

Abstract
Objective
Mitogen-inducible gene 6 (MIG-6) regulates epidermal growth factor receptor (EGFR) signaling in synovial joint tissues. Whole-body knockout of the Mig6 gene in mice has been shown to induce osteoarthritis and joint degeneration. To evaluate the role of chondrocytes in this process, Mig6 was conditionally deleted from Col2a1-expressing cell types in the cartilage of mice.
Methods
Bone and cartilage in the synovial joints of cartilage-specific Mig6-deleted (knockout [KO]) mice and control littermates were compared. Histologic staining and immunohistochemical analyses were used to evaluate joint pathology as well as the expression of key extracellular matrix and regulatory proteins. Calcified tissue in synovial joints was assessed by micro–computed tomography (micro-CT) and whole-skeleton staining.
Results
Formation of long bones was found to be normal in KO animals. Cartilage thickness and proteoglycan staining of articular cartilage in the knee joints of 12-week-old KO mice were increased as compared to controls, with higher cellularity throughout the tissue. Radiopaque chondro-osseous nodules appeared in the knees of KO animals by 12 weeks of age and progressed to calcified bone–like tissue by 36 weeks of age. Nodules were also observed in the spine of 36-week-old animals. Erosion of bone at ligament entheses was evident by 12 weeks of age, by both histologic and micro-CT assessment.
Conclusion
MIG-6 expression in chondrocytes is important for the maintenance of cartilage and joint homeostasis. Dysregulation of EGFR signaling in chondrocytes results in anabolic activity in cartilage, but erosion of ligament entheses and the formation of ectopic chondro-osseous nodules severely disturb joint physiology.
Osteoarthritis (OA) is a degenerative joint disease that affects more than 10% of the adult population of North America (1). There are currently no clinically approved disease-modifying treatments for OA. Consequently, patients experience a general loss in their quality of life, with increased health care costs and lost work time causing considerable socioeconomic damage (2). While the precise cause and pathobiology of this disease are not fully understood, both mechanical and genetic factors play a key role in specific subtypes of OA (2).
The major hallmark of OA is the degeneration of the articular cartilage and the altered phenotype or death of the only cell type in cartilage, chondrocytes (2). Chondrocytes are essential for maintaining articular cartilage, which is largely composed of a highly complex network of extracellular matrix (ECM) proteins (2). In an anabolic state, chondrocytes produce and lay down these structural proteins into the surrounding matrix to repair damage and build up the cartilage (2). In a catabolic state, chondrocytes release factors to turn over surrounding cartilage through proteolytic digestion (e.g., matrix metalloproteinases) and through release of cytokines (e.g., tumor necrosis factor α and interleukin-1β), which induce catabolic activity in surrounding cells and tissues (synovium, bone, meniscus) (2). In healthy cartilage, anabolic and catabolic processes are in balance, while in OA, this balance is disrupted toward a net increase in catabolic activity (2).
Bone is important in supporting the overlying cartilage but has also been implicated in the initiation and progression of OA (3). Both sclerosis of subchondral bone and the formation of bony osteophyte outgrowths are common in the mid-to-late stages of OA in response to local growth factors (such as transforming growth factor β [TGFβ] and bone morphogenetic protein) (2). Abnormal geometry of the bone in a joint can affect the distribution of forces and joint stability and can lead to cartilage degeneration (4). The ligaments, tendons, and the meniscus in the knee further stabilize the joint and prevent malalignment and overextension. Many animal models of OA exploit the stabilizing function of the ligaments in the knee joint by using surgical techniques to reproducibly induce a destabilized joint, with subsequent development of articular cartilage degeneration and other OA-like phenotypes (5–8).
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that signals through multiple downstream pathways in a ligand- and context-specific manner (9,10). EGFR ligands include epidermal growth factor (EGF), TGFα, amphiregulin, heparin-binding EGF-like growth factor (HB-EGF), and others (11). Evidence shows that EGFR signaling is involved in bone and cartilage physiology during both development and homeostasis in adulthood. EGFR signaling induced by EGF and TGFα is highly important in the regulation of endochondral ossification during the formation of bone (9,12–14). We have shown that TGFα/EGF can stimulate catabolic activity in chondrocytes in vitro and cartilage degeneration ex vivo (15,16). TGFα is also up-regulated in some forms of human OA and rheumatoid arthritis, as well as in animal models of osteoarthritis (15,17–19). EGFR signaling regulates the differentiation of osteoclasts, an important cell type in the turnover of bone (20). Tight regulation of EGFR signaling is therefore required for the maintenance of cartilage and bone health.
