide medin, thought to be cleaved from lactadherin3. Molecular information regarding the structure of medin, the mechanisms that trigger medin aggregation and the formation of the insoluble fibrils is not currently available.
Amyloid formation occurs through an assembly pathway often described simplistically in successive stages of nucleation followed by elongation4. The nucleation point represents the trigger for progression from a ‘non-amyloid or native’ conformation into an ‘amyloid-prone’ conformation. This transition is therefore a key event in the conversion from a functional protein into a pathologically relevant state that is responsible for disease initiation and progression. Molecular information about amyloid proteins in their monomeric, intermediate or fibrillar form and regarding their interaction and aggregation to form the insoluble fibrils is sparse. This is because amyloid proteins are notoriously difficult to study in their soluble forms due to their inherent propensity to aggregate along with the heterogeneous and transient nature of the key soluble intermediates. Structural bioinformatics techniques of modelling and molecular dynamics (MD) simulations provide tools to investigate the structure and stability of these critical protein species and were employed here to generate a model for the monomeric structure of medin.
Several approaches including NMR, have been applied to probe soluble structures and the role of intermediate conformations for a range of amyloid proteins. NMR is an invaluable tool to probe individual atomic nuclei in a macromolecule in solution and can be applied to investigate the molecular structure, dynamics and kinetics of proteins. To date, many solution state studies of amyloid proteins have utilised truncated constructs5,6 and/or non-physiological conditions, often employing structure-inducing additives, detergents or lipids which often contradict or bear little resemblance to structures thought to exist in the cell7,8,9,10,11. However recent advances in NMR techniques are enabling more studies to be carried out on these poorly tractable, but extremely biologically important aggregating systems12. In this work we used direct-detected 13C NMR to probe soluble medin structure employing conditions that are more physiologically relevant, avoiding stabilising components. The benefits of utilising 13C-direct detect NMR in this study are threefold; enabling backbone assignment in shorter experimental time than conventional 1H-detect triple resonance experiments, larger chemical shift dispersion and shorter transverse relaxation rates that enable the observation of resonances that relax too fast in conventional 1H-detect experiments. Although not providing data sufficient for 3D structure determination, the experiments supported a strong fold prediction resulting from ab initio structure modelling.
Previous literature on medin has identified the C-terminal portion of medin, in particular residues 42–49 as highly amyloidogenic. Short peptides composed only of these eight residues are capable of rapid aggregation into microcrystalline fibres3,13. Furthermore, removal of this C-terminal region from medin abolishes amyloid forming potential14. It is essential to identify and understand the interactions that occur early in the aggregation process that result in formation of amyloid fibrils. Here we use a combined experimental and computational approach to elucidate the early stages of medin nucleation, information that is critical for understanding of the initiation and progression of AMA and protein aggregation in general.
Results
Ab initio modelling
Models were generated online using QUARK15 and locally in two runs of ROSETTA16 using the sequence for medin (Fig. 1) obtained from UniProt17 (ID:Q08431). The QUARK and ROSETTA methods are broadly comparable in overall methodology, each assembling tertiary structures from structure fragments derived from the PDB, but differ significantly in some aspects such as the fragment libraries, which are fixed in length 3- and 9-residues in ROSETTA but are variable in QUARK and in their scoring functions. Consistency between the two methods would therefore enhance confidence in the results. The two ROSETTA runs varied in their fragment libraries: in one, fragments of full length lactadherin C2 domain crystal structures and homologous sequences were excluded (Fig. 1c, NHC). Although fragment assembly methods are based on a correlation between sequence and local secondary structure, we wished to test whether similar results were obtained when fragments of C2 domains - in which medin sequence is found in full length lactadherin - were excluded from the model building, thereby avoiding any bias towards the local conformations adopted by medin in its C2 domain context.
Figure 1. Ab initio fragment assembly protein modelling of human monomeric medin.

Medin models generated online using QUARK (a) and locally in two runs of ROSETTA including (b) or excluding (c) fragments deriving from structures of lactadherin C2 domain and homologues (NHC). β-sheet positioning in the three models (d). Secondary structure assignment was performed using DSSP64 as throughout this work.
The results of all three modelling efforts agree to a remarkable degree (Fig. 1) and are also consistent, as expected, with secondary structure predictions (Fig. S1). Figure 1 shows the most favoured model returned by QUARK (a) alongside representatives of the largest clusters from the ROSETTA runs (b and c). In each, medin forms a four-stranded β-sheet (d). Terminal regions vary somewhat in conformation but the central portion from residues 21–35 consistently folds into a β-hairpin. This hairpin is also present in all the returned QUARK models, as well as smaller, less-favoured clusters of structures from ROSETTA modelling despite differences in terminal regions and is therefore a strongly predicted feature of the models. We have previously suggested that a similar β -hairpin may be present in insoluble medin fibrils18. Searches of the PDB revealed the nearest structural neighbours of QUARK and ROSETTA models to be small 3- or 4-stranded β-sheet structures in, respectively, the WW domain of human PRPF40A and Bacillus subtilis YmzC protein (Fig. S2). The folding pathways of WW domains have been extensively studied and implicated in amyloid protofibril generation19,20,21,22. A similar triple strand monomer structure has also been observed in MD simulations on amyloid-β23,24 suggesting that this strand arrangement may be conserved across different amyloid-prone proteins.
