

Abstract
Pheochromocytomas and paragangliomas (PPGLs) are rare and frequently heritable neural-crest derived tumours arising from the adrenal medulla or extra-adrenal chromaffin cells respectively. The majority of PPGL tumours are benign and do not recur with distant metastases. However, a sizeable fraction of these tumours secrete vasoactive catecholamines into the circulation causing a variety of symptoms including hypertension, palpitations and diaphoresis. The genetic landscape of PPGL has been well characterized and more than a dozen genes have been described as recurrently mutated. Recent studies of DNA-methylation have revealed distinct clusters of PPGL that share DNA methylation patterns and driver mutations, as well as identified potential biomarkers for malignancy. However, these findings have not been adequately validated in independent cohorts. In this study we use an array-based genome-wide approach to study the methylome of 39 PPGL and 4 normal adrenal medullae. We identified two distinct clusters of tumours characterized by different methylation patterns and different driver mutations. Moreover, we identify genes that are differentially methylated between tumour subcategories, and between tumours and normal tissue.
Pheochromocytoma and paraganglioma (PPGL) are neural crest derived chromaffin cell tumours of the adrenal medulla and paraganglia, respectively. The tumours may secrete catecholamines such as epinephrine, norepinephrine and dopamine into the systemic circulation, leading to symptoms of sympathetic hyperactivity. The incidence is 2–8/1,000,000 per year1,2, and up to 40% of all PPGL are known to be part of inherited syndromes, including Neurofibromatosis type 1, Multiple Endocrine Neoplasia type 2A and 2B (MEN2A/MEN2B) as well as von Hippel-Lindau syndrome3. To date, more than a dozen genes have been identified as conferring susceptibility to PPGL: SDHA, SDHB, SDHC, SDHD, SDHAF2 (from here on abbreviated as SDHx), FH, VHL, RET, EPAS1, TMEM127, MAX, NF13,4,5,6,7,8,9,10,11,12,13 and MDH214. In addition, recurrent somatic mutations have been found in the HRAS15 in sporadic PPGLs. In total these genetic aberrations are the cause of more than 60% of PPGL cases. The histone methyl transferase KMT2D has also been reported recurrently mutated16, as has the chromatin modulator ATRX17. However, the functional implications of these mutations remain to be elucidated. PPGL tumours can be sorted into two main clusters, based on driver mutation and gene expression patterns. Cluster 1 contains tumours with mutations in VHL, EPAS1 and SDHx/FH/MDH2, and is characterized by a pseudohypoxic gene expression signature18 caused by the stabilization of EPAS-1 which drives angiogenesis and tumour development19,20. Cluster 2 contains tumours with mutations in RET, TMEM127, MAX, NF1 and HRAS and is characterized by aberrant activation of kinase signalling pathways21. The vast majority of cases are benign and do not cause distant metastases. Approximately ten percent relapse with distant metastases, and metastatic pheochromocytoma is currently lacking an effective treatment. There are no reliable predictors of metastatic recurrence, but extra-adrenal location and SDHB mutations have been associated with increased risk of metastasis and poor outcome22. However, adrenal tumours without SDHx mutations can also give rise to metastatic disease. Patients with hereditary forms of the disease are at risk of developing multiple primary tumors.
Aberrant DNA methylation has been identified as an important feature of human cancers. In normal tissue, intergenic CpG sites are typically methylated, while CpG-islands in promoter regions exhibit tissue-dependent methylation levels. In tumour tissue, intergenic regions are often hypomethylated, while certain promoter regions are hypermethylated, causing transcriptional changes23 by interfering with transcription factor binding and affecting the chromatin structure. Molecular subgroups characterized by different DNA methylation patterns have been described in several tumour types, and have been reported to be associated with differences in survival24,25,26. Moreover, DNA-methylation of specific genes may be used as biomarkers for response to chemotherapy, such as methylation of MGMT and response to alkylating agents27.