Fine tuning of EGFR signaling takes place at multiple levels, from gene transcription to ligand binding (21). Mitogen-inducible gene 6 (MIG-6), which is also known as ERBB receptor–feedback inhibitor 1, receptor-associated late transducer, and Gene 33, is a scaffold protein that binds to EGFR and impedes downstream signaling while targeting it for internalization and degradation (22,23). Pathways downstream of EGFR activation stimulate the induction of Mig6, which then acts as a negative-feedback response and attenuates EGFR signaling (24).
Whole-body deletion of Mig6 in mice results in a complex set of phenotypes, including the degeneration of articular joints, breakdown of articular cartilage observed at a relatively early age, and severely diseased joints in an injury-induced model of OA (8,25–27). Mig6 deletion targeted to the developing mouse limb mesenchyme using the Prx1-Cre driver line results in the transient anabolic buildup of cartilage and the development of an OA-like phenotype with increasing age (28). Since Mig6 is deleted from all mesenchymal limb tissues in these mice, it is unclear whether bone, cartilage, synovium, or multiple tissue types acting together are involved in the development of cartilage degeneration in these knockout (KO) models.
To evaluate the contribution of MIG-6 in the regulation of EGFR activity in cartilage homeostasis and OA, we used a Col2a1 promoter–driven Cre/lox system to selectively delete Mig6 from chondrocytes in mice.
MATERIALS AND METHODS
Animals
All mice were bred and housed in accordance with the Animal Care and Use Guidelines of Western University. To conditionally delete Mig6 in chondrocytes, MIG-6fl/fl (26,28) mice were bred with Col2a1-Cre mice (29–32) to generate knockout (Mig-6fl/flCol2a1-Cre+/−), heterozygous (Mig-6fl/+ Col2a1-Cre+/−), and control (Mig-6fl/flCol2a1-Cre−/−or Mig-6fl/+Col2a1-Cre−/−) animals. Mice were weighed prior to killing by CO2 asphyxiation. Genotype frequency was determined by polymerase chain reaction (PCR) analysis using DNA processed from biopsy samples of ear tissue from mice surviving to at least 21 days of age. Genomic excision of Mig6 was assessed using standard PCR, with primers flanking the loxP-flanked (floxed) region of the gene (exons 2–4) (26). Gross morphology of the joint was imaged using a Leica EC3 camera and a Leica S6-D microscope.
Skeletal preparation and long bone measurement
Animals were euthanized, skinned, and eviscerated on postnatal day 40. Overnight fixation in 70% ethanol was followed by 2 days in acetone. Animals were stained in a solution of 0.05% alizarin red, 0.015% Alcian blue, and 5% glacial acetic acid in 70% ethanol for 10–12 days, with clearing of extraskeletal tissues using a graded series of 2–0.5% KOH (31). Long bones (femur, tibia, humerus, radius, and ulna) were measured using Leica Application Suite software (v3.8.0) on images obtained using a Leica EC3 camera and a Leica S6-D microscope.
Histologic assessment
Limbs were fixed in 4% paraformaldehyde for 24 hours and decalcified in 5% EDTA in phosphate buffered saline (PBS), pH 7.0, for 10–12 days. Joints were processed and embedded in paraffin in sagittal or frontal orientation, with serial sections taken at a thickness of 5 μm. All sections were deparaffinized in xylene, rehydrated in a graded series of 100–70% ethanol in water, followed by 100% water, and stained as follows. Glycosaminoglycan content and general histology were assessed in sections stained with Safranin O–fast green (0.02% fast green for 30 minutes, 1% acetic acid for 10 seconds, and 1.5% Safranin O for 3 minutes) or toluidine blue (0.04% toluidine blue in 0.2M acetate buffer, pH 4.0, for 10 minutes). Staining of collagen content was performed using picrosirius red (0.1% sirius red in saturated picric acid solution for 60 minutes, with 0.5% acetic acid washes). The size and organization of collagen fibrils were determined using polarized light microscopy (33). Light intensity and tissue angle (45°) relative to polarizing filter (Leica no. 11505087) and analyzer (Leica no. 11555045) were kept identical between samples.
