

Abstract
Reduction of mitochondrial complex I activity is one of the major hypotheses for dopaminergic neuron death in Parkinson’s disease. However, reduction of complex I activity in all cells or selectively in dopaminergic neurons via conditional deletion of the Ndufs4 gene, a subunit of the mitochondrial complex I, does not cause dopaminergic neuron death or motor impairment. Here, we investigated the effect of reduced complex I activity on non-motor symptoms associated with Parkinson’s disease using conditional knockout (cKO) mice in which Ndufs4 was selectively deleted in dopaminergic neurons (Ndufs4 cKO). This conditional deletion of Ndufs4, which reduces complex I activity in dopamine neurons, did not cause a significant loss of dopaminergic neurons in substantia nigra pars compacta (SNpc), and there was no loss of dopaminergic neurites in striatum or amygdala. However, Ndufs4 cKO mice had a reduced amount of dopamine in the brain compared to control mice. Furthermore, even though motor behavior were not affected, Ndufs4 cKO mice showed non-motor symptoms experienced by many Parkinson’s disease patients including impaired cognitive function and increased anxiety-like behavior. These data suggest that mitochondrial complex I dysfunction in dopaminergic neurons promotes non-motor symptoms of Parkinson’s disease and reduces dopamine content in the absence of dopamine neuron loss.
Parkinson’s disease is the second most common progressive neurodegenerative disorder. It is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) of the brain and by motor dysfunction including bradykinesia, rigidity, and tremor1,2,3. Although the mechanisms of dopaminergic neuron death in Parkinson’s disease are not completely established, inhibition of mitochondrial complex I has been a leading hypothesis4. This hypothesis was originally based on the observation that accidental exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) caused Parkinsonism in humans, as well as the discovery that a metabolite of MPTP, MPP+, is a mitochondrial complex I inhibitor5,6. The complex I inhibition hypothesis was further supported by the finding of reduced complex I activity in various tissues from Parkinson’s disease patients, including familial forms of Parkinson’s disease with the loss-of-function mutation of PTEN-induced putative kinase 1 (PINK1)7,8,9,10,11,12,13,14. Furthermore, treatment of rodents with MPTP or rotenone, another complex I inhibitor, induces key features of Parkinson’s disease15,16,17,18,19,20,21,22,23,24,25.
We tested the mitochondrial complex I hypothesis using mouse strains in which the Ndufs4 gene was deleted. Ndufs4 encodes an 18 kDa protein that is one of 46 subunits of the mitochondrial complex I; it is required for the complete assembly and function of complex I26,27,28,29,30,31. Systemic deletion of the Ndufs4 gene starting from embryonic development abolishes complex I activity but does not cause dopaminergic neuron death in culture or in young mice32,33. Furthermore, inducible deletion of the Ndufs4 gene in all the cells of adult mice reduces complex I activity by 71% but does not cause dopamine neuron death34. We also generated conditional Ndufs4 knockout mice (cKO) in which Ndufs4 is specifically deleted in dopaminergic neurons. Complex I activity was reduced by 84% in dopaminergic synaptosomes isolated from the striatum of the Ndufs4 cKO mice, but these mice do not show dopamine neuron loss, motor deficits, or altered sensitivity to MPTP over a 24-month life-span34. These results suggest that reduced mitochondrial complex I activity alone does not cause dopaminergic neuron death during aging nor does it contribute to dopamine neuron toxicity in the MPTP model of Parkinson’s disease.
In addition to motor impairments, many Parkinson’s disease patients also suffer from non-motor symptoms, including anxiety, depression, as well as impairment of olfaction and cognition35,36,37,38. These non-motor symptoms may start in the early stages of the Parkinson’s disease prior to the onset of motor symptoms39,40,41. Though the degeneration of serotonergic and noradrenergic neurons may underlie these non-motor symptoms, dopamine transporter-deficient mice exhibit hyperactivity and anxiety responses, suggesting that dysfunction of the dopamine pathway may also contribute42,43,44,45,46. In this study, we used Ndufs4 cKO mice to investigate the effects of complex I dysfunction on non-motor symptoms associated with Parkinson’s disease34.
Results
Loss of Ndufs4 selectively in dopaminergic neurons does not cause loss of dopaminergic neuron cell bodies or nerve terminals in the substantia nigra
We previously generated mice in which the Ndufs4 gene was selectively inactivated in dopaminergic neurons (Ndufs4 cKO) to achieve selective reduction of mitochondrial complex I activity in dopaminergic neurons34. We showed that this conditional Ndufs4 deletion impaired the activity of mitochondrial complex I in dopaminergic synaptosomes of Ndufs4 cKO mice34. We report here that the number of tyrosine hydroxylase (TH)-positive, dopaminergic neurons is not significantly different in the SNpc of control vs. Ndufs4 cKO mice (Fig. 1a,b), confirming our previous findings34. Furthermore, there was no increase in basal levels of terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL, Fig. 1c) in TH+ dopaminergic neurons. Additionally, there was no significant difference in the distribution or intensity of TH immunostaining in the striatum or amygdala, target areas of nigrostriatal or mesolimbic pathways of dopamine neurons in the substantia nigra, respectively (Fig. 1d–g). Together with our previous reports32,33,34, these data suggest that mitochondrial complex I dysfunction does not induce the loss of the cell body or nerve terminals of dopaminergic neurons of substantial nigra.
Figure 1. Dopaminergic neuron-specific Ndufs4 knockout does not cause dopamine neuron death.