13C-direct detection NMR
Fragment assembly ab initio modelling methods have developed to a point that their overall performance on small proteins is good25 yet the reliability of their results varies and final model quality is not as predictable as homology modelling. Validation of key model characteristics was therefore sought using solution state NMR. Due to the highly dynamic and often rapid aggregation kinetics of amyloid proteins, obtaining structural information about the soluble early stages of aggregation presents a considerable challenge. In this study we employed a variety of measures to obtain the first structural information about medin in its soluble form. Previously we assigned H-N backbone resonances of medin using a combination of fast-pulsing NMR experiments and 3D triple resonance experiments acquired under denaturing conditions26 but were unable to assign the other backbone atoms required to elucidate secondary structure in non-denaturing conditions. Conventional 3D experiments were unfeasible due to the high protein concentration required to maintain sensitivity during a 3D pulse sequence, (under these concentrations medin aggregates rapidly within a few hours) coupled with the coherence transfer efficiency required in the longer coherence transfers of 3D pulse sequence. In contrast, 13C-direct detection experiments use shorter pulse sequences to reduce the loss of signal from low concentration samples. In addition the length of time required for acquisition at sufficient resolution for chemical shift dispersion between 13C signals, even using fast-pulsing methods was unobtainable due to the inherent instability of medin in its soluble form. As an alternative we sought to reduce dimensionality (and thus coherence transfer steps) and increase chemical shift dispersion using 2D 13C direct detection experiments (Fig. 2a), enabling the use of lower protein concentrations and increasing the available window of study before aggregation. Moreover, all experiments were collected at 288 K to further slow the aggregation process and extend the acquisition window. SOFAST 1H15N HMQC experiments were interleaved throughout all experiments to monitor aggregation (Fig. S3).
Figure 2. NMR data overview.

(a) Schematic representation of the NMR workflow incorporating the use of 13C-direct detection experiments. (b) CON spectrum of human medin collected at 288 K on a Bruker Avance III NMR spectrometer operating at 14.1T using a TXO probe. (c) SSP27 output using C’ and Cα shifts for medin. (d) Structural propensity of backbone ΔC’, ΔCα and ΔHα chemical shifts towards either α-helix or β-strand from random coil calculated using ncIDP with reference chemical shift libraries as shown (Tamiola et al.28, Wang and Jardetsky76, and Schwarzinger et al.77) and CCPN29. Structural propensities of 10% or more are shown and summarised in Table S2. Light grey regions indicate areas of secondary structure from computational modelling as shown in Fig. 1.
CON and CACO experiments yielded good chemical shift dispersion with adequate signal/noise ratios (Fig. 2b). Using this strategy it was possible to assign 98% NH, 98% HN, 98% C’, 96% Cα and 94% Hα shifts (see BMRB ID: 27021). These shifts were used to probe secondary structure propensities using Secondary Structure Propensity (SSP)27, neighbour corrected Intrinsically Disordered Protein Library (ncIDP)28 and the Collaborative Computing Project for NMR (CCPN) software Analysis29. SSP provides a score for secondary structure propensity for carbon shifts with negative values indicating β-strand and positive values α-helix. The backbone chemical shifts of medin indicate three areas of β-strand (residues 8–14, 21–23 and 39–50) with the C-terminal region suggesting up to 30% structural propensity (Fig. 2c). Deviation in chemical shift for medin when compared to random coil values from different chemical shift libraries including those which correct for sequence specificity was calculated using ncIDP. Structural propensities greater than 10% were observed from the ΔC’, ΔCα and ΔHα chemical shifts, with ΔNH and ΔHN not considered due to the susceptibility of HN chemical shifts to be sensitive to temperature30 and thus may produce spurious deviations from library values (Fig. 2d and Table S2)31. In addition, CCPN analysis of secondary structure propensity was also carried out (Fig. 2d). Overall the secondary structure analysis suggests medin has a propensity for β-strand elements (Fig. 2c,d) in good agreement with the positioning of the strands in the ab initio models (Fig. 1).
Chemical shift data was used to investigate the flexibility of the protein backbone using the random coil index (RCI)32 within the CSI 2.0 server33. Results showed increased flexibility in regions centred on residues 10 and 30, and at the C-terminus (Fig. 3). MD simulations performed on the QUARK model also indicated increased flexibility at the C-terminus and around residue 30; however, the root mean square fluctuations (RMSF) profiles are dominated by flexibility at the N-terminus which is not reflected in the RCI calculated from the NMR data (Fig. 3). It should be noted that the RCI values for the N-terminal region must be considered intrinsically less reliable than those of the rest of the protein due to the relative paucity of assignments in this region (BMRB ID: 27021).
Figure 3. Comparison of medin backbone dynamics using random coil index and root mean square fluctuation.