DNA methylation has been investigated in PPGLs. Early studies used sequencing- and PCR-based methods to investigate the methylation status of carefully selected candidate genes28,29, while more recent studies have used array-based technologies to investigate genome-wide methylation30,31. SDHx-mutated tumours have been found to exhibit a hypermethylator phenotype, due to allosteric inhibition of the demethylating TET family of enzymes by the build-up of succinate. Another subset of tumours has been found to exhibit global hypomethylation31. Recently, the methylation level of a number of CpG-sites was proposed as markers of malignancy in PPGL, independent of genetic background30, with high levels of methylation of a specific CpG-residue in the promoter of the RDBP gene pinpointed as a strong candidate biomarker for metastatic disease. In this paper we report the results from a global methylation analysis of 39 tumours from 35 patients and evaluate the set of proposed malignancy markers in this cohort.
Results
Hierarchical clustering
Unsupervised hierarchical clustering with respect to the 10% most differentially methylated probes, as determined by standard deviation, revealed two clusters (Fig. 1, extended heatmap including normal samples in Supplemental Figure 1), from here on referred to as clusters A and B. Cluster A (n = 28) contained the majority of tumours with mutations in the RET, NF1 and HRAS genes and most of those without known mutations, whereas cluster B (n = 11) contained the majority of the tumours with mutations in the VHL gene. Cluster A contained all malignant tumours in the cohort (n = 10), however, no difference in survival could be demonstrated (Supplemental Fig. 2). Principal Components Analysis (PCA) (Fig. 2a) was consistent with the results from the cluster analysis and separated the two clusters along the first principal component. Tumours in cluster A had an average methylation index (MI) of 0.242 while tumours in Cluster B had a higher average MI of 0.277 (p < 0.0001, Mann-Whitney test). Normal adrenal medulla had an average MI of 0.273, which was not statistically different from that of cluster B (p = 0.4, Kolmogorov-Smirnov), but significantly higher than that of Cluster A (p < 0.0001, Kolmogorov-Smirnov test, Fig. 2b). Malignant tumours were found to have a lower methylation index than benign tumours (0.237 vs 0.256, p = 0.0012 by the Mann-Whitney test, Fig. 2c).
Figure 1. Heatmap and hierarchical clustering of tumour samples based on DNA methylation.

Two distinct clusters are observed. The symbols *, #, and § denote samples originating from the same cases.
Figure 2.

(a) Principal components analysis of the tumour samples shows the two clusters separated along the first principal component. (b) Methylation index of tumours in the two clusters, and of normal adrenal medulla. Tumours in cluster A have a lower global methylation level than tumours in cluster B and normal tissue. (c) Methylation index of benign tumours, malignant tumours and normal tissue.
Differential methylation
Comparing methylation in pheochromocytomas to methylation in normal adrenal medulla, 642 probes were differentially methylated. Of these, 64 were more methylated and 578 were less methylated in tumours (Supplementary Table 1). Comparing the individual clusters to normal adrenal we found 1429 probes in Cluster A and 70 probes in Cluster B that were differentially methylated (Supplementary Tables 2 and 3), supporting the notion that tumours in cluster B are more similar to normal adrenal medulla in methylation. In total 34 of these probes were differentially methylated in both clusters (Fig. 3a). When the benign and the malignant tumours in cluster A were separately compared to normal medulla, 1254 probes were found differentially methylated in the benign tumours and 2904 were found differentially methylated in the malignant tumours (Supplementary Tables 4 and 5). Of these, 1006 were differentially methylated in both the benign and the malignant tumours.
Figure 3.
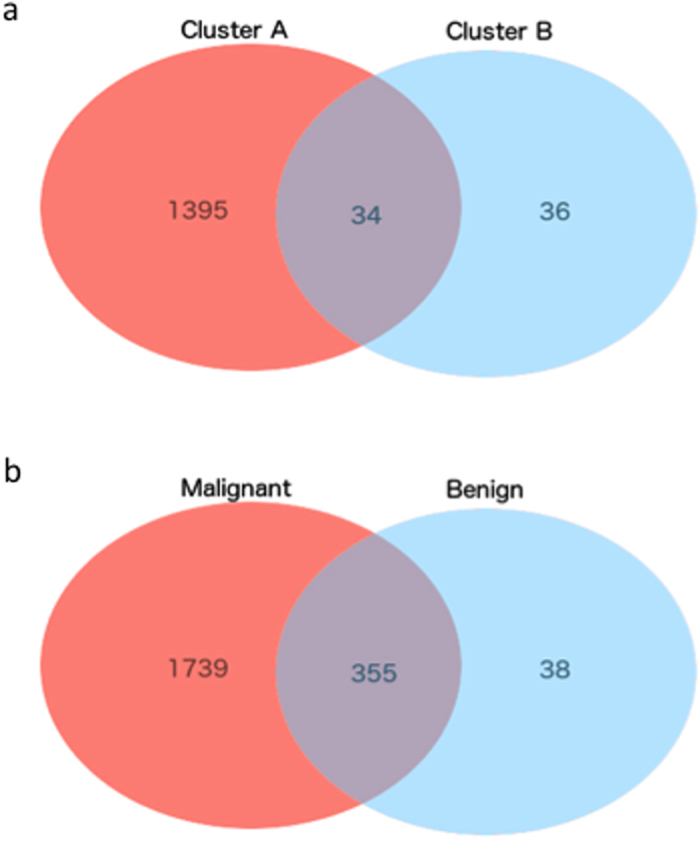
(a) Venn diagram of probes aberrantly methylated in the two clusters (compared to normal tissue). (b) Venn diagram of probes aberrantly methylated in benign and malignant tumours, compared to normal tissue.