Following antigen retrieval and blocking with 5% serum in PBS, sections were stained overnight at 4°C by immunohistochemistry using primary antibody for SOX9 (R&D Systems), phospho-EGFR (phosphoTyr-1173; Cell Signaling Technology), type II collagen (Santa Cruz Biotechnology), or proliferating cell nuclear antigen (PCNA; Cell Signaling Technology). Sections were incubated with secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology). Following DAB+ chromogen (Dako Canada) exposure, sections were counterstained with methyl green (0.5% methyl green in 0.1M sodium acetate buffer, pH 4.2, for 5 minutes). All sections were dehydrated in a graded series of 70–100% ethanol in water, followed by 100% xylene, and mounted using xylene-based mounting media. Tartrate-resistant acid phosphatase (TRAP; Sigma Canada) staining was performed on sections of 12-week-old knee joints according to the manufacturer’s instructions, with minor alterations (i.e., 60 minutes in Triton X-100). All images were taken with a Leica DFC295 digital camera and a Leica DM1000 microscope.
Articular cartilage measures
Knee articular cartilage thickness and chondrocyte cell density were determined from Safranin O–fast green–stained frontal sections by 2 observers (MAP and BAR) who were blinded with regard to the tissue source. Leica Application Suite software (v3.8.0) was used to evaluate the average articular cartilage thickness by measuring from the articular surface to the subchondral bone interface across 3 points in each quadrant of the knee joint (medial and lateral tibia and femur), in 4 sections spanning at least 500 μm. Cell density of knee articular cartilage chondrocytes was determined by counting all lacunae with evidence of nuclear staining in the medial femur/tibia using a centered region of interest measuring 200 μm wide and 100 μm deep from the articular surface.
Micro–computed tomography (micro-CT)
Whole-body scans of 36-week-old MIG-6–KO mice were conducted using a GE eXplore speCZT micro-CT scanner at a resolution of 50 μ/voxel. High-resolution scans of the left knee of 12-week-old mice were conducted using a GE eXplore RS micro-CT scanner at a resolution of 20 μ/voxel. Mice were evaluated for morphologic changes using GE Healthcare MicroView software (v2.2) to generate 2-dimensional (2-D) maximum intensity projection (MIP) and 3-D isosurface images (34,35). Abnormal tissue was manually highlighted in red in Adobe Photoshop CC.
Statistical analysis
Data are presented as the mean ± SEM. All statistical analyses were performed using GraphPad Prism software (v6.0).
RESULTS
Normal early bone development in cartilage-specific MIG-6–KO mice
Excision of Mig6 from the Col2a1-expressing tissues of Mig-6fl/flCol2a1-Cre+/−(KO) and Mig-6fl/+Col2a1-Cre−/− (heterozygous) animals was confirmed using PCR amplification for an excision-specific product (433 bp) (Figure 1A). Xiphoid cartilage, meniscus, and ectopic growths (described below) demonstrated excision of Mig6, which was not seen in control liver tissue (Figure 1A) or in the brain or heart (results not shown). MIG-6–KO mice appeared outwardly normal and healthy at weaning (Figure 1B). Genotype analyses based on 644 pups in 86 litters demonstrated no evidence of embryonic or postnatal lethality (Figure 1C). The weights of animals were comparable from birth through adulthood (Figure 1D). Measurement of the femur, tibia, humerus, radius, and ulna showed no statistically significant differences in long bone length on postnatal day 40 in female animals (Figure 1E).
Figure 1.
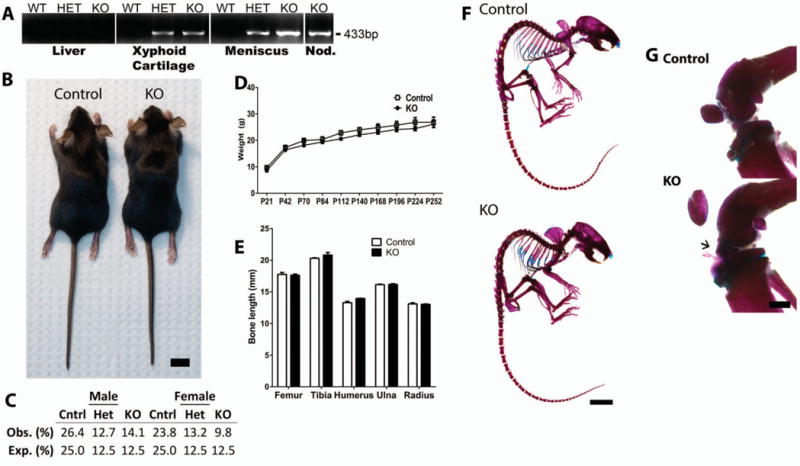
Normal skeletal development in cartilage-specific mitogen-specific gene 6 (MIG-6)–knockout (KO) mice and control mice. A, Presence of 433-bp polymerase chain reaction product (indicating recombination) only in joint tissues, but not control tissues (such as the liver), from heterozygous (HET) and KO mice as compared with wild-type (WT) control mice. Nod. = nodule (ectopic growth). B, Photograph of representative 28-day-old (postnatal day 28 [P28]) female control and KO mice used in the studies. Bar = 1 cm. C, Observed (Obs.) genotype frequencies of male and female weanling (P21) mice, showing no statistically significant difference from expected (Exp.) values (P = 0.280), by chi-square test. D, Changes in the weights of mice from P21 to P252, showing no statistically significant differences. E, Lengths of long bones obtained from 40-day-old mice, showing no statistically significant differences. Values in D and E are the mean ± SEM. F, Representative alizarin red/Alcian blue–stained skeletons of 40-day-old mice. Bar = 1 cm. G, Alizarin red/Alcian blue–stained knee joints from 40-day-old mice, showing normal joint morphology but abnormal staining of the tissue anterior to the meniscus (arrow) in the KO mouse. Images are representative of 5 or more mice per group. Bar = 1 mm.