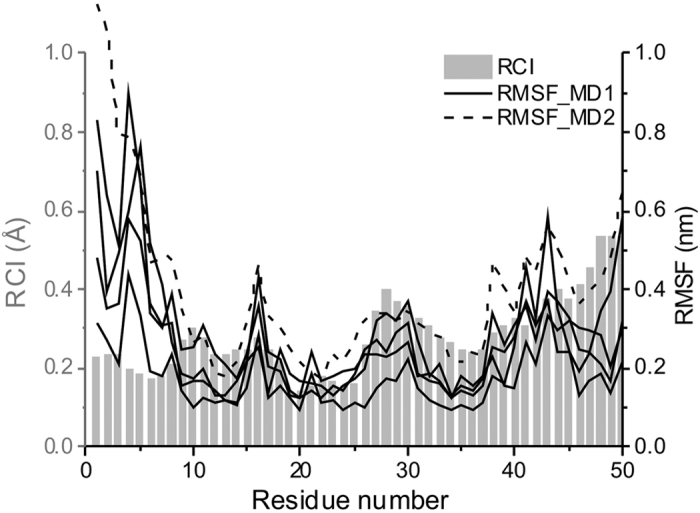
All atom RMSF data from OPLS-AA/L force field MD simulations (four independent trajectories)(MD1) and AMBER99SB force field with the TIP3P water model (MD2) performed on the top QUARK ab initio model are shown as black lines, solid and dashed respectively. RCI data derived from the NMR chemical shift data using the online server CSI 2.033.
Molecular Dynamics simulations
We used MD simulations with the OPLS-AA/L force field and the SPC/E water model to investigate the stability of the medin model, the potential role of the C-terminus in amyloid formation and to propose a possible mechanism for amyloid initiation. Four independent MD simulations were ran using GROMACS (Fig. 3, solid lines). They indicated good overall model stability with the three central β-strands persisting throughout all simulations as indicated by the DSSP analysis (Figs 4a and S4). Interestingly, in one of the simulations the C-terminal region, initially forming the fifth β-strand, adopts a flexible random coil conformation at around 180 ns of the simulation, coincident with dissociation of the region from the central β-sheet. To explore the dynamics of this region further this trajectory was extended to 1 μs in which time repeated events of dissociation and re-association of this C-terminal region were observed (Fig. S5). A subset of the conformations sampled in which the region was detached from the main β-sheet are shown in Fig. 4c. In some frames the C-terminal strand disengages but remains nearby; in others, such as the 185 ns frame, the strand moves far from central sheet (Fig. 4d). This key observation was replicated in a 1 μs trajectory of the same system carried out with the AMBER99SB force field and the TIP3P water model. Although still a relatively rare event, the C-terminal strand repeatedly detached and reattached from the core 3-strand β-sheet (Figs S5 and S6). This trajectory also reproduced the RMSF profiles of the initial MD runs (Fig. 3, dashed line).
Figure 4. Instability of the C-terminal strand is predicted to trigger amyloidogenesis.

(a) Secondary structure matrix of the initial OPLS-AA/L force field MD trajectory in which C-terminal strand detachment was observed. Snapshots sampled every 250ps are colour coded according to secondary structure as assigned by DSSP. Arrows on the left indicate secondary structure (β-strands) present in the starting model. (b) Starting QUARK model used in the simulations. (c) Ensemble of structures at 185, 310, 400, 600 and 900 ns from the MD simulation shown in (a) in which the C-terminal strand is detached from the central sheet. (d) Snapshot of medin at 185 ns illustrating loss of the C-terminal strand. (e) Per-residue aggregation propensity prediction data for the starting model (left) and the 185 ns snapshot (right) generated by Aggrescan3D34. Blue indicates higher aggregation propensity (>0), and grey lower values (<0).
The apparent discrepancy between lifetime of secondary structure elements in the MD simulations (strands retained throughout the majority of the simulations, Figs S4 and S5) and the subtle NMR chemical shift deviation from random coil towards β-strand (between 10% and 20% typically, Fig. 4c,d and Table S2) are reflective of the differing sampling rates of the two techniques. The chemical shift measured is an average of the conformational space sampled over micro to milliseconds thus the secondary structure elements visible in the structural models may be averaged over the NMR HSQC timescale.
Probing the instability of the C-terminal strand
The Aggrescan3D server34 was used to investigate how detachment of this C-terminal strand could affect the aggregation potential of the structure (Fig. 4e). Overall, the 185 ns structure following C-terminal detachment showed an increase in amyloid propensity compared to the starting structure with total score values of −2.00 and −29.87 respectively. This difference is larger than that between wild-type β2-microglobulin with low amyloid propensity at neutral pH (−73.54) and its well-characterised amyloidogenic ΔN6 variant which aggregates at neutral pH (−52.32)34. Furthermore, the total score of medin following C-terminal detachment (−2.00) is greater than that of the highly amyloidogenic β2-microglobulin variant ΔN6 (−52.32), suggesting an extremely high propensity for medin to self-assemble.
Aggrescan3D showed that the first and third β-strands, along with the C-terminal tail region exhibited increased amyloidogenicity following C-terminal detachment (Fig. 4e). Flexibility observed in the C-terminal region exposes two key regions of the medin sequence that are predicted to be highly amyloidogenic and were previously shown to form fibrils instantly as isolated peptides (residues 32–41 and 42–50)14,35. Larsson et al., also investigated fibrillation of the longer peptide 31–50 which showed slower fibril formation than the smaller peptide fragments, possibly due to rearrangement and dissociation of latent strands that may be present. In addition prediction servers also suggest the N-terminal strand in our model to be amyloidogenic36: at the 185 ns time-point, for example, this strand is fully exposed (Fig. 4e).