In malignant tumours, 2094 probes were differentially methylated, while 393 probes were differentially methylated in benign tumours. In total 355 of these probes were found differentially methylated in both benign and malignant tumours compared to normal adrenal medulla (Fig. 3b). Notable genes found to be differentially methylated between tumour subgroups and normal tissue include CTSZ, a lysosomal enzyme which has found to be overexpressed in hepatocellular carcinoma32 and which was hypomethylated in tumours in Cluster A, as well as SPRR3 which encodes the protein Esophagin which has been implicated in tumour cell proliferation and invasion in glioblastoma multiforme33, and has been shown to be upregulated in colorectal cancer34. Several cancer-testis antigens, including CTAG2 and CT45-2 were also found to be less methylated in tumour tissue than in normal tissue. Analysis of the expression of CTAG2 and CT45-2 in the TCGA Pheochromocytoma and Paraganglioma RNA-Seq datasest revealed that CTAG2 was expressed in 47% (89/187) of the samples. However, CT45-2 expression was not detected in any of the samples. Gene Ontology-enrichment analysis revealed genes involved in different immune response and cell differentiation pathways to be overrepresented in the list of genes differentially methylated in Cluster A (Supplementary Table 6). Whether or not this is related to a higher level of infiltrating immune cells in this subgroup should be further studied. No category was overrepresented among the genes differentially methylated in Cluster B.
A total of 275 probes were differentially methylated between benign and malignant tumours in our cohort (Supplementary Table 8). Of these, 14 were more methylated in malignant tumours and 261 were more methylated in benign tumours. Gene-ontology analysis of all the probes differentially methylated between benign and malignant tumours revealed overrepresentation of genes involved in immune response (Supplementary Table 9). A total of 119 probes were differentially methylated between benign and malignant tumours within Cluster A (Supplementary Table 10). Of these the majority (98 probes) had lower methylation levels in the malignant samples.
In the cases were sufficient samples were available tumours sharing a mutated gene were compared with normal medulla (Supplementary Tables 11–14). The highest number of differentially methylated probes were detected in tumours with NF1-mutations (1651 probes) followed by those without known mutations (1184 probes), those with RET-mutations (432 probes) and those with VHL-mutations (339 probes). The overlap of differentially methylated probes in these groups are shown as a Venn Diagram in Supplementary Figure 3. 100 probes were differentially methylated in all four groups and are listed in Supplementary Table 15.
Correlation with Somatic Copy Number Aberrations
DNA hypomethylation is known to be associated with chromosomal instability. We compared the number of Somatic Copy-Number Aberrations (SCNAs) as determined by manual inspection of plotted SNP array data between cluster A (hypomethylated) and cluster B (non-hypomethylated) and found that tumours in cluster A had a greater number of chromosomal aberrations than tumours in cluster B (9.5 vs. 5.3, p = 0.0027 by the Mann-Whitney test, Fig. 4a). Similarly, malignant tumours carried a greater number of SCNAs than benign tumours (Fig. 4b 14.9 vs., 7.6, p = 0.0004 by the Mann-Whitney test). This remained true when only tumours in Cluster A were considered (14.9 vs. 8.9, p = 0.0198 by Kolmogorov-Smirnov). Linear regression of SCNA on methylation index revealed a significant negative correlation (R2 = 0.21, p = 0.0056, Supplementary Figure 4).