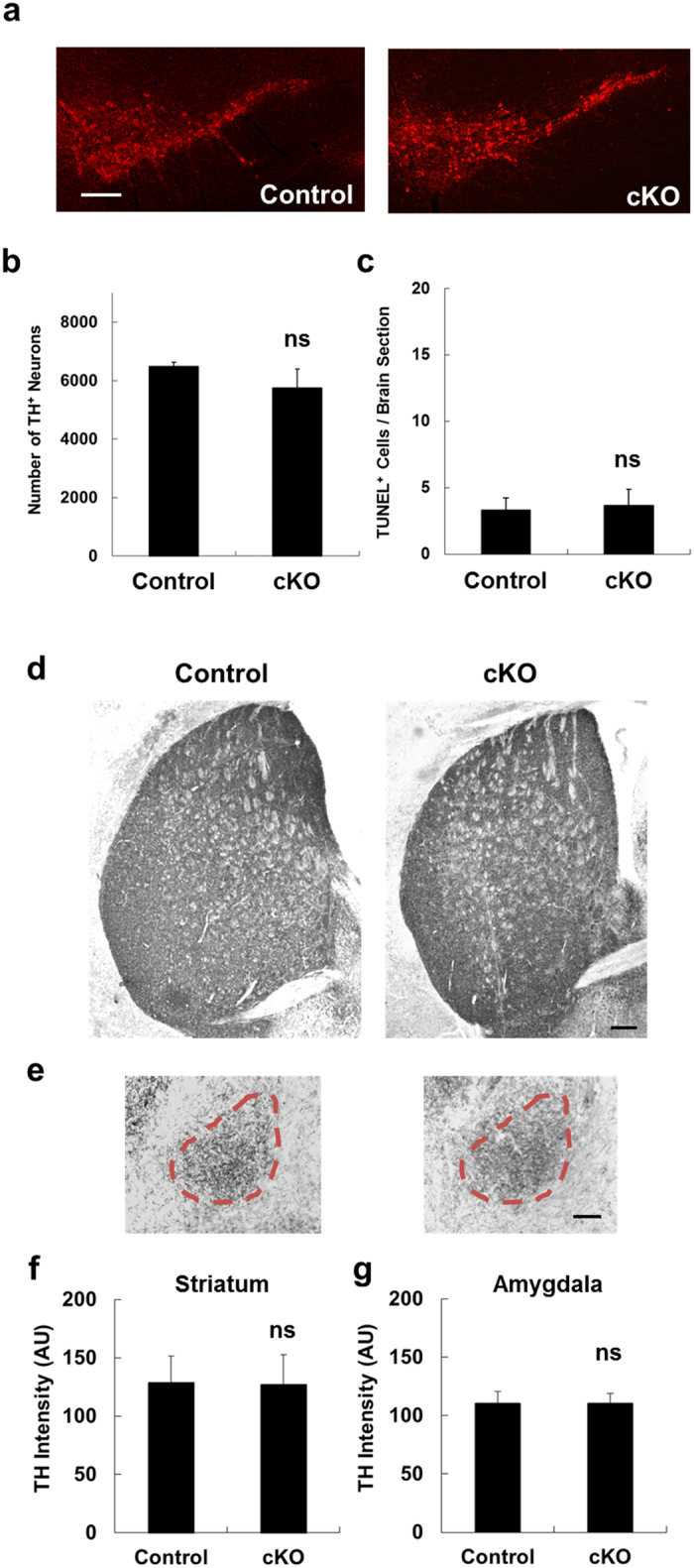
(a) Representative photomicrographs of TH immunostaining of SNpc. cKO: conditional knockout of Ndufs4 (Dat-cre/Ndufs4lox/lox) mice; control: Ndufs4lox/lox littermates. Bar, 200 μm. (b) Quantification of the total number of TH+ neurons in the SNpc. (c) Quantification of the total number of apoptotic, TUNEL+ cells in the SNpc. n.s. not statistically significant. (e,f) Representative TH staining of striatal (e) and amygdalar (f) tissue from Dat-cre/Ndufs4lox/lox (cKO) mice and Ndufs4lox/lox (Control) mice. Bar, 200 μm. (g,h) Quantification of TH staining intensity in the striatum (g) and amygdala (h). AU, arbitrary units. n.s. not statistically significant.
Ndufs4 cKO mice have reduced dopamine in the striatum and amygdala but the level of serotonin is not affected
We reported that Ndufs4 cKO mice exhibited age-dependent decline of dopamine content in the striatum34. This observation was confirmed using 8-month-old mice (Fig. 2a). There was also a significant decline of dopamine content in the amygdala of Ndufs4 cKO mice compared to control littermates (Fig. 2b). However, the serotonin content was not reduced in either the striatum (Fig. 2c) or amygdala (Fig. 2d), suggesting that conditional deletion of Ndufs4 in dopaminergic neurons specifically causes dopamine deficiency.
Figure 2. Dopamine content, not serotonin content, is decreased in the striatum and amygdala of Ndufs4 cKO mice.

The concentrations of dopamine (a,c) and serotonin (b,d) in the striatum (a,b) and amygdala (c,d) from both Ndufs4 cKO and control mice were measured. *p < 0.05; n.s. not statistically significant.
Conditional deletion of Ndufs4 does not impair motor behavior
Ndufs4 cKO and their littermate controls were subjected to behavioral tests to assess their motor function. Ndufs4 cKO mice behaved similarly to control mice in the open field test, including the total distance traveled (Fig. 3a) and the total time moving (Fig. 3b). The latency to fall in the rotarod test was also similar (Fig. 3c). These results are consistent with our previous report34 and suggest that conditional Ndufs4 deletion in dopaminergic neurons does not lead to motor deficits.
Figure 3. Motor behavior is not affected by conditional Ndufs4 deletion.

Locomotor activity in the open field test was quantified as total distance traveled (a), and total moving time (b). n.s. not statistically significant.
Ndufs4 inactivation in dopaminergic neurons leads to cognitive impairment
Impaired cognitive function is one of the common non-motor symptoms associated with Parkinson’s disease. To investigate the effect of conditional reduction of complex I activity on cognitive behavior, control and Ndufs4 cKO mice were subjected to a novel object recognition test to evaluate both short- and long-term memory. At 3 months of age, both control and Ndufs4 cKO mice remembered the familiar object “A” equally well 1 h after training and spent significantly more time investigating the novel object “C” during testing, indicating that they had normal short-term memory (Fig. 4a,b). However, unlike control mice, Ndufs4 cKO did not remember the novel object recognition when the test was performed 24 h after training (Fig. 4c,d). Furthermore, by the age of 6 months, Ndufs4 cKO mice had lost their 1-h short-term memory as well (Fig. 4e–h). These data suggest that conditional Ndufs4 deletion in dopaminergic neurons leads to progressive memory loss.
Figure 4. Ndufs4 cKO mice show progressive memory loss in the novel object recognition assay.

(a–h) Ndufs4 cKO and littermate control mice were tested for the novel object recognition assay at 3 months and then again at 6 months of age. The same cohort of mice was subjected to both 1-h and 24-h memory tests on two different days using different sets of objects. (a,b)Training and 1 h memory test in 3-month-old mice. (c,d) Training and 24-h memory in 3-month-old mice. (e,f) Training and 1-h memory in 6-month-old mice. (g,h) Training and 24-h memory in 6-month-old mice. n = 7–10 mice/genotype. (i–p) Ndufs4 cKO and littermate control mice were tested for the short-term spatial memory using the novel object location test at 3 months and then again at 6 months of age. Mice were subjected to both 1-h and 24-h memory tests on two different days. (i,j) Training and 1-h test using 3-month-old mice. (k,l) Training and 24-h test using 3-month-old mice. (m,n) Training and 1-h test using 6-month-old mice. (o,p) Training and 24-h test using 6-month-old mice. n = 7–10 mice/genotype. *p < 0.05; **p < 0.01; ***p < 0.001.