Discussion
Medin is a particularly demanding protein to study under physiological conditions since it aggregates in the typical time window and concentrations required for the majority of structural techniques. To address this challenge and obtain a structure of monomeric medin we employed two complementary techniques. Fragment assembly ab initio protein modelling was first used to predict a tertiary structure for monomeric medin. This was followed by 13C-direct detection NMR to rapidly obtain CSI and RCI information that together revealed secondary structure information in good agreement with the modelling data. We then investigated the stability of the generated structure and proposed a mechanism for nucleation of medin association to form dimers as a nucleation point for fibrillation.
In all models generated from bioinformatic methods and NMR data the key structural elements of a core consisting of three central strands incorporating residues N7-A13, W21-D25 and K30-I36 are well maintained. Other less favored fold predictions still maintain the key strands with differences at the termini suggesting a lack of stable structure at the termini. We utilized MD simulations to probe this hypothesis and to test the stability of the proposed structural model for medin. MD simulations showed that the C-terminus was prone to detachment from the central core which we hypothesize to be required for initiation of amyloidogenesis.
Previous studies investigating the structure of medin using circular dichroism (CD) spectroscopy suggested that medin was a mixture of random coil and β-sheet37, with approx. 30% β-sheet18,38. NMR data presented here also suggests a propensity for β-sheet (Fig. 2c,d). Results from intrinsic disorder prediction servers suggest that medin is predominantly ordered, with only the termini predicted to be disordered Fig. S7. This is consistent with RMSF and RCI data showing increased flexibility in the N-terminus (Fig. 3), and MD simulations suggesting flexibility in the C-terminus (Fig. 4). This is comparable to most human proteins that are reported to have some disordered residues within their terminal regions39, with 97% having predicted disorder in the N- or C-terminal residues40. We propose that medin has an intrinsic propensity to adopt β-sheet structure, and transiently populates aggregation-prone conformations with a β-sheet core in which the highly amyloidogenic C-terminal residues initiate nucleation and amyloid growth. The transient nature of the strand-detached amyloidogenic conformation is demonstrated throughout the 1 μs MD simulation (Fig. S5) where although β-sheet is the predominant state of the C-terminal residues from 47–49, there are episodes where this strand is completely or partially detached and subsequently reanneals.
Here we propose that the detachment of the C-terminal strand and subsequent movement away from the central strands may act as the initiating event in self-assembly. Movement of the C- terminal strand opens up possibilities for dimerisation as shown in Fig. 5. We present six arrangements for dimerisation of medin involving association of the key amyloidogenic regions identified above, which maintain the β-sheet arrangement of strand two (W21-D25) and strand three (K30-I36). Four of the orientations assemble via strand three (green) pairing with either strand one (blue) or three (green) of a neighbouring molecule in either a parallel or antiparallel orientation (Fig. 5, models 1–4). Multiple studies reveal models for self-association of transthyretin41, insulin42, mutant β2-microglobulin43 and a WW domain from formin binding protein 2844 all favour an antiparallel orientation of the same strand (as depicted in model 4, Fig. 5). In the latter work a detachment of one strand from the monomeric structure is required for fibril initiation as posited here for medin.
Figure 5. Schematic representation of possible medin dimers.
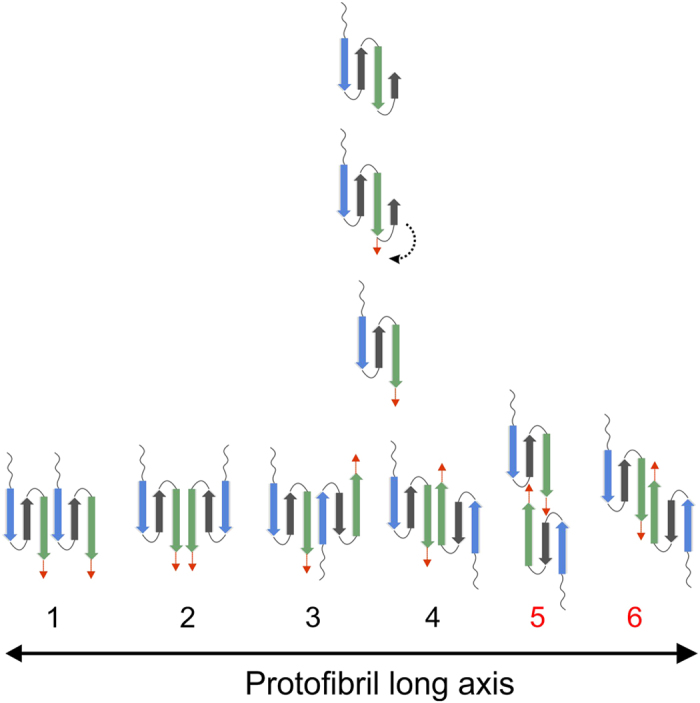
Cartoon illustration of proposed C-terminal strand detachment and subsequent dimer formation through interaction of the amyloidogenic first and third strands (blue and green respectively) as defined by Aggrescan3D (see Fig. 4e). Options 1–4 could involve assembly through pre-formed strands; blue strand (N7-A13) and green strand (K30-I36) with optional involvement of the C-terminal residues 36–50 (red arrow), whereas options 5 and 6 requires stabilisation by the ‘zipping back’ of the C-terminus to form a longer strand encompassing residues 30–50 (green strand and red arrow).