Figure 4.

(a) Number of Somatic Copy Number Aberrations (SCNAs) in the two clusters. Tumours in cluster B carry more SCNAs than tumours in cluster A. (b) Number of SCNAs in malignant and benign tumours. Malignant tumours carry more SCNAs.
Epigenetic heterogeneity
Three patients had multiple tumours included in the study. In two cases there was a high degree of concordance between different tumours from the same patient with 325 and 500 probes showing a difference in methylation of more than 0.2, respectively. However, in the third case, which included a primary tumour, a local recurrence, and a distant metastasis, a great discordance was found between the primary tumours and the two recurrences. The metastasis and the local recurrence had 2567 and 2677 probes with a methylation level differing more than 0.2 from the primary tumour. Moreover, they exhibited a methylation profile radically different from all other tumours in the cohort, clustering together in the outskirts of cluster A. The difference in MI between the primary tumour and the recurrences was negligible. Heatmaps for these three cases are given in Supplemental Figures 5–7. Tumour purity as estimated by ASCAT (19% contamination in the primary compared to 17% and 25% in the recurrent tumours) suggests that it is unlikely that these differences are explained by infiltrating non-tumoural cells.
PNMT expression
Expression of PNMT, Phenylethanolamine-N-methyltransferase, the enzyme which converts norepinephrine to epinephrine is known to be regulated by promotor methylation31. In order to validate methylation analysis, PNMT expression was correlated with the observed methylation. A good concordance between the two was observed, with virtually abolished expression in tumours with high methylation level of the promoter. (Fig. 5). Linear regression revealed that the expression was significantly correlated to the probe cg12894984 (R2 = 0.14, p = 0.025) but not to the probe cg11896923. There was no significant difference in PNMT expression between the two clusters in our cohort; however, ANOVA revealed that the expression differed significantly between tumours with different mutation (p = 0.0024, Supplementary Figures 8 and 9).
Figure 5.

(a) PNMT mRNA expression is decreased in tumours with high levels of PNMT promoter methylation. (b) Tumours in cluster B tend to have higher levels of PNMT promoter methylation than tumours in cluster A.
Evaluation of biomarkers
A recent study proposed 52 probes as methylation-based markers of malignancy. We calculated Δβ- values for all these probes and were not able to replicate these findings (a |Δβ|- value of more than 0.2) in any of the probes. The probes associated with RDBP (cg04710641 and cg06351503) had Δβ- values of 0.007 and −0.098 between benign and malignant tumours, respectively. No significant differences in beta-values were found between benign and malignant tumours (p > 0.1 for both probes). In order to further assess the potential of the probes as predictors of metastasis Receiver Operating Characteristic (ROC) curves were generated (Fig. 6) and the Area Under the Receiver Operating Characteristic curve (AUROC) were calculated. For the two RDBP probes the AUROCs were 0.55 and 0.6, respectively.
Figure 6.

(a) Receiver Operating Characteristic (ROC) curve for a CpG probe associated with RDBP previously suggested as a biomarker of metastatic disease. (b) ROC curve for the other probes associated with RDBP on array.
Next we assessed the viability of the other 51 probes as predictors of metastasis. The AUROC was calculated for each of the probes. The mean AUROC of the probes was 0.60 (range 0.43–0.81). The best performance was by cg00626119 (NTRK1), which exhibited a Δβ of −0.15 in our cohort. A total of nine probes exhibited AUROCs > 0.7, which could indicate viability as biomarkers (Supplementary Table 16). All of these were less methylated in malignant tumours than in benign tumours.
Discussion
We report here a global methylation analysis of thirty-nine PPGLs. We were able to reproduce many of the results previously published regarding methylation in PPGL. Namely, we found that tumours cluster according to mutated driver gene, and that tumours with mutations activating kinase pathways have global hypomethylation. The reason for this hypomethylation remains to be elucidated. Letouzé et al. identified three distinct clusters based on methylation data: M1–3. M1 contains tumours with SDHx mutations and hypermethylation, M2 VHL-mutated tumours, and M3 tumours with NF1 and RET mutations and hypomethylation31. Roughly, our cluster A corresponds to the M3 cluster and cluster B to the M2 cluster. Unlike Letouzé et al., we were not able to identify a hypermethylator phenotype, possibly due to a paucity of SDHx-mutated tumours.