Further evaluation of overall skeletal morphology by alizarin red/Alcian blue staining showed no overt differences in the development of the axial or appendicular skeleton on postnatal day 40 in female MIG-6–KO animals (Figure 1F). Despite a lack of overall skeletal phenotype, evidence of early ectopic growth development in knee joint tissue was seen on postnatal day 40 in some female KO animals, as indicated by diffuse alizarin red/Alcian blue staining of tissue from the anterior meniscus (Figure 1G). The majority of male and female KO mice developed ectopic growths in one or both knee joints, with few exceptions (data available upon request from the corresponding author). Based on these data and on the similar knee joint histopathologic features between the sexes (data not shown), females were chosen for detailed analyses for the remainder of this study.
Anabolic buildup of articular cartilage in KO mice
Proteoglycan staining (Safranin O) of tissue sections appeared to be increased in the articular cartilage of KO mice at 12 weeks, but decreased at 36 weeks (Figures 2A–D). Picrosirius red staining for collagens showed increased staining in the highly cellular areas in the articular cartilage of KO mice (Figures 2E and F). Further examination of picrosirius red–stained sections under polarized light revealed strong red birefringence of collagen fibers in subchondral bone, with weaker green/yellow birefringence of the articular cartilage in control mice (Figures 2G and H). Interestingly, the superficial and mid-zone articular cartilage of KO animals tended to show more red birefringence as compared to that in control mice, particularly at 12 weeks of age. This may indicate that the ECM organization in these areas is altered compared to controls (Figures 2G and H).
Figure 2.
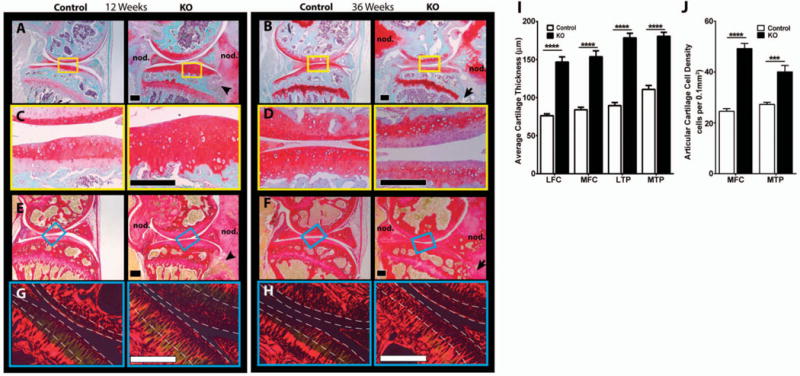
Histologic evaluation of knee articular cartilage in 12-week-old and 36-week-old cartilage-specific mitogen-specific gene 6–knockout (KO) mice and control mice. A–D, Safranin O–fast green–stained sagittal sections of knee joints from 12-week-old (A and C) and 36-week-old (B and D) mice show large chondro-osseous nodules (nod.) and bone infiltration (arrowhead and arrow) in the KO mice. Images in C and D are higher-magnification views of the boxed areas in A and B, respectively. E–H, Picrosirius red–stained sagittal sections of the knee joints from 12-week-old (E and G) and 36-week-old (F and H) mice show tissue infiltrating into bone (arrowhead and arrow) in the KO mice. Images in G and H are higher-magnification views of the boxed areas in E and F, respectively, under polarized light. Areas between the broken lines mark superficial and mid-zone cartilage. Images are representative of 5 mice per group. Bars = 200 μm. I, The average thickness of the articular cartilage in the lateral femoral condyle (LFC), medial femoral condyle (MFC), lateral tibial plateau (LTP), and medial tibial plateau (MTP) is significantly increased in the KO mice. J, Cell density in the medial femoral condyle and medial tibial plateau are significantly increased in the KO mice. Values in I and J are the mean ± SEM. *** = P<0.001; **** = P<0.0001 by two-way analysis of variance with Bonferroni multiple comparisons test.