The detached C-terminal strand provides further options for dimer formation through interaction of this region with the same portion of another monomer (Fig. 5, model 5), or extension of strand three to the terminus (Fig. 5, model 6). Model 5 shares some similarities with a model recently proposed for β2-microglobulin45. Model 6 is reminiscent of domain swapped dimers observed for immunoglobulin light chains46, prion proteins47, β2-microglobulin48,49 and other proteins50. In Fig. 5 dimerisation scenarios are shown with strand three initiating dimerisation, where subsequent monomer/dimer addition would involve strand one self-associating in order to extend the sheet. Although there is no experimental confirmation for which strand initiates dimerisation, several structural observations support our hypothesis.
We propose that the C-terminal region is important for initiating aggregation by first moving away from the sheet to expose key amyloidogenic regions and then engaging in amyloid formation, potentially ‘zipping’ back from the C terminus to extend the β-strand along the entire sequence to include residues 30–50 as shown in the dimer models, Fig. 5. Aromatic interactions are often implicated in amyloid assembly through π-π stacking51. The proposed structural model for medin positions the aromatic residues (W11, W22) within the core β-sheet region (Fig. S8) in a manner that would allow for stacking within and between sheets. In addition phenylalanine residues previously implicated in assembly of medin52,53 are readily accessible within flexible regions (F8 in the flexible N-terminus, F42 and F49 in the extended C-terminus (red region in Fig. 5)), and could assist in driving the extension of the C-terminal β-strand as indicated in Figs 4 and 5. In addition, we recently showed that medin can be nitrated at positions Y16 and W11/W22 with subsequent alterations in aggregation properties38. The model proposed here is consistent with all of these sites being accessible for modification altering the ability of the protein to assemble through intermolecular facial β-sheet interactions (Fig. S8).
Furthermore, the models presented here are consistent with formation of a β-hairpin and an extended C-terminal β-sheet as described in our previous fibrillar model for medin using solid state NMR18. The fibrillar model was proposed based on solid state NMR data suggesting medin fibrils are composed of a mixture of β-sheet and random coil, the possibility of stabilisation of the hairpin with a salt-bridge between D25 and K30, and an interaction between I35 or I36 and W11 or W21. This study probing soluble medin and early stages in aggregation initiation has autonomously generated models that are consistent with the experimental findings of the previous work on stable mature amyloid-like fibres (see Fig. S8), suggesting that structural rearrangement of the protofibrils could occur to produce mature fibrils with extended β-sheet structure.
We present here the first model for the amyloidogenic protein medin in its transient monomeric form determined using a complementary approach of structural bioinformatics and NMR. The concept of a protective loop shielding aggregation prone regions has also been proposed for other self-assembling proteins54,55 and as a method of ‘negative design’ to prevent aggregation56. This mechanism of the loop moving out of the way to allow dimerisation resembles models previously proposed for several other amyloidogenic proteins suggesting that despite variations in sequence and initial structure, these proteins may share a common pathway for amyloid nucleation and subsequent protofibril and fibril formation.
Methods
Structure prediction
A consensus secondary structure prediction for medin, to which 18 different methods contributed, was obtained from the Genesilico Metaserver57 (Fig. S1). The structure of soluble monomeric medin was predicted by fragment assembly ab initio methods using both the QUARK server15 and locally installed ROSETTA16,58. The QUARK server returns ten models, each a representative of a cluster of similar structures found within the modelled set of predictions. Likewise, 5000 predictions were generated and clustered with ROSETTA, and cluster representatives identified. These cluster representatives are considered candidate fold predictions59. The 3- and 9-residue fragment libraries required for ROSETTA were obtained from the ROBETTA server60. Libraries of fragments were generated with (NHC) or without the exclusion of homologous sequences i.e. to exclude (NHC) or not fragments of the full length lactadherin C2 domain sequence and homologues. ROSETTA modelling was done with each set of libraries and the results compared. A secondary structure prediction in PSIPRED file format with reliability scores is also required for Rosetta. The prediction from the PSIPRED server61,62, matching closely the consensus prediction which in turn which agreed well with experimental data, was used. THESEUS63 was used for superposition of structures and PyMOL (http://www.pymol.org) for their visualisation. Secondary structure within the models was detected using DSSP64 and STRIDE65 which agreed exactly in their specification of β-strands. eFOLD66 was used to search the Protein Data Bank (PDB)67 for structural neighbours of the models.