Within the hypomethylated cluster A we observe a greater number of chromosomal instability, as measured by number of chromosomal aberrations, in malignant tumours compared to benign tumours. This represents a possible mechanism through which aberrant DNA methylation might affect diverse disease progression course in pheochromocytoma in addition to altering gene expression.
Genomic hypomethylation has been found to be causally linked to chromosomal instability. In the hypomethylated cluster A we observe increased chromosomal instability as measured by number of chromosomal aberrations. This represents a possible mechanism through which aberrant DNA methylation can affect disease progression in pheochromocytoma in addition to altering gene expression. Indeed, malignant tumours were found to harbour a greater number of copy number aberrations than benign tumours, even within Cluster A, supporting the idea that hypomethylation can contribute to malignant progression.
In three cases we were able to study DNA methylation in multiple (2–3) tumours from the same individual. While this number is too small to draw any general conclusions, we observe a wide range in the number of differentially methylated probes between tumours from the same patient. The extent and impact of epigenetic heterogeneity in pheochromocytoma is an interesting topic for future studies. The importance of genetic heterogeneity in cancer is well established and has recently been studied in pheochromocytoma. Epigenetic heterogeneity, on the contrary, remains largely uninvestigated. Pioneering studies have reported that epigenetic heterogeneity is abundant and potentially plays an important role35.
The malignancy biomarkers proposed by other studies could not be reproduced in our cohort. In particular, RDBP, which has been suggested to be hypermethylated in metastasizing PPGL regardless of mutational status, was not hypermethylated in the malignant tumours of our cohort. A likely explanation for this is the differences in composition that exist between our cohort and the cohorts investigated in other studies. Notably, other studies investigating DNA methylation in PPGL have had an abundance of SDHx-mutated tumours, while only one such tumour is part of the cohort studied here. SDHx- and especially SDHB-mutated tumours have high metastatic potential, while also exhibiting a hypermethylator phenotype. This could explain why hypermethylation of RDBP is found to be associated with malignancy in other cohorts, but not in our cohort, where the probes for RDBP performed marginally better than chance as predictors of metastatic disease. The other probes that were proposed were also evaluated. While a number of them showed promise as markers of malignancy, closer examination revealed that they showed different methylation pattern in our cohort than in the cohorts in which they were discovered; in the present study they were less methylated in malignant tumours than in benign tumours, while the opposite relationship has previously been reported. This highlights the difficulties inherent in designing malignancy markers for a tumour type as diverse as PPGL. Since the different groups of PPGL exhibit striking differences in genetic background and tumorigenic pathways, reliable malignancy markers valid for all PPGL remain undiscovered. Future studies of large cohorts, incorporating multiple types of omics data may enable more reliable risk stratification of cases.
Limitations of this study
A potential pitfall of this study is the presence of normal cell contamination (e.g. vascular cells, immune cells and fibroblasts) in the tumour samples. We have previously shown that tumour cell purity differs between mutational subgroups36. This may contribute to the differences noted between different subgroups by the present study and others. Tumour purity estimated by ASCAT (as reported previously) did not differ significantly between the methylation clusters in this study (Supplementary Table 17). However, an influence of infiltrating stromal cells on the results reported in this study cannot be excluded.
PPGLs are rare. This study included 39 tumours and 4 normal adrenal medullas. This is the only independent large cohort to date investigating and corroborating the findings by de Cubas et al.30. Even though we present statistically significant results and have employed several statistical methods to ensure solidity, much larger cohort or a meta analysis based on several reports are needed to confirm clinical significance of these findings.
In conclusion, previously discovered molecular subgroups of pheochromocytoma defined by DNA methylation were validated while proposed markers of malignancy could not be validated in our cohort.