We performed the novel object location test to evaluate spatial learning and memory47,48. Two identical objects were used in training; one of them was moved to a novel location during tests conducted 1 h or 24 h after training. Control mice, at both 3 and 6 months of age, remembered the object placed at the familiar location 1 h or 24 h after training, spending significantly more time investigating the object in the novel location (Fig. 4i–p). However, the littermate Ndufs4 cKO mice, at both 3 and 6 months of age, spent an equal amount of time exploring both objects when tested either 1 h or 24 h after training. These results suggest that unlike control mice, the Ndufs4 cKO mice did not form any short- or long-term spatial memory in this test.
Animals were also subjected to the Morris water maze test to further evaluate their spatial learning and memory function (Fig. 5). The Ndufs4 cKO mice learned to locate the hidden platform as well as control mice over the course of a 7-day training period (Fig. 5a), and had comparable spatial memory as control mice in the probe test conducted 24 h after training (Fig. 5b). However, following a 6-day period of reversal training in which the hidden platform was moved to the opposite location, the Ndufs4 cKO mice swam longer distances than control mice to find the new platform (Fig. 5c). Furthermore, they spent significantly less time in the new target quadrant than control mice in the probe test after reversal training (Fig. 5d). In addition, although both groups of mice spent more time in the new target quadrant (Q3) than in the old one (Q1) in the reversal probe test, the ratio of time spent in Q3 over that in Q1 was higher for control than for Ndufs4 cKO mice (Fig. 5e). Two days after the reversal probe test, mice were also subjected to a visible platform test where both groups of mice had similar latencies to reach the platform (Fig. 5f). Thus, the differences observed in the reverse hidden platform training and testing were not caused by differences in vision, ability to swim, or motivation to escape. Reversal training is more demanding and complex than the initial hidden platform training because the mice need to actively forget the old location of the hidden platform and learn to find it in a new location. These results provide further evidence that the Ndufs4 cKO mice exhibit impaired spatial memory formation.
Figure 5. Ndufs4 cKO mice show impaired spatial learning and memory in the reversal Morris water maze test.

(a) Target (in quadrant 1) acquisition in the hidden platform, quantified as swim distance to platform, was similar in control and Ndufs4 cKO mice. (b) Both control and Ndufs4 cKO mice spent more time in the virtual target quadrant (quadrant 1) in the probe test conducted 24 h after training, suggesting that they retained the spatial memory. (c) Swim distance to escape in the reverse hidden platform training where the hidden platform was moved to quadrant 3. Ndufs4 cKO mice swam longer distances to locate the platform, suggesting impaired learning of newer spatial information. (d) Ndufs4 cKO mice spent less time in the virtual target quadrant (quadrant 3) than control mice during the reversal probe test. (e) The ratio of time spent in Q3 versus Q1 in the reversal probe test was significantly lower for cKO mice than control mice. (f) Control and cKO mice performed equally well in the visible platform test; they showed comparable latency to reach the platform. *p < 0.05; **p < 0.01; ***p < 0.001; n.s. not statistically significant.
We also performed auditory-cued and contextual fear conditioning as well as contextual fear extinction tests. Auditory cued-fear conditioning requires amygdala function, whereas contextual fear conditioning is hippocampus dependent. Contextual fear extinction is an active form of forgetting in which animals learn to dissociate the context from the foot shock; it is distinct from and may be more complex than contextual fear conditioning49. The Ndufs4 cKO mice manifested normal contextual fear memory (Fig. 6a) and cued-fear memory 24 h after training (Fig. 6b). The contextual memory for both Ndufs4 cKO and control mice was context specific because the mice did not show freezing when placed in a novel context (Fig. 6c). However, in contrast to control mice, the Ndufs4 cKO mice did not exhibit fear extinction (Fig. 6d). The contextual memory in the Ndufs4 cKO mice, which persisted after fear extinction trials, was still context-dependent because the mice did not show freezing when exposed to a novel context after the 8-day fear extinction training (Fig. 6e).
Figure 6. Ndufs4 cKO mice are deficient in contextual fear memory extinction.

(a) Control and Ndufs4 cKO mice have similar levels of contextual fear memory 24 h post-training. (b) Control and Ndufs4 cKO mice showed similar cued-fear response 24 h post-training. (c) Neither control nor Ndufs4 cKO mice froze when placed in a novel context 24 h after training. (d) However, fear extinction was impaired in Ndufs4 cKO mice. (e) The freezing response that persisted in Ndufs4 cKO mice after 8-days of fear extinction training was context specific because the mice did not freeze in a novel context after fear extinction trials. **p < 0.01.
Loss of Ndufs4 in dopaminergic neurons induces anxiety-like behavior
Several behavior tests were performed with 3-, 6-, and 9-month-old mice to investigate the effect of complex I dysfunction on anxiety-like behavior, another common non-motor symptom of Parkinson’s disease. In the elevated-plus maze test, 9-month-old Ndufs4 cKO mice spent significantly more time in the closed arms (Fig. 7a,b) and significantly less time in the open arms (Fig. 7a,c) compared to control mice. The frequency of entry to the open arms (Fig. 7d) and more specifically, to the open ends (Fig. 7e) was also significantly reduced in 9-month-old Ndufs4 cKO mice. Starting at 6 months of age, Ndufs4 cKO mice traveled less distance in the center of the open field (Fig. 8a,b), although their total distance traveled in the entire field was unchanged (Fig. 8c). These mice also made fewer entries to the light box and fewer transitions between the light and dark boxes in the dark/light test starting from 6 months of age (Fig. 8d). In the social interaction test, Ndufs4 cKO mice showed fewer interactions with other mice (Fig. 8e). Collectively, these data suggest that Ndufs4 cKO mice exhibit age-dependent, anxiety-like behavior.
Figure 7. Anxiety-related behavior is enhanced in Ndufs4 cKO mice in the elevated plus maze test.

(a) Representative activity traces from a control and an Ndufs4 cKO mouse in the elevated plus maze test. C, closed arm; O, open arm. (b) Time spent in closed arms of an elevated plus maze. (c) Time spent in open arms of an elevated plus maze. (d) Number of entries to the open arms of an elevated plus maze of 9-month-old mice. (e) Number of entries to the open ends of an elevated plus maze of 9-month-old mice. *p < 0.05; **p < 0.01; n.s. not statistically significant.
Figure 8. Anxiety-related behavior in the open field.

(a) Representative activity traces from a control and an Ndufs4 cKO mouse in the open field test. (b) Distance traveled to the center in the open field test. (c) Total distance traveled in the entire open field. (d) Ndufs4 cKO mice show increased anxiety in a light/dark exploration test. Data shown are the number of transitions made between the light and dark chamber in a light and dark box. (e) Social interaction is reduced in Ndufs4 cKO mice. Data shown are the time spent in social interactions. *p < 0.05; **p < 0.01; n.s. not statistically significant.