Molecular Dynamics
The stability and dynamic properties of the top QUARK model were initially explored using the GROMACS package, release 5.0.668 in conjunction with the OPLS-AA/L all-atom force field69. The model was placed in a cubic box of water so that it was at least 1 nm from any edge. With amino acid protonation states assigned for pH 7, the system was neutral so no counterions were added. The SPC/E water model was used70. The system was then energy minimised using the steepest descents algorithm. Four independent 200 ns trajectories with different starting velocities were then calculated, with one extended to 1 μs. A further 1 μs trajectory was generated using the AMBER99SB force field71 and the TIP3P water model72. For long-range electrostatic terms, the Particle-Mesh Ewald (PME) algorithm73 was used with non-bonded and van der Waals cut-offs of 1 nm. The LINCS algorithm74 was used to constrain all bond lengths. The temperature of the simulations was set to 310.15 K to represent the physiological temperature at which medin aggregation occurs. A pressure of 1bar was used with an integration time step of 2fs and periodic boundary conditions applied. Programs of the GROMACS suite were also used to calculate per-residue, all atom RMSF during the trajectories and to characterise the variation in secondary structure visible through the trajectory by DSSP64 assignments.
NMR methods
13C, 15N uniform isotope labelled medin was expressed as previously described38. Medin concentration was adjusted to 80 μM in 20 mM sodium phosphate (NaPhos), 20 mM sodium chloride (NaCl) pH 6.5, containing 10% 2H20 for all NMR experiments. All solution-state NMR experiments were performed at 288 K to reduce the rate of medin aggregation. Two dimensional 13C-direct detected (CON and CACO) spectra were acquired on a Bruker AVANCE III 600 MHz spectrometer equipped with an observe 5 mm triple resonance (TXO) cryoprobe optimised for 13C detection (Wellcome Trust NMR facility at the University of Birmingham). All other experiments were collected at the University of Liverpool Centre for Structural Biology on a Bruker AVANCE III 800 MHz spectrometer equipped with an inverse 5 mm triple resonance (TCI) cryoprobe. A schematic of the experimental workflow used for backbone assignment is detailed in Fig. 2a. In brief, 2D CON and CACO 13C-direct detected experiments75 were recorded alongside 1H detected 2D experiments to establish 1HN, 13Cα, 13CO, 1HCα and 15NH assignments. Pulse sequences and parameter sets for these experiments are detailed in Table S1. 1H-15N Band-selective optimised flip-angle short-transient heteronuclear multiple quantum coherence (SOFAST HMQC) experiments were interleaved throughout to monitor the effect of aggregation as described previously26. Fast pulsing methods were not employed for 13C-direct detection due to the inherent low sensitivity of the technique. Spectra were processed using Topspin 3.1 (Bruker) and assignment and secondary structure analysis carried out using CCPN software Analysis29. The assigned chemical shifts were submitted to CSI 2.033 server to interrogate backbone dynamics. The shift between medin backbone assignment and predicted random coil chemical shift were calculated for C’, Cα and Hα using three reference libraries (Tamiola et al.28, Wang and Jardetsky76, and Schwarzinger et al.77) within ncIDP and secondary structure propensities calculated using this data and independently from SSP (C’ and Cα shifts)27. Propensities were calculated to be 10, 20 or 50% towards either α-helix or β-strand from random coil chemical shift values as reported in Wishart and Sykes31 when two out of the three backbone shifts were in agreement. Strands and helices were inferred when three or more consecutive residues have greater than 10% structure propensity. The chemical shifts have been deposited in the BioMagRes-Bank accession No 27021.
Additional Information
How to cite this article: Davies, H. A. et al. Probing Medin Monomer Structure and its Amyloid Nucleation Using 13C-Direct Detection NMR in Combination with Structural Bioinformatics. Sci. Rep. 7, 45224; doi: 10.1038/srep45224 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the British Heart Foundation (JM/HAD, FS/12/61/29877). We thank Dr Sara Whittaker for assistance at the Wellcome Trust NMR facility at the University of Birmingham.
Footnotes
The authors declare no competing financial interests.
Author Contributions Conceptualisation, J.M. and H.A.D.; Methodology, H.A.D., D.J.R. and M.M.P.; Investigation, H.A.D. and D.J.R.; Writing – Original Draft, J.M., H.A.D., D.J.R. and M.M.P.; Writing – Review and Editing, J.M. and H.A.D.; Funding Acquisition, J.M.