Materials and Methods
Cohort
Thirty-nine histologically verified PPGLs (38 pheochromocytomas and 1 paraganglioma) from 35 individuals, as well as 4 normal adrenal medullae were included in this study. The cohort characteristics are outlined in Table 1. The mean age at diagnosis was 50.5 years (range 17–75 years) and the gender ratio (female:male) was 1.69. Two patients had MEN2A, two had MEN2B, an additional three patients had von Hippel-Lindau syndrome, and one had Neurofibromatosis type 1 while one patient had paraganglioma syndrome type 4. One patient with von Hippel-Lindau disease had bilateral disease with both tumours included in the cohort. The tumours had been previously studied for mutations in known driver genes and using high-density SNP array (data available for 35 of the tumours)36,37. Tumour purity was estimated from SNP array data using ASCAT v. 2.138 as implemented in Nexus Copy-number Variation 7.5 (Biodiscovery Inc., CA).
Table 1. Cohort characteristics.
| Tumour characteristics | Complete cohort | Cluster A | Cluster B |
|---|---|---|---|
| PCC/PGL | 38/1 | 28/0 | 10/1 |
| Benign/Malignant | 29/10 | 18/10 | 11/0 |
| Mutation: NF1/RET/HRAS/VHL/SDHB/- | 7/7/2/7/1/15 | 6/5/2/2/0/13 | 1/2/0/5/1/2 |
| Patient characteristics | |||
| Male/Female | 13/22 | 12/13 | 1/9 |
| Age at diagnosis | 50.5 (17–75) | 53.2 (17–75) | 43.3 (27–69) |
| Metastatic disease | 7 | 7 | 0 |
| Syndrome: VHL/NF1/MEN2/PGL4 | 3/1/4/1 | 0/1/3/0 | 3/0/1/1 |
DNA extraction and methylation array analysis
All tissues were snap-frozen in liquid nitrogen perioperatively and were stored at −70 °C. The tumours were cryosectioned, and DNA was extracted using Qiagen DNA Blood and Tissue kit (QIAGEN, Redwood city) in accordance with the manufacturer’s instructions. The extracted DNA was treated using the QIAGEN EpiTect Bisulfite kit and fragmented. The final product was analyzed using the Illumina Infinium HumanMethylation27 (Illumina Inc. San Diego) microarray at the Bioinformatics and Expression Analysis Core Facility at the Karolinska Institute. The array has probes for 27,578 CpG-sites, located in the promoters of 14,497 protein-coding genes spread across the genome.
RNA extraction and RT-PCR
Similarly to DNA, RNA was extracted from cryosections of fresh frozen tumour tissue using the Qiagen RNEasy Mini- and Midi-kits (QIAGEN, Redwood city). Extracted RNA was converted to cDNA using RevertAid First strand cDNA Synthesis kit (ThermoFisher scientific, MA,USA). RT-PCR was performed using SYBR Green on the Bio-Rad CFX96 Real-Time System (Bio-Rad, Hercules, California). Beta-actin was used as a reference gene, and the 2−ΔΔCT method was used for calculations. Primer sequences are available upon request.
Analysis of TCGA PCPG Expression data
Upper Quantile normalized FPKM (FPKM-UQ) gene expression data for the TCGA Pheochromocytoma&Paraganglioma cohort were obtained from the Genomics Data Commons portal (https://gdc-portal.nci.nih.gov). Expression values for genes of interest were extracted and summarized using custom python scripts. Genes with an FPKM-value > 1 in a given sample were considered to be expressed.
Further data processing and statistics
Generated microarray images were imported into Illumina GenomeStudio (Illumina Inc., San Diego, US) where beta-values were calculated and exported. Differential Methylation analysis was performed in GenomeStudio using the Illumina Custom Error Model. Probes with |DiffScores| > 30 together with |Δβ| > 0.2 were considered differentially methylated. Further analysis was performed using the R statistical environment and GraphPad Prism 7 (GraphPad Software Inc, CA). Hierarchical clustering of the samples was performed after removal of probes on the sex chromosomes and of probes with missing values using the 10% most variably methylated probes, as determined by standard deviation, with the Euclidean distance as the distance metric. Principal Components Analysis (PCA) was performed using the R function prcomp. The Mann-Whitney U test and Fisher’s exact test were used. When sample-sizes were small, the two-sample Kolmogorov-Smirnov test was used in place of Mann-Whitney U. The Benjamini-Hochberg39 method was used to correct for multiple hypothesis testing when required. The Benjamini-Hochberg procedure is an alternative approach to monitor false discovery rates. Briefly, individual variables p-values are sorted in ascending order and are designated a numerical rank. Numerical rank divided by total number of variables (p-values) multiplied by the desired false discovery rate, gives the Benjamini-Hochberg critical value. The largest p-value that is numerical larger than its Benjamini-Hochberg critical value as well as all p-values smaller than it are then considered as significant, even those larger than their Benjamini-Hochberg critical value40,41. This procedure is fully implemented in Illumina GenomeStudio software. Two-tailed p-values of less than 0.05 were considered to be statistically significant. The CRAN package “pROC” was used to generate Receiver Operating Characteristics (ROCs) for the purpose of evaluating specific probes as predictors of metastatic disease. The Venn Diagram in Supplemental Figure 3 was generated using http://bioinformatics.psb.ugent.be/cgi-bin/liste/Venn/calculate_venn.htpl.