Discussion
Parkinson’s disease patients suffer from both motor and non-motor symptoms. However, the contribution of complex I deficiency to the non-motor symptoms of Parkinson’s disease has not been established and, thus, was the subject of this study. To this end, we utilized a conditional Ndufs4 knockout mouse we recently generated by crossing Ndufs4loxP/loxP mice31 with Slc6a3iCre/+ mice50 in which the codon-improved Cre recombinase gene (iCre) is expressed under the control of the regulatory elements for the dopamine transporter (DAT) gene, encompassed in a Bacterial Artificial Chromosome (Slc6a3). Thus, the Ndufs4 gene is conditionally deleted in dopamine neurons in our Ndufs4 cKO mice34. In a prior study, Sterky et al. crossed the same Ndufs4loxP/loxP mice31 to mice that express the cre-recombinase in heart and skeletal muscle51. It was reported that the loss of Ndufs4 causes only mild reduction of complex I activity in intact mitochondria prepared from heart of these mice52. Immortalized mouse embryonic fibroblasts prepared from Ndufs4−/− mice showed 35–60% reduction of complex I activity in intact cells53. These results led some researchers to argue that Ndufs4 deletion causes only mild complex I deficiency in vivo52.
However, we reported that, as a result of Ndufs4 deletion, complex I-dependent O2 consumption is almost completely lost in intact mesencephalic neurons cultured from Ndufs4−/− mice32. Furthermore, purified DAT+ synaptosomes with intact mitochondria from the brains of Ndufs4 cKO mice showed 84% loss of complex I activity, measured by complex I inhibitor-sensitive oxygen consumption rate using the polarography assay34. It is possible that these seemingly different results were obtained by different research groups because different Cre mice were used to delete Ndufs4, and different methods were employed to quantify complex I activity in intact mitochondira (ATP produciton vs. O2 consumption). Another important difference is the types of tissues used for the assays—heart vs. brain or cultured neurons. It has been documented by an independent group that all investigated tissues of the Ndufs4−/− mice show a significant decrease in complex I activity in intact mitochondria compared with the control animals54. However, there are significant differences in the residual complex I activities in the different Ndufs4−/− tissues; 44% in heart, 26% in brain, and 9% in lung tissues54. This 74% decrease of complex I activity in the brain of Ndufs4−/− mice is a substantial reduction and consistent with our findings32,34.
The Ndufs4 cKO mice did not show loss of dopaminergic neurons in the SNpc or dopaminergic nerve terminals in the striatum and amygdala, and their motor behavior was also normal. These data confirm our previous report that reduction of complex I activity in dopaminergic neurons per se is not sufficient to cause dopaminergic neuron degeneration or dopamine-dependent motor deficits34. Our data are consistent with other studies suggesting that deficiency of mesencephalic complex I activity does not directly correlate with Parkinson’s disease in mice52 or in humans55.
Both axons and cell bodies of dopaminergic neurons degenerate during the progression of Parkinson’s disease56. At the onset of diagnosis, while only approximately 30% of dopaminergic neuron cell bodies are lost, there is a 50% loss of dopaminergic axon terminals and up to 68~82% decrease of dopamine content in the striatum57,58,59,60,61. These findings suggest a dying back mechanism; the loss of dopamine precedes the structural degeneration of dopaminergic innervation and cell bodies in the nigrostriatal pathway. Interestingly, mitochondrial complex I deficiency in the Ndufs4 cKO mice also led to reduced dopamine in the striatum and amygdala without affecting the dopamine nerve terminals in these regions or dopamine neuron cell bodies in the substantia nigra. We do not know the molecular mechanism underlying the reduction of striatal dopamine in the absence of dopamine neuron death in Ndufs4 cKO mice. The loss is specific to dopamine since serotonin is not affected. Since the reduction of complex I activity leads to reduced ATP synthesis in Ndufs4 null mice31, one possibility is that dopamine synthesis or transport to the nerve terminals in the striatum is inhibited as a result of reduced ATP. Though Ndufs4 cKO mice have a significant decline (approximately 35% in the striatum) of dopamine content compared to control littermates, this decline was not sufficient to cause motor dysfunction. This is consistent with the literature that much more serve loss of striatal dopamine is required before motor deficits appears in rodents62,63,64,65,66,67,68. For example, mice exposed to MPTP can lose up to 50% of dopamine without apparent motor deficits67,68. Similarly, up to 80% of striatal dopamine is already lost at the onset of Parkinson’s disease diagnosis69.
Many Parkinson’s disease patients experience anxiety and depression35,36,37,70. Dopamine is important for emotional responses. For example, deletion of VMAT2, which transports cytosolic dopamine into vesicles, increases anxiety in mice71. Dopamine signaling during development critically affects aggressive and affective behaviors72. Interestingly, dopamine content in the striatum and amygdala of Ndufs4 cKO mice is lower than that in their controls. The Ndufs4 cKO mice also showed anxiety-like behaviors in the open field, elevated plus maze, light/dark exploration, and social interaction tests.
Cognitive impairment is also a common non-motor symptom of many Parkinson’s patients35,36,37,70. Dopamine is important for learning and memory73,74. Ndufs4 cKO mice exhibit impaired learning and memory in several commonly used behavioral tests, including novel object recognition, novel object location, reversal hidden platform Morris water maze, and contextual fear extinction. The defects in hippocampus-dependent spatial memory and extinction of contextual fear exhibited by the Ndufs4 cKO mice may reflect loss of dopamine and the fact that dopamine receptors in the hippocampus regulate synaptic plasticity and hippocampus-dependent memory75,76. For example, activation of dopamine receptors in the hippocampus increases long term potentiation (LTP) presumably by stimulation of adenylyl cyclase activity77 which is known to be required for hippocampus-dependent memory and LTP78.
Disruption of mitochondrial complex I has been implicated in diverse mental disorders including depression79,80. Down regulation or oxidative damage of complex I subunits and reduced complex I activity were observed in the brains from bipolar disorder patients81,82. In animal models, complex I inhibitor induced anxiety-like behavior83. Because Ndufs4 cKO mice do not show loss of dopamine neurons or impaired motor functions associated with Parkinson’s disease, our findings that Ndufs4 cKO mice exhibit cognitive impairment and anxiety may have broader implications for mitochondrial deficiencies in mental disorders in non-Parkinson’s disease patients.
In summary, data presented here provide evidence that reduced activity of mitochondrial complex I in dopaminergic neurons contributes to dopamine loss and non-motor behavior deficits associated with Parkinson’s disease including cognitive impairment and anxiety-like behavior. Furthermore, the Ndufs4 cKO mice may be a useful model to study the role of mitochondrial deficiencies in mental disorders.