The thickness of the articular cartilage was increased ~1.5-fold in MIG-6–KO mice at 12 weeks of age in all 4 quadrants of the knee joint (Figure 2I). This increase in cartilage thickness in KO mice was reduced at 36 weeks (data not shown). Cellular density was increased in the articular cartilage of the medial tibial plateau and medial femoral condyle of 12-week-old KO mice as compared to control mice (Figure 2J). No abnormalities in cell number, organization, or phenotype were noted in the growth plate of MIG-6–KO animals.
Immunohistochemistry for SOX9, a key regulator of chondrocyte gene expression, showed clear nuclear staining in the expanded articular cartilage of the knee joints of 12-week-old KO animals (Figure 3A). COL2A1 staining in the superficial and mid zone of the knee cartilage from 12-week-old KO animals was similar to that in the controls (Figure 3B). Immunohistochemical staining for phospho-EGFR (Tyr-1173) was increased in the articular cartilage of 12- and 36-week-old KO animals (Figures 3C and D), as expected upon deletion of a negative regulator. Immunostaining for the proliferative marker PCNA in articular cartilage was increased in 12-week-old KO animals (data available upon request from the corresponding author). In the growth plate, staining for phospho-EGFR, SOX9, and PCNA (data available upon request from the corresponding author) in KO animals appeared similar to that in the controls. TRAP staining for osteoclasts was increased in the subchondral (Figure 3E) and trabecular bone (Figure 3F) of 12-week-old KO animals.
Figure 3.
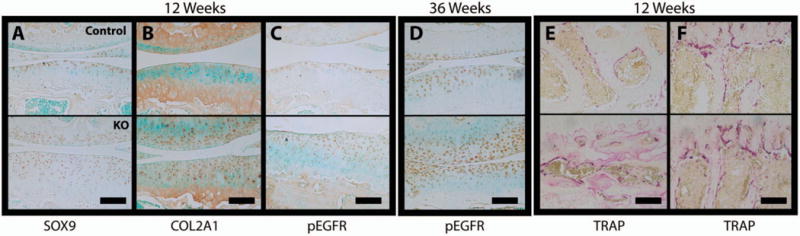
Staining for anabolic/catabolic markers in the knees of 12-week-old and 36-week-old cartilage-specific mitogen-specific gene 6–knockout (KO) mice and control mice. Immunohistochemistry for SOX9 (A), Col2a1 (B), and phosphorylated epidermal growth factor receptor (pEGFR; Tyr-1173) in 12-week-old animals, for pEGFR in 36-week-old animals (D), and for tartrate-resistant acid phosphatase (TRAP) in the subchondral (E) and trabecular (F) bone in 12-week-old animals was performed on joint sections from control (top) and KO (bottom) mice. Images are representative of 5 mice per group. Bars = 100 μm.
Elbow joints from 36-week-old female and male KO mice also demonstrated thickened articular cartilage, increased cell density, and SOX9 staining (data available upon request from the corresponding author).
Formation of chondro-osseous nodules upon cartilage-specific deletion of Mig6
The formation of osteophyte-like chondro-osseous nodules in the knee joints was observed in nearly all skeletally mature KO mice examined (data available upon request from the corresponding author). At 4 weeks of age, knee joints from control and KO mice showed no discernible differences by micro-CT (Figure 4A). However, by 12 weeks of age, all female KO mice had developed visible nodules in at least 1 knee joint (data available upon request from the corresponding author). These nodules appeared as enlarged growths associated with the patella and patellar tendon (data available upon request from the corresponding author). By 36 weeks of age, most KO animals had developed nodules in both knees that prevented normal ambulation (data available upon request from the corresponding author); we were unable to bend and extend these joints harvested from KO mice, but were able to easily bend and extend the joints harvested from control mice.
Figure 4.