References
- Mucchiano G., Cornwell G. G. I. & Westermark P. Senile aortic amyloid. Evidence for two distinct forms of localized deposits. Am. J. Pathol. 140, 871–877 (1992). [PMC free article] [PubMed] [Google Scholar]
- Peng S. et al. Role of aggregated medin in the pathogenesis of thoracic aortic aneurysm and dissection. Lab. Invest. 87, 1195–1205 (2007). [DOI] [PubMed] [Google Scholar]
- Haggqvist B. et al. Medin: An integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proc. Nat. Acad. Sci. USA 96, 8669–8674 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. D. & Lansbury P. T. Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Ann. Rev. Biochem. 66, 385–407 (1997). [DOI] [PubMed] [Google Scholar]
- Talafous J., Marcinowski K. J., Klopman G. & Zagorski M. G. Solution structure of residues 1–28 of the amyloid-beta peptide. Biochem. 33, 7788–7796 (1994). [DOI] [PubMed] [Google Scholar]
- Zhang S. et al. The Alzheimer’s peptide A-beta adopts a collapsed coil structure in water. J. Struct. Biol. 130, 130–141 (2000). [DOI] [PubMed] [Google Scholar]
- Crescenzi O. et al. Solution structure of the Alzheimer amyloid beta peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 269, 5642–5648 (2002). [DOI] [PubMed] [Google Scholar]
- Tomaselli S. et al. The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of beta conformation seeding. ChemBioChem 7, 257–267 (2006). [DOI] [PubMed] [Google Scholar]
- Nanga R. P., Brender J. R., Vivekanandan S. & Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta 1808, 2337–2342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S. M., Xu S., Sheftic S. R. & Alexandrescu A. T. Dynamic alpha-helix structure of micelle-bound human amylin. J. Biol. Chem. 284, 11982–11991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer T. S., Bax A., Cole N. B. & Nussbaum R. L. Structure and dynamics of micelle-bound human a-synuclein. J. Biol. Chem. 280, 9595–9603 (2005). [DOI] [PubMed] [Google Scholar]
- Karamanos T. K., Kalverda A. P., Thompson G. S. & Radford S. E. Mechanisms of amyloid formation revealed by solution NMR. Prog. Nucl. Magn. Reson. Spectrosc. 88–89, 86–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madine J., Copland A., Serpell L. C. & Middleton D. A. Cross-b spine architecture of fibrils formed by the amyloidogenic segment NFGSVQFV of medin from solid-state NMR and X-ray fiber diffraction measurements Biochem. 48, 3089–3099 (2009). [DOI] [PubMed] [Google Scholar]
- Larsson A. et al. Unwinding fibril formation of medin, the peptide of the most common form of human amyloid. Biochem. Biophys. Res. Commun. 361, 822–828 (2007). [DOI] [PubMed] [Google Scholar]
- Xu D. & Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl C. A., Strauss C. E., Misura K. M. & Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 383, 66–93 (2004). [DOI] [PubMed] [Google Scholar]
- UniProt: The universal protein knowledgebase. Nucleic Acids Res. 45, D158–d169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. A., Madine J. & Middleton D. A. Comparisons with amyloid-beta reveal an aspartate residue that stabilizes fibrils of the aortic amyloid peptide medin. J. Biol. Chem. 290, 7791–7803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisuradze G. G. et al. Preventing fibril formation of a protein by selective mutation. Proc. Natl. Acad. Sci. USA 112, 13549–13554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson N. et al. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA 103, 16248–16253 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager M. et al. The folding mechanism of a beta-sheet: the WW domain. J. Mol. Biol. 311, 373–393 (2001). [DOI] [PubMed] [Google Scholar]
- Nguyen H. et al. Tuning the free-energy landscape of a WW domain by temperature, mutation, and truncation. Proc. Nat. Acad. Sci. USA 100, 3948–3953 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul J. A. & Bevan D. R. The role of molecular simulations in the development of inhibitors of amyloid beta-peptide aggregation for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 3, 845–856 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melquiond A., Dong X., Mousseau N. & Derreumaux P. Role of the region 23–28 in Abeta fibril formation: insights from simulations of the monomers and dimers of Alzheimer’s peptides Abeta40 and Abeta42. Curr. Alzheimer Res. 5, 244–250 (2008). [DOI] [PubMed] [Google Scholar]
- Kinch L. N. et al. Evaluation of free modeling targets in CASP11 and ROLL. Proteins 84, 51–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. A., Phelan M. M. & Madine J. 1H, 15N and 13C assignment of the amyloidogenic protein medin using fast-pulsing NMR techniques. Biomol. NMR Assign. 10, 75–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A., Singh V. K., Jia Z. & Forman-Kay J. D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: Implications for fibrillation. Protein Sci. 15, 2795–2804 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiola K., Acar B. & Mulder F. A. A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Che. Soc. 132, 18000–18003 (2010). [DOI] [PubMed] [Google Scholar]
- Vranken W. F. et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- Merutka G., Jane Dyson H. & Wright P. E. ‘Random coil’ 1H chemical shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J. Biomol. NMR 5, 14–24 (1995). [DOI] [PubMed] [Google Scholar]
- Wishart D. S. & Sykes B. D. Chemical shifts as a tool for structure determination. Methods Enzym. 239, 363–392 (1994). [DOI] [PubMed] [Google Scholar]
- Berjanskii M. V. & Wishart D. S. A simple method to predict protein flexibility using secondary chemical shifts. J. Am. Chem. Soc. 127, 14970–14971 (2005). [DOI] [PubMed] [Google Scholar]
- Hafsa N. E. & Wishart D. S. CSI 2.0: a significantly improved version of the Chemical Shift Index. J. Biomol. NMR 60, 131–146 (2014). [DOI] [PubMed] [Google Scholar]
- Zambrano R. et al. AGGRESCAN3D (A3D): server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 43, W306–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. A., Madine J. & Middleton D. A. Solid-state NMR reveals differences in the packing arrangements of peptide aggregates derived from the aortic amyloid polypeptide medin. J. Pep. Sci. 18, 65–72 (2011). [DOI] [PubMed] [Google Scholar]