Methods
Generation of Ndufs4 cKO mice
The experimental Ndufs4 cKO mice (Ndufs4loxP/loxP:: Gt(ROSA)SorfsLacZ/ fsLacZ:: Slc6a3iCre/+) were generated as described34. Ndufs4loxP/loxP::Gt(ROSA)SorfsLacZ/fsLacZ littermates were used as controls. Mice were housed and maintained in standard laboratory conditions of 12:12 h light:dark cycle. Regular chow and water were provided ad libitum. All animal experiments were performed in accordance with the guidelines of the University of Washington and Chonnam National University Institutional Animal Care and Use Committee. All procedures were approved by the University of Washington and Chonnam National University Institutional Animal Care and Use Committee. Male mice were used for behavior experiments.
Immunohistochemistry and quantification of TH+ neurons in the SNpc
Mice were perfused, and the brains were harvested and post-fixed as described34,84. Brain sections (40 μm) were labeled with either mouse or rabbit antibodies against tyrosine hydroxylase (TH, 1:5,000, Pel-Freez Biologicals, Rogers, AR) in PBS containing 0.1% Triton X-100, 5% BSA, and 5% goat serum. Sections were washed and incubated with Alexa Fluor 488 or 568 goat anti-mouse or anti-rabbit IgG respectively (1:200; Molecular Probes, Eugene, OR) for 1 h at room temperature. Photomicrographs were captured using a fluorescence microscope equipped with a digital camera (Axiovert 200 M, Zeiss, Oberkochen, Germany).
TH+ neurons were counted manually, blinded of genotype. Beginning from the first slide of the SNpc section when TH+ neurons were visible, all TH+ neurons were counted on every fourth slide through the entire SNpc in one brain hemisphere. The estimated total number of TH+ neurons in each brain was calculated by multiplying the TH+ cell counts by 8 and presented as the total number of TH+ neurons in the SNpc of each brain. Six mice from each group were analyzed at 9 months of age.
TUNEL staining
TdT-mediated dUTP-biotin nick end labeling (TUNEL) staining labels apoptotic cells by detecting 3′-OH ends of broken DNA. DeadEnd™ TUNEL system (Promega, Madison, WI, USA) was used for TUNEL labeling32. Briefly, sections were rinsed in 0.1 M PBS 3 times and then incubated in isopropanol solutions, rinsed in 0.1 M PBS, incubated in TUNEL dilution buffer for 2 min, followed by another hour at 37 °C after the addition of terminal deoxynucleotidyl transferase (TdT, 1:3 dilution with TUNEL buffer). Finally the sections were incubated in stop buffer at room temperature for 10 min and rinsed in 0.1 M PBS. Three mice from each group were analyzed at 9 months of age.
Dopamine and serotonin measurement
The tissue from the striatum or amygdala was dissected, frozen on dry ice, and sent to the Neurochemistry Core Laboratory at Vanderbilt University’s Center for Molecular Neuroscience Research for dopamine quantification. Briefly, the tissues were homogenized in 0.1 M trichloroacetic acid containing 10 mM sodium acetate/0.1 mM EDTA/1 mM isoproterenol (internal standard)/10.5% methanol, pH 3.8, and centrifuged at 10,000 g for 20 min. Total dopamine content in the supernatant was quantified by HPLC coupled with electrochemical detection (0.7 V). The HPLC system (Antec Leyden, Zoeterwoude, Netherlands) consisted of a 515 HPLC pump, a 717 plus autosampler, an electrochemical detector (Decade II; Antec Leyden), and an HPLC column (150 × 4.6 mm; Nucleosil C18; Phenomenex, Torrance, CA). The homogenization buffer was used as the mobile phase (0.7 ml/min), and 20 μl of the sample was injected into an HPLC column (3.9 × 300 mm; Nova-Pak C18, Waters Milford, MA). Dopamine or serotonin content was normalized to the protein concentration in each tissue. Four mice from each group were analyzed at 9 months of age.
Behavior tests
Animals were habituated to the behavior test room for a week before testing. All behavior tests were carried out with male mice between 10:00 am and 6:00 pm and analyzed by experimenters without the knowledge of the genotype and/or treatment.
Open field test
The open field test was performed to measure locomotor activity as described85. Mice were tested at 3, 6 and 9 months of age (Control n = 8, 14 and 8 respectively; cKO n = 8, 13 and 9 respectively). A TruScan Photo Beam Tracking arena (Coulbourn, Whitehall, PA) was used for the test and TruScan 2.02 software (Coulbourn) was used for data analysis.
Rotarod test
The rotarod test was performed as described84. Control (n = 16) and cKO (n = 15) mice (9 months old) were placed on the stationary cylinder of the rotarod apparatus (San Diego Instruments, San Diego, CA) and habituated on the apparatus for at least four consecutive trials in which the rod was kept at a constant speed (4 rpm). Once the animals were able to stay on the rod rotating at 4 rpm for at least 60s, they were subjected to the rotarod test. Mice were placed on the rod rotating at an accelerating speed from 4 to 29 rpm in 300s. The time before animals fell off the rod was recorded with a maximum cut-off of 300s. Mice were tested for eight consecutive trials with at least 5-min intervals. The data from the last four trials were averaged as the latency to fall.
Novel object recognition test
This assay was done with mice at 3 and 6 months of age (Control n = 7; cKO n = 9 and 10 for 3 and 6 months respectively) as described48,85. Mice were introduced to 2 objects (A and B, or D and E) during training, and then to one familiar (A or D) and one novel object (C or F) at 1 or 24 h later during the test session. A 5-min session was used for both training and testing. Exploratory activity towards each toy was recorded for both training and testing sessions and analyzed by experimenters blinded to genotype. Significantly more time spent exploring the novel object than the familiar object during testing indicates memory retention for the familiar object.
Novel object location memory test
This assay was done with mice at 3 and 6 months of age (Control n = 7 and 9 respectively; cKO n = 9 and 10 respectively) as described48. Briefly, a 5-min session was used for both training and testing. Mice were allowed to explore two objects during training. The same two objects were used during testing except that one of them (randomly chosen) was moved to a different location. Exploratory activity towards each object was recorded for both training and testing sessions and analyzed by experimenters blinded to genotype. Increased time spent exploring the object in the novel location during testing indicates retention of spatial memory for the relative locations of the two objects presented during training.
Morris water maze assay
This was done as described85. Briefly, control (n = 9) and cKO (n = 10) mice (9 months old) were placed in a 1.2-meter-diameter, 25-cm-deep pool of water at room temperature (25 °C). Non-toxic white paint was added to make the water opaque. Three extra-maze cues, different in shape and size were placed on the wall surrounding the water tank. A small escape platform (13 cm x 8 cm) made of clear plexiglass was submerged just below the surface of the water and maintained in a fixed location for the entire training session. A total of 28 trials (4 trials per day for 7 consecutive days) were performed during the training session. Mice were allowed to swim for 40 s to find the platform, or guided to the platform after 40 s of the allotted maximum swim time was reached. Each trial ended after mice were allowed to stay on the escape platform for 15 s. A probe test was performed 24 h after the last training trial in which the escape platform was removed and mice were allowed to swim for 60 s in search of the escape platform. Reversal training ensued 24 h after the probe test for a total of 24 trials (4 trials per day for 6 consecutive days) where the escape platform was placed in the opposite quadrant from the initial training session. A reversal probe test was performed 24 h after the last trial of reversal training. Inter-trial interval was 30 min for all sessions and tests. All data were collected and analyzed using ANYmaze software (San Diego Instruments).