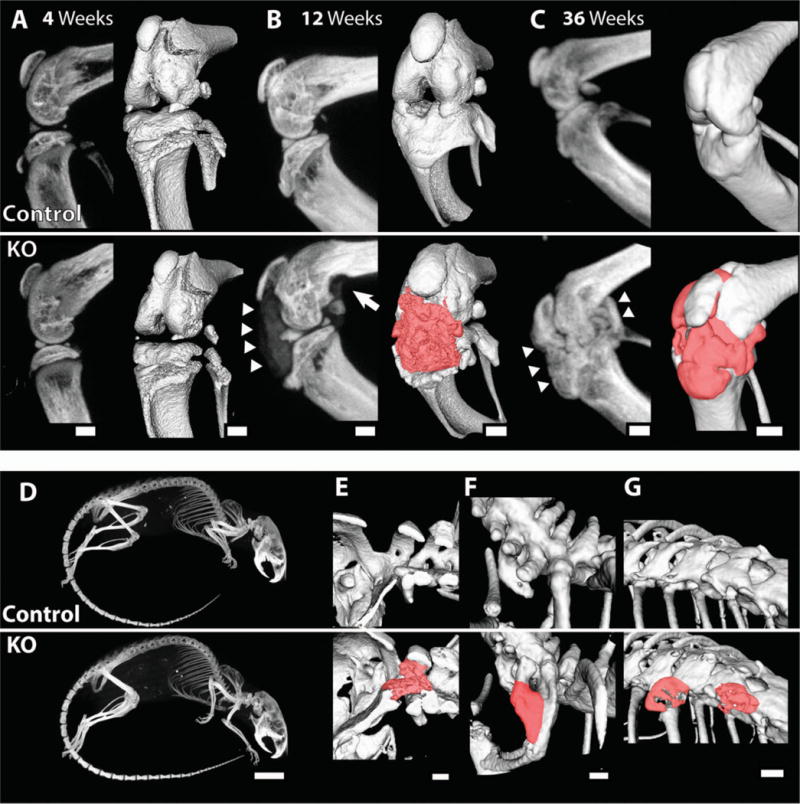
Calcified nodules in the knee and spine of 12-week-old and 36-week-old cartilage-specific mitogen-specific gene 6–knockout (KO) mice and control mice. Control and KO mice were scanned by micro–computed tomography at the indicated ages. A–C, Sagittal plane maximum-intensity projection (MIP) images (left) and 3-dimensional isosurface images (right) obtained at age 4 weeks (A), 12 weeks (B), and 36 weeks (C), showing ectopic calcified tissue (arrowheads and red manual contrast versus white for bone) and bone erosion (arrow) in the KO mice. D, MIP images of 36-week-old mice. E–G, Three-dimensional isosurface images, showing the presence of calcified material (red manual contrast versus white for bone) in some of the 36-week-old KO animals at the base of the skull (E), as a fusion between the C7 and T1 vertebrae (F), and between the T10 and T11 vertebrae (G). Images are representative of 3 or more mice per group. Bars = 1 mm in A–C and E–G; 1 cm in D.
At 12 weeks of age, micro-CT analyses identified calcified tissue in the joint space of KO mice as a diffusely radiopaque material (Figure 4B). This tissue appeared to show less intensity than bone and filled the anterior joint space and posterior areas of the joint (Figure 4B). By 36 weeks, the nodules had developed an intensity similar to bone and had integrated fully with the surrounding joint structure (Figure 4C).
Further examination of similar synovial joints by micro-CT showed no evidence of calcified tissue in the elbow, ankle, or temporomandibular joint (TMJ) (Figure 4D). However, growths along the spine were detected in multiple animals (Figures 4E–G) (additional data available upon request from the corresponding author). Nodules at the base of the skull were observed in KO animals, one of which seemed to integrate with the transverse ligament of C1 (atlas) (Figure 4E). Fusion of the C7 vertebra with the T1 vertebra was observed in 1 of the KO mice (Figure 4F). Another KO mouse developed nodules extending laterally from the transverse processes of T10 and T11 (Figure 4G).
Histologically, the nodules appeared to be composed largely of highly cellular cartilaginous tissue at earlier time points (12 weeks) (Figure 2A), staining strongly with Safranin O. As the animals aged, histologic analysis showed a transition to a mixed chondro-osseous tissue (36 weeks) (Figures 2B and 5A). Examination of sections under polarized light showed strong red birefringence in the meniscus that transitioned to less intense birefringence in the proteoglycan-rich regions of the nodules (Figure 5B). However, calcified regions of the nodule showed strong birefringence similar to that of bone (data not shown). Highly positive TRAP staining was observed in the areas bordering cartilaginous and ossified tissues in the nodular growths from the knees of 12-week-old animals (Figure 5C).
Figure 5.