Gene ontology analysis
Gene ontology analysis was performed using the PANTHER Overrepresentation test (http://www.pantherdb.org, release 20160715) with the PANTHER GO Ontology dataset. Bonferroni-correction was used to correct for multiple testing and only results with corrected p-values < 0.05 were considered significant.
Ethics
All patients had provided written informed consent for the preservation and subsequent analysis of tissues. Ethical approval was obtained from the regional ethics committee in Uppsala. Experiments were conducted in accordance with relevant guidelines and regulations.
Additional Information
How to cite this article: Backman, S. et al. Global DNA Methylation Analysis Identifies Two Discrete clusters of Pheochromocytoma with Distinct Genomic and Genetic Alterations. Sci. Rep. 7, 44943; doi: 10.1038/srep44943 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Footnotes
The authors declare no competing financial interests.
Author Contributions S.B., R.M., A.F.-D. and J.C. performed experiments. S.B. carried out statistical and bioinformatical analysis and drafted the manuscript. S.B., R.M., A.F.-D., J.C., K.C., P.S., P.H. and P.B. participated in the acquisition, analysis, or interpretation of data. All authors contributed with critical revision of the manuscript and all authors performed administrative, technical or material support. P.B. obtained funding. S.B. and P.B. designed the study. P.B. conceived and supervised the study.
References
- Stenström G. & Svärdsudd K. Pheochromocytoma in Sweden 1958–1981. An analysis of the National Cancer Registry Data. Acta Med. Scand. 3, 225–232 (1986). [PubMed] [Google Scholar]
- Beard C., Sheps S., Kurland L., Carney J. & Lie J. Occurence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin. Proc. 12, 802–804 (1983). [PubMed] [Google Scholar]
- Favier J., Amar L. & Gimenez-Roqueplo A.-P. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nature Reviews Endocrinology 11, 101–111 (2015). [DOI] [PubMed] [Google Scholar]
- Zhuang Z. et al. Somatic HIF2A Gain-of-Function Mutations in Paraganglioma with Polycythemia. New England Journal of Medicine 367, 922–930 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y. et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nature genetics 42, 229–233 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comino-Méndez I. et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nature genetics 43, 663–667 (2011). [DOI] [PubMed] [Google Scholar]
- Burnichon N. et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Human Molecular Genetics 21, 5397–5405 (2012). [DOI] [PubMed] [Google Scholar]
- Clark G. et al. Germline FH Mutations Presenting With Pheochromocytoma. Journal of Clinical Endocrinology and Metabolism 99, E2046–E2050 (2014). [DOI] [PubMed] [Google Scholar]
- Burnichon N. et al. SDHA is a tumor suppressor gene causing paraganglioma. Human molecular genetics 19, 3011–3020, doi: 10.1093/hmg/ddq206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti D. et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. American journal of human genetics 69, 49–54, doi: 10.1086/321282 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S. & Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nature genetics 26, 268–270, doi: 10.1038/81551 (2000). [DOI] [PubMed] [Google Scholar]
- Baysal B. E. et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 (2000). [DOI] [PubMed] [Google Scholar]
- Bayley J. P. et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. The Lancet. Oncology 11, 366–372, doi: 10.1016/S1470-2045(10)70007-3 (2010). [DOI] [PubMed] [Google Scholar]
- Cascon A. et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst. 107 (2015). [DOI] [PubMed] [Google Scholar]
- Crona J. et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. Journal of Clinical Endocrinology and Metabolism 7, 1266–1271 (2013). [DOI] [PubMed] [Google Scholar]
- Juhlin C. C. et al. Whole-exome sequencing defines the mutational landscape of pheochromocytoma and identifies KMT2D as a recurrently mutated gene. Genes, chromosomes & cancer 54, 542–554, doi: 10.1002/gcc.22267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein L. et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nature communications 6, 6140, doi: 10.1038/ncomms7140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnichon N. et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Human Molecular Genetics 20, 3974–3985 (2011). [DOI] [PubMed] [Google Scholar]