Contextual fear conditioning and contextual fear extinction
This was done as described with slight modifications85. Briefly, control (n = 8) and cKO (n = 8) mice (9 months old) were placed in a 10″(W) x 10″(D) x 16″(H) square-shaped arena fitted with a metal grid shock floor (Coulbourn). On the day of training, each mouse was placed in the training context (with striped wallpaper) and allowed to freely explore for 2 min. A 90 dB tone, the conditioned stimulus (CS), was then presented for 30s. During the final 2s of tone presentation, a 0.7 mA foot shock, the unconditioned stimulus (US), was delivered. CS and US were delivered automatically using tone generator and shocker controlled by TruScan software (Coulbourn). Mice were then returned to their home cages. Twenty-four hours later, the contextual fear conditioning test was performed in the training room, where mice were placed in the same context arena without any foot shock for 2 min. Two hours after the contextual test, mice were subjected to a cued test. Mice were placed in a novel context (hexagonal shaped arena with clear plastic walls) in a different room and allowed to freely explore for 2 min. The CS (tone) was then presented for 2 min. Two hours after the cued test, mice were subjected to a control test in which they were placed in another novel context (triangular shaped arena with solid grey plastic walls) in a third room for 2 min with no presentation of either tone or foot shock. Freezing behavior was recorded manually every 5s for each of the 2 min assessment periods for the three tests. Freezing behavior was defined as lack of bodily movement with all 4 paws remaining stationary on the floor except normal respiration.
One day following cued and contextual fear conditioning tests, mice were placed in the training context without the foot shock or tone for one extinction trial per day for 8 consecutive days. Each extinction trial lasted a total of 10 min and freezing behavior was recorded during the final 2 min of each trial. One day after the completion of the 8 d fear extinction trials, mice were placed in a novel context (trapezoid shaped arena with solid grey plastic walls) and freezing behavior was recorded for 2 min.
Elevated plus maze test
The elevated plus maze test was done as described86. The apparatus consisted of two open arms and two enclosed arms arranged in a plus-sign orientation. The arms were elevated 30 inches above the floor, with each arm projecting 12 inches from the center. Mice were tested at 3, 6 and 9 months of age (Control n = 7, 8 and 11 respectively; cKO n = 9, 10 and 11 respectively). Exploratory activities in both open and closed arms were recorded and analyzed using ANYmaze system (Stoelting, Wood Dale, IL) by experimenters blinded to genotype. Because rodents naturally prefer dark and enclosed compartments, a greater willingness to explore the open arms indicates less anxiety while more time spent in the closed arms is indicative of increased anxiety87,88,89.
Light/dark box assay
This test was performed as described90. A 2-chamber box (10″ width × 5″ depth × 16″ height for each chamber) was used for this test (Light/dark chamber, San Diego Instruments Co). Mice were tested at 3, 6 and 9 months of age (Control n = 7, 8 and 7 respectively; cKO n = 9, 10 and 8 respectively). Mice were habituated in the room for 1 h before the test with the room lights off. During the test, a mouse was placed in the dark chamber and allowed to freely move for 5 min. The time spent in each chamber was scored.
Social Interaction Test
The social interaction test was performed as described91 in a regular cage. Each of control (n = 8) and cKO (n = 7) mice (9 months old) was placed in the cage for 10 min for habituation. On test day, pairs of unfamiliar mice were placed in the cage for 10 min. Mouse behavior and the time of social interaction was recorded by a computer and the video-tracking system (Stoelting).
Statistical analysis
All data are expressed as mean ± standard error of the means (s.e.m.). Statistical analysis was performed using Two-way ANOVA with repeated measures to analyze data for the water maze tests and fear extinction assays. One-Way ANOVA was used to analyze the other behavior data. Student’s t-test was used for all the other data, n.s. not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
Additional Information
How to cite this article: Choi, W.-S. et al. Conditional deletion of Ndufs4 in dopaminergic neurons promotes Parkinson’s disease-like non-motor symptoms without loss of dopamine neurons. Sci. Rep. 7, 44989; doi: 10.1038/srep44989 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
This work was supported by NIH grants ES012215 and ES013696 (ZX), Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A3B03931710, WSC) and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013R1A1A1059258, WSC, 2014R1A1A2055836, HWK, and 2016R1D1A1B03933283, HWK), and facilitated by grant P30 HD02274 from the National Institute of Child Health and Human Development. We thank members of the Xia laboratory for technical assistance on behavior tests, and for critical reading of the manuscript.
Footnotes
The authors declare no competing financial interests.
Author Contributions W.S.C., H.W.K., and Z.X. designed the experiments, analyzed data, and contributed to the main text; W.S.C. and H.W.K. performed experiments; W.S.C. and Z.X. designed the project and wrote the text. R.D.P. generated Ndufs4lox/lox mice and contributed to the text. F.T. generated Slc6a3iCre/+ mice; D.S. contributed to the design of experiments and writing of the manuscript; Z.X. supervised the project.