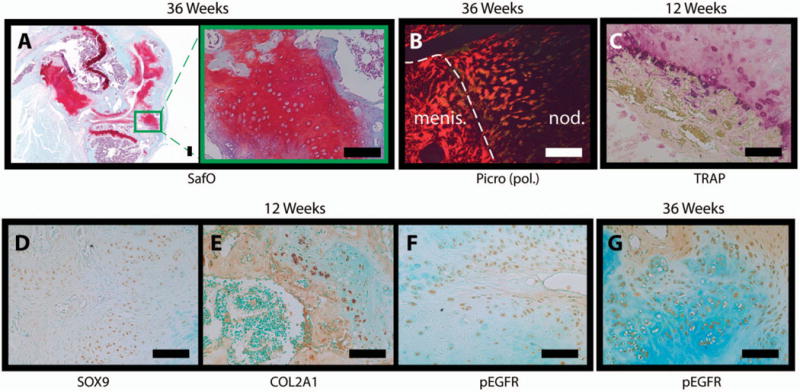
Histology of nodular growths in 12-week-old and 36-week-old cartilage-specific mitogen-specific gene 6–knockout (KO) mice. A, Safranin O (SafO)–fast green stained sections of knee joints from a 36-week-old KO mouse with a nodule. Image at the right is a higher-magnification view of the boxed area in the image at the left. B, Polarized light microscopy [pol.] of a picrosirius red (Picro)–stained nodule (nod.) adjacent to the anterior meniscus (menis.; delineated by the broken line) in section from a 36-week-old KO mouse. C, Tartrate-resistant acid phosphatase (TRAP)–stained nodule in a section from a 12-week-old KO mouse. D–G, Immunohistochemistry for SOX9 (D), Col2a1 (E), and phosphorylated epidermal growth factor receptor (pEGFR; Tyr-1173) in nodule sections from a 12-week-old female KO mouse and for phospho-EGFR in a nodule section from a 36-week-old female KO mouse. Images are representative of 5 mice per group. Bars = 100 μm.
Cells composing the nodules stained strongly for SOX9 (Figure 5D), Col2a1 (Figure 5E), and PCNA (data available upon request from the corresponding author) at 12 weeks. Nodule cells also showed intense staining for phospho-EGFR (Tyr-1173) at both 12 weeks (Figure 5F) and 36 weeks (Figure 5G).
Erosion of bone at ligament entheses in KO mice
Large lesions presenting as radiotransparent noncalcified or partially calcified tissue were visible in the knee joints of MIG-6–KO mice at the age of 12 and 36 weeks, but not at the age of 4 weeks (Figures 6A–D). Histologic analysis revealed that the lesions were associated with ligament entheses, particularly of the anterior and posterior cruciate ligaments (Figure 6E). In the anterior portion of the knee joint, highly cellular, fibrocartilage-like tissue with poor staining for glycosaminoglycan content appeared to invade the subchondral bone via the ligament entheses and did not seem to originate from the articular cartilage (Figures 2A, B, E, and F and Figure 6E). Consistent with these observations, TRAP staining also appeared to be increased in the areas of erosion associated with the ligament entheses (Figure 6F). The growth plate may also be involved in nodular growths and erosion of bone, as invading tissue was associated with a locally disrupted growth plate in severely affected KO mice (Figures 2B, E, and F).
Figure 6.
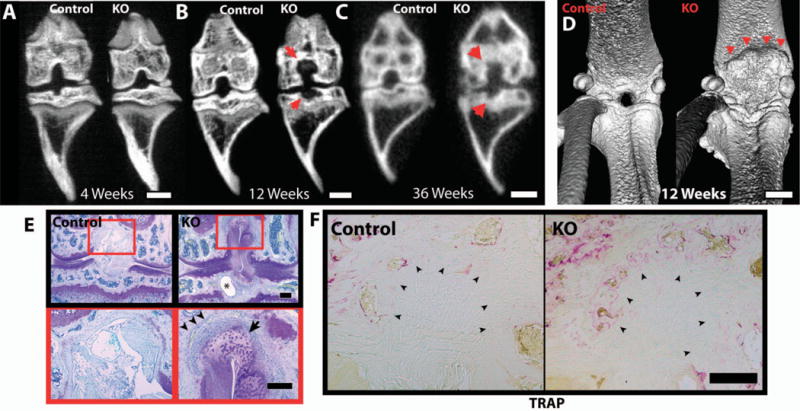
Bone erosion in the knee joints of female 12-week-old and 36-week-old cartilage-specific mitogen-specific gene 6–knockout (KO) mice and control mice. Control and KO mice were scanned by micro–computed tomography at the indicated ages. A–C, Frontal maximum intensity projection (MIP) images obtained at age 4 weeks (A), 12 weeks (B), and 36 weeks (C), showing erosions at the insertion of the cruciate ligament (arrows) in KO mice. D, Three-dimensional isosurface images of 12-week-old control and KO mice, showing erosion of the femur (arrowheads) in the posterior portion of the knee joint of the KO mouse. E, Toluidine blue–stained frontal knee joint sections from 12-week-old control and KO mice, showing a subchondral cyst (asterisk), bone erosion (arrowheads), and abnormal tissue (arrow) at the insertion of the femoral cruciate ligament in the KO mouse. Images at the bottom are higher-magnification views of the boxed areas in the images at the top. F, Tartrate-resistant acid phosphatase (TRAP)–stained section of ligament enthesis from 12-week-old control and KO mice, showing increased staining in the areas of erosion associated with the ligament entheses (arrowheads). Images are representative of 3 mice per group in A–D and 5 mice per group in E and F. Bars = 1 mm in A–D; 200 μm in E and F.