- Kim W. & Kaelin W. Role of VHL Gene Mutation in Human Cancer. Journal of Clinical Oncology 22, 4991–5004 (2004). [DOI] [PubMed] [Google Scholar]
- Selak M. et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7, 77–85 (2005). [DOI] [PubMed] [Google Scholar]
- Dahlia P. L. M. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nature Reviews Cancer 14, 108–119 (2014). [DOI] [PubMed] [Google Scholar]
- Amar L. et al. Succinate Dehydrogenase B Gene Mutations Predict Survival in Patients with Malignant Pheochromocytomas or Paragangliomas. Journal of Clinical Endocrinology and Metabolism 92 (2007). [DOI] [PubMed] [Google Scholar]
- Jones P. & Baylin S. The Epigenomics of Cancer. Cell 128, 683–692 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- *Barreau O. et al. Identification of CpG Island Methylator Phenotype in Adrenocortical Carcinomas. Journal of Clinical Endocrinology and Metabolism 98, E174–E184 (2012). [DOI] [PubMed] [Google Scholar]
- Toyota M. et al. CpG island methylator phenotype in colorectal cancer. Proceedings of the National Academy of Sciences 96, 8681–8686 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H. et al. Identification of CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 17, 510–522 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegi M. et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. New England Journal of Medicine 352, 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
- Geli J. et al. The Ras effectors NORE1A and RASSF1A are frequently inactivated in pheochromocytoma and abdominal paraganglioma. Endocrine-related cancer 14, 125–134 (2007). [DOI] [PubMed] [Google Scholar]
- Muscarella P. et al. Expression of the p16INK4A/Cdkn2a gene is prevalently downregulated in human pheochromocytoma tumor specimens. Gene Expr 14, 207–216 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cubas A. A. et al. DNA Methylation Profiling in Pheochromocytoma and Paraganglioma Reveals Diagnostic and Prognostic Markers. Clinical cancer research: an official journal of the American Association for Cancer Research 21, 3020–3030, doi: 10.1158/1078-0432.CCR-14-2804 (2015). [DOI] [PubMed] [Google Scholar]
- Letouzé E. et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23, 749–752 (2013). [DOI] [PubMed] [Google Scholar]
- Wang J., Chen L., Li Y. & Guan X. Y. Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PloS one 6, e24967, doi: 10.1371/journal.pone.0024967 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Zhang C., Ma G. & Zhang Q. Expression of SPRR3 is associated with tumor cell proliferation and invasion in glioblastoma multiforme. Oncology Letters 7, 427–432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D. et al. Upregulation of SPRR3 promotes colorectal tumorigenesis. Molecular Medicine 16, 271–277 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocks D. et al. Intratumour DNA methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Repots 8, 798–806 (2014). [DOI] [PubMed] [Google Scholar]
- Crona J. et al. Spatiotemporal Heterogeneity Characterizes the Genetic Landscape of Pheochromocytoma and Defines Early Events in Tumorigenesis. Clinical cancer research: an official journal of the American Association for Cancer Research 21, 4451–4460, doi: 10.1158/1078-0432.CCR-14-2854 (2015). [DOI] [PubMed] [Google Scholar]
- Crona J. et al. Integrative genetic characterization and phenotype correlations in pheochromocytoma and paraganglioma tumours. PloS one 9, e86756, doi: 10.1371/journal.pone.0086756 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loo P. et al. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences of the United States of America 107, 16910–16915, doi: 10.1073/pnas.1009843107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. & Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 57, 289–300 (1995). [Google Scholar]
- Klipper-Aurbach Y. et al. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med Hypotheses 45, 486–490 (1995). [DOI] [PubMed] [Google Scholar]
- Hochberg Y. & Benjamini Y. More powerful procedures for multiple significance testing. Stat Med 9, 811–818 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.