References
- Dawson T. M. & Dawson V. L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302, 819–822 (2003). [DOI] [PubMed] [Google Scholar]
- Hirsch E. C., Jenner P. & Przedborski S. Pathogenesis of Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 28, 24–30 (2013). [DOI] [PubMed] [Google Scholar]
- Olanow C. W. & Tatton W. G. Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci 22, 123–144 (1999). [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman P. M., Muqit M. M. & Wood N. W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci 7, 207–219 (2006). [DOI] [PubMed] [Google Scholar]
- Langston J. W., Ballard P., Tetrud J. W. & Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 (1983). [DOI] [PubMed] [Google Scholar]
- Lang A. E. & Blair R. D. Parkinson’s disease in 1984: an update. Can Med Assoc J 131, 1031–1037 (1984). [PMC free article] [PubMed] [Google Scholar]
- Haas R. H. et al. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Annals of neurology 37, 714–722 (1995). [DOI] [PubMed] [Google Scholar]
- Mizuno Y. et al. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 163, 1450–1455 (1989). [DOI] [PubMed] [Google Scholar]
- Parker W. D. Jr., Boyson S. J. & Parks J. K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Annals of neurology 26, 719–723 (1989). [DOI] [PubMed] [Google Scholar]
- Schapira A. H. et al. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269 (1989). [DOI] [PubMed] [Google Scholar]
- Morais V. A. et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344, 203–207 (2014). [DOI] [PubMed] [Google Scholar]
- Vilain S. et al. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet 8, e1002456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais V. A. et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med 1, 99–111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. et al. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci USA 108, 12920–12924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz G. P. et al. Membrane-permeable Bcl-xL prevents MPTP-induced dopaminergic neuronal loss in the substantia nigra. J Neurochem 104, 757–765 (2008). [DOI] [PubMed] [Google Scholar]
- Gibrat C. et al. Differences between subacute and chronic MPTP mice models: investigation of dopaminergic neuronal degeneration and alpha-synuclein inclusions. J Neurochem 109, 1469–1482 (2009). [DOI] [PubMed] [Google Scholar]
- Huang E. et al. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat Cell Biol 12, 563–571 (2010). [DOI] [PubMed] [Google Scholar]
- Kim S. T. et al. The effect of electroaucpuncture for 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced proteomic changes in the mouse striatum. J Physiol Sci 60, 27–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroske E., Meredith G. E., Callen S., Totterdell S. & Lau Y. S. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience 106, 589–601 (2001). [DOI] [PubMed] [Google Scholar]
- Pinna A. et al. A new ethyladenine antagonist of adenosine A(2A) receptors: behavioral and biochemical characterization as an antiparkinsonian drug. Neuropharmacology 58, 613–623 (2010). [DOI] [PubMed] [Google Scholar]
- Zhao Q., Gao J., Li W. & Cai D. Neurotrophic and neurorescue effects of Echinacoside in the subacute MPTP mouse model of Parkinson’s disease. Brain Res 1346, 224–236 (2010). [DOI] [PubMed] [Google Scholar]
- Betarbet R. et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3, 1301–1306 (2000). [DOI] [PubMed] [Google Scholar]
- Sherer T. B., Kim J. H., Betarbet R. & Greenamyre J. T. Subcutaneous Rotenone Exposure Causes Highly Selective Dopaminergic Degeneration and alpha-Synuclein Aggregation. Exp. Neurol. 179, 9–16 (2003). [DOI] [PubMed] [Google Scholar]
- Inden M. et al. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J Neurochem 101, 1491–1504 (2007). [DOI] [PubMed] [Google Scholar]
- Pan-Montojo F. et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5, e8762 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacco S. et al. Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J Biol Chem 278, 44161–44167 (2003). [DOI] [PubMed] [Google Scholar]
- van den Heuvel L. et al. Demonstration of a new pathogenic mutation in human complex I deficiency: a 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am J Hum Genet 62, 262–268 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde S. M. et al. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem Biophys Res Commun 275, 63–68 (2000). [DOI] [PubMed] [Google Scholar]
- Petruzzella V. & Papa S. Mutations in human nuclear genes encoding for subunits of mitochondrial respiratory complex I: the NDUFS4 gene. Gene 286, 149–154 (2002). [DOI] [PubMed] [Google Scholar]
- Vogel R. O. et al. Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Mol Genet Metab 91, 176–182 (2007). [DOI] [PubMed] [Google Scholar]
- Kruse S. E. et al. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metabolism 7, 312–320 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W. S., Kruse S. E., Palmiter R. D. & Xia Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc Natl Acad Sci USA 105, 15136–15141 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W. S., Palmiter R. D. & Xia Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J Cell Biol 192, 873–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. W. et al. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiology of aging 36, 2617–2627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya M., Kubu C. S. & Giroux M. L. Parkinson disease: not just a movement disorder. Cleve Clin J Med 75, 856–864 (2008). [DOI] [PubMed] [Google Scholar]
- Aarsland D., Taylor J. P. & Weintraub D. Psychiatric issues in cognitive impairment. Movement disorders: official journal of the Movement Disorder Society 29, 651–662 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima M. M. et al. Motor and non-motor features of Parkinson’s disease - a review of clinical and experimental studies. CNS Neurol Disord Drug Targets 11, 439–449 (2012). [DOI] [PubMed] [Google Scholar]
- Hawkes C. Olfaction in neurodegenerative disorder. Adv Otorhinolaryngol 63, 133–151 (2006). [DOI] [PubMed] [Google Scholar]
- Ward C. D., Sanes J. N., Dambrosia J. M. & Calne D. B. Methods for evaluating treatment in Parkinson’s disease. Advances in neurology 37, 1–7 (1983). [PubMed] [Google Scholar]
- Aarsland D. et al. The rate of cognitive decline in Parkinson disease. Archives of neurology 61, 1906–1911 (2004). [DOI] [PubMed] [Google Scholar]
- Chaudhuri K. R. & Schapira A. H. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. The Lancet. Neurology 8, 464–474 (2009). [DOI] [PubMed] [Google Scholar]
- Braak H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of aging 24, 197–211 (2003). [DOI] [PubMed] [Google Scholar]
- Kish S. J. et al. Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain: a journal of neurology 131, 120–131 (2008). [DOI] [PubMed] [Google Scholar]
- Frisina P. G., Haroutunian V. & Libow L. S. The neuropathological basis for depression in Parkinson’s disease. Parkinsonism & related disorders 15, 144–148 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisina P. G., Tse W., Halbig T. D. & Libow L. S. The pattern of cognitive-functional decline in elderly essential tremor patients: an exploratory-comparative study with Parkinson’s and Alzheimer’s disease patients. Journal of the American Medical Directors Association 10, 238–242 (2009). [DOI] [PubMed] [Google Scholar]
- Carpenter A. C., Saborido T. P. & Stanwood G. D. Development of hyperactivity and anxiety responses in dopamine transporter-deficient mice. Developmental neuroscience 34, 250–257 (2012). [DOI] [PubMed] [Google Scholar]
- Oliveira A. M., Hawk J. D., Abel T. & Havekes R. Post-training reversible inactivation of the hippocampus enhances novel object recognition memory. Learn Mem 17, 155–160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. Genetic Activation of ERK5 MAP Kinase Enhances Adult Neurogenesis and Extends Hippocampus-Dependent Long-Term Memory. J. Neurosci. 34, 2130–2147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. et al. Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior. Neurobiol Learn Mem 87, 149–158 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turiault M. et al. Analysis of dopamine transporter gene expression pattern – generation of DAT-iCre transgenic mice. Febs J 274, 3568–3577 (2007). [DOI] [PubMed] [Google Scholar]