- Gursky O. & Aleshkov S. Temperature-dependent beta-sheet formation in beta-amyloid Abeta(1–40) peptide in water: uncoupling beta-structure folding from aggregation. Biochim. Biophys. Acta 1476, 93–102 (2000). [DOI] [PubMed] [Google Scholar]
- Olofsson A., Borowik T., Gröbner G. & Sauer-Eriksson A. E. Negatively charged phospholipid membranes induce amyloid formation of medin via an alpha-helical intermediate. J. Mol. Biol. 374, 186–194 (2007). [DOI] [PubMed] [Google Scholar]
- Davies H. A. et al. Oxidative stress alters the morphology and toxicity of aortic medial amyloid. Biophys. J. 109, 2363–2370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. The most important thing is the tail: multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 587, 1891–1901 (2013). [DOI] [PubMed] [Google Scholar]
- Pentony M. M. & Jones D. T. Modularity of intrinsic disorder in the human proteome. Proteins 78, 212–221 (2010). [DOI] [PubMed] [Google Scholar]
- Schormann N., Murrell J. R. & Benson M. D. Tertiary structures of amyloidogenic and nonamyloidogenic transthyretin variants: new model for amyloid fibril formation. Amyloid 5, 175–187 (1998). [DOI] [PubMed] [Google Scholar]
- Whittingham J. L. et al. Insulin at pH 2: structural analysis of the conditions promoting insulin fibre formation. J. Mol. Biol. 318, 479–490 (2002). [DOI] [PubMed] [Google Scholar]
- Mendoza V. L. et al. Structure of the preamyloid dimer of β-2-microglobulin from covalent labeling and mass spectrometry. Biochem. 49, 1522–1532 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y., Nordenskiöld L. & Tam J. P. Folding, misfolding, and amyloid protofibril formation of WW domain FBP28. Biophys. J. 90, 3983–3992 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z., Schmidt C. & Politis A. Uncovering the early assembly mechanism for amyloidogenic β2-microglobulin using cross-linking and native mass spectrometry. J. Biol. Chem, 291, 4626–4637(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen A. F. P. et al. Domain metastability: A molecular basis for immunoglobulin deposition? J. Mol. Biol. 399, 207–213 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus K. J. et al. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat. Struct. Mol. Biol. 8, 770–774 (2001). [DOI] [PubMed] [Google Scholar]
- Liu C., Sawaya M. R. & Eisenberg D. β2-microglobulin forms three-dimensional domain-swapped amyloid fibrils with disulfide linkages. Nat. Struct. Mol. Biol. 18, 49–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanska K. et al. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic beta2-microglobulin variant. Proc. Natl. Acad. Sci. USA 108, 1314–1319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Gotte G., Libonati M. & Eisenberg D. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Struct. Mol. Biol. 8, 211–214 (2001). [DOI] [PubMed] [Google Scholar]
- Gazit E. A possible role for p-stacking in the self-assembly of amyloid fibrils. FASEB J. 16, 77–83 (2002). [DOI] [PubMed] [Google Scholar]
- Reches M. & Gazit E. Amyloidogenic hexapeptide fragment of medin: homology to functional islet amyloid polypeptide fragments. Amyloid 11, 81–89 (2004). [DOI] [PubMed] [Google Scholar]
- Gazit E. Self-assembled peptide nanostructures: the design of molecular building blocks and their technological utilization. Chem. Soc. Rev. 36, 1263–1269 (2007). [DOI] [PubMed] [Google Scholar]
- Hennig J. et al. Local destabilization of the metal-binding region in human copper–zinc superoxide dismutase by remote mutations is a possible determinant for progression of ALS. Biochem. 54, 323–333 (2015). [DOI] [PubMed] [Google Scholar]
- Chan Sze & Wah S. et al. Mechanism of amyloidogenesis of a bacterial AAA + chaperone. Structure 24, 1095–1109 (2016). [DOI] [PubMed] [Google Scholar]
- Richardson J. S. & Richardson D. C. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 99, 2754–2759 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowski M. A. & Bujnicki J. M. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31, 3305–3307 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. T., Kooperberg C., Huang E. & Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225 (1997). [DOI] [PubMed] [Google Scholar]
- Simons K. T. et al. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins 34, 82–95 (1999). [DOI] [PubMed] [Google Scholar]
- Kim D. E., Chivian D. & Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–531 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999). [DOI] [PubMed] [Google Scholar]
- Buchan D. W. et al. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theobald ,. D. L. & Wuttke D. S. THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics 22, 2171–2172 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. & Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- Frishman D. & Argos P. Knowledge-based protein secondary structure assignment. Proteins 23, 566–579 (1995). [DOI] [PubMed] [Google Scholar]
- Krissinel E. & Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 (2004). [DOI] [PubMed] [Google Scholar]
- Rose P. W. et al. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 43, D345–356 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005). [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L., Maxwell D. S. & Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996). [Google Scholar]
- Berendsen H. J. C., Grigera J. R. & Straatsma T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987). [Google Scholar]
- Hornak V. et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. et al. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- Darden T., Perera L., Li L. & Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 7, R55–60 (1999). [DOI] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H. J. C. & Fraaije J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- Bermel W. et al. Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J. Am. Chem. Soc. 128, 3918–3919 (2006). [DOI] [PubMed] [Google Scholar]
- Wang Y. & Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11, 852–861 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzinger S. et al. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 123, 2970–2978 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.