- Hansson A. et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci USA 101, 3136–3141 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky F. H. et al. Altered dopamine metabolism and increased vulnerability to MPTP in mice with partial deficiency of mitochondrial complex I in dopamine neurons. Hum Mol Genet 21, 1078–1089 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsecchi F. et al. Metabolic consequences of NDUFS4 gene deletion in immortalized mouse embryonic fibroblasts. Biochim Biophys Acta 1817, 1925–1936 (2012). [DOI] [PubMed] [Google Scholar]
- Calvaruso M. A. et al. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum Mol Genet 21, 115–120 (2012). [DOI] [PubMed] [Google Scholar]
- Palin E. J., Paetau A. & Suomalainen A. Mesencephalic complex I deficiency does not correlate with parkinsonism in mitochondrial DNA maintenance disorders. Brain: a journal of neurology 136, 2379–2392 (2013). [DOI] [PubMed] [Google Scholar]
- Cheng H. C., Ulane C. M. & Burke R. E. Clinical progression in Parkinson disease and the neurobiology of axons. Annals of neurology 67, 715–725 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley J. M. & Lees A. J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain: a journal of neurology 114 (Pt 5), 2283–2301 (1991). [DOI] [PubMed] [Google Scholar]
- Greffard S. et al. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Archives of neurology 63, 584–588 (2006). [DOI] [PubMed] [Google Scholar]
- Ma S. Y., Roytta M., Rinne J. O., Collan Y. & Rinne U. K. Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J Neurol Sci 151, 83–87 (1997). [DOI] [PubMed] [Google Scholar]
- Riederer P. & Wuketich S. Time course of nigrostriatal degeneration in parkinson’s disease. A detailed study of influential factors in human brain amine analysis. Journal of neural transmission 38, 277–301 (1976). [DOI] [PubMed] [Google Scholar]
- Scherman D. et al. Striatal dopamine deficiency in Parkinson’s disease: role of aging. Annals of neurology 26, 551–557 (1989). [DOI] [PubMed] [Google Scholar]
- Gao Z. G., Cui W. Y., Zhang H. T. & Liu C. G. Effects of nicotine on 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced depression of striatal dopamine content and spontaneous locomotor activity in C57 black mice. Pharmacol Res 38, 101–106 (1998). [DOI] [PubMed] [Google Scholar]
- Ghosh A. et al. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 29, 13543–13556 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden J. P. et al. Dopamine-dependent compensation maintains motor behavior in mice with developmental ablation of dopaminergic neurons. J Neurosci 33, 17095–17107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung L. W. et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J Exp Med 209, 837–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. et al. Selective expression of Parkinson’s disease-related Leucine-rich repeat kinase 2 G2019S missense mutation in midbrain dopaminergic neurons impairs dopamine release and dopaminergic gene expression. Hum Mol Genet 24, 5299–5312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Manchado A. B. et al. Chronic and progressive Parkinson’s disease MPTP model in adult and aged mice. J Neurochem 136, 373–387 (2016). [DOI] [PubMed] [Google Scholar]
- Rojas P. et al. EGb761 protects against nigrostriatal dopaminergic neurotoxicity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice: role of oxidative stress. Eur J Neurosci 28, 41–50 (2008). [DOI] [PubMed] [Google Scholar]
- Dauer W. & Przedborski S. Parkinson’s Disease: Mechanisms and Models. Neuron 39, 889–909 (2003). [DOI] [PubMed] [Google Scholar]
- Blonder L. X. & Slevin J. T. Emotional dysfunction in Parkinson’s disease. Behav Neurol 24, 201–217 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor T. N. et al. Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci 29, 8103–8113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q. et al. Dopamine and serotonin signaling during two sensitive developmental periods differentially impact adult aggressive and affective behaviors in mice. Mol Psychiatry 19, 688–698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvas M., Henschen C. W. & Palmiter R. D. Contributions of signaling by dopamine neurons in dorsal striatum to cognitive behaviors corresponding to those observed in Parkinson’s disease. Neurobiol Dis 65, 112–123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvas M. & Palmiter R. D. Contributions of striatal dopamine signaling to the modulation of cognitive flexibility. Biol Psychiatry 69, 704–707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarinana J., Kitamura T., Kunzler P., Sultzman L. & Tonegawa S. Differential roles of the dopamine 1-class receptors, D1R and D5R, in hippocampal dependent memory. Proc Natl Acad Sci USA 111, 8245–8250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva W. C., Kohler C. C., Radiske A. & Cammarota M. D1/D5 dopamine receptors modulate spatial memory formation. Neurobiol Learn Mem 97, 271–275 (2012). [DOI] [PubMed] [Google Scholar]
- Otmakhova N. A. & Lisman J. E. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci 16, 7478–7486 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. T. et al. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23, 787–798 (1999). [DOI] [PubMed] [Google Scholar]
- Rezin G. T. et al. Inhibition of mitochondrial respiratory chain in brain of rats subjected to an experimental model of depression. Neurochem Int 53, 395–400 (2008). [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D. & Karry R. Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS One 3, e3676 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scola G., Kim H. K., Young L. T. & Andreazza A. C. A fresh look at complex I in microarray data: clues to understanding disease-specific mitochondrial alterations in bipolar disorder. Biol Psychiatry 73, e4–5 (2013). [DOI] [PubMed] [Google Scholar]
- Andreazza A. C., Shao L., Wang J. F. & Young L. T. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry 67, 360–368 (2010). [DOI] [PubMed] [Google Scholar]
- Hollis F. et al. Mitochondrial function in the brain links anxiety with social subordination. Proc Natl Acad Sci USA 112, 15486–15491 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W. S., Abel G., Klintworth H., Flavell R. A. & Xia Z. JNK3 mediates paraquat- and rotenone-induced dopaminergic neuron death. J. Neuropathol. Exp. Neurol. 69, 511–520 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y. W., Chan G. C. K., Kuo C. T., Storm D. R. & Xia Z. Inhibition of Adult Neurogenesis by Inducible and Targeted Deletion of ERK5 Mitogen-Activated Protein Kinase Specifically in Adult Neurogenic Regions Impairs Contextual Fear Extinction and Remote Fear Memory. J Neurosci 32, 6444–6455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J. et al. Conditional Inhibition of Adult Neurogenesis by Inducible and Targeted Deletion of ERK5 MAP Kinase Is Not Associated with Anxiety/Depression-Like Behaviors. eneuro 2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- File S. E., Kenny P. J. & Ouagazzal A. M. Bimodal modulation by nicotine of anxiety in the social interaction test: role of the dorsal hippocampus. Behav Neurosci 112, 1423–1429 (1998). [DOI] [PubMed] [Google Scholar]
- Paine T. A., Jackman S. L. & Olmstead M. C. Cocaine-induced anxiety: alleviation by diazepam, but not buspirone, dimenhydrinate or diphenhydramine. Behav Pharmacol 13, 511–523 (2002). [DOI] [PubMed] [Google Scholar]
- Pellow S., Chopin P., File S. E. & Briley M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods 14, 149–167 (1985). [DOI] [PubMed] [Google Scholar]
- Zou J., Storm D. R. & Xia Z. Conditional Deletion of ERK5 MAP Kinase in the Nervous System Impairs Pheromone Information Processing and Pheromone-Evoked Behaviors. PLoS One 8, e76901 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S. et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci USA 105, 1710–1715 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]