DISCUSSION
Data from this study suggest that disruption of Mig6 function in mouse cartilage leads to a complex mixture of anabolic and catabolic effects in the joints. MIG-6 has been studied extensively for its role as a tumor suppressor in cancer (36–38), but its role in joint development and homeostasis is not well understood, despite the striking joint phenotypes of mice with ubiquitous or Prx1-Cre–driven Mig6 deletion (8,25–28). In the present study, we selectively deleted Mig6 from chondrocytes using a Col2a1-driven Cre/lox-transgenic mouse to examine the effects of Mig6 deletion in cartilage. Based on the available information, we believe that all or most of the described phenotype in our KO mice was due to increased EGFR signaling. We cannot, however, exclude the possibility that other cellular pathways (e.g., signaling from other receptor tyrosine kinases) contribute to some degree.
Suppression of cellular proliferation by Mig6 has previously been shown under multiple experimental conditions in vitro and in vivo (39–41), for example, in keratinocytes (25–27). Our previous work demonstrated that EGFR activation by TGFα promotes proliferation in cultured chondrocytes (15,18). Here, we demonstrated an increase in cellular density in the articular cartilage and high cellularity in chondro-osseous nodules in the cartilage-specific MIG-6–KO animals. Consistent with this finding, the increased staining for PCNA in KO animals points to an increase in proliferation in the articular cartilage. Greater numbers of cells expressing SOX9 and displaying EGFR activation (e.g., phospho-EGFR staining) were present in both the articular cartilage and nodules from KO animals. An increase in SOX9 has also been shown in response to EGFR ligands (amphiregulin, HB-EGF, and others) in human urethral cancer cells (42). Furthermore, a recent study has demonstrated that Prx1-Cre–driven deletion of Mig6 causes increased SOX9 and EGFR activation in cartilage as well as increased cell proliferation in the knee joint (28), similar to what we found in the present studies. In contrast, in our in vitro studies, TGFα was found to decrease Sox9 expression (15,18).
One potential explanation for these seemingly contradictory findings is that our previous studies examined Sox9 messenger RNA levels on a per cell base, whereas here, we studied the expression domain of SOX9 protein in tissue sections. It is therefore feasible to speculate that the increased expression of SOX9 in our MIG-6–KO mice is secondary to increased chondrocyte proliferation, which results from activation of EGFR signaling.
With an increase in chondrocyte number in the articular cartilage of MIG-6–KO animals, an increase in cartilage thickness is not unexpected. Examination of picrosirius red staining under polarized light revealed that the structure of the articular cartilage ECM in KO mice was altered compared to that of control mice. The noncalcified articular cartilage of control animals showed little red-orange birefringence, while that from KO animals showed increased intensity, which may indicate a disruptive shift toward larger, organized collagen fibers (33). The resultant altered stiffness of the cartilage may have an impact on the structural integrity of the cartilage (43). Even so, we were surprised to find that even at 36 weeks of age, the degeneration of articular cartilage from the MIG-6–KO animals was far milder than in previously described whole-body– and limb mesenchyme–targeted KO animals (25,28). Increased subchondral bone stiffness can contribute to the degeneration of overlying cartilage by subjecting it to increased load (2,44), and synovial tissue may also secrete catabolic factors (2). Normal levels of MIG-6 in the bone and other joint tissues of our animals could explain the lack of articular cartilage damage compared to whole-joint levels in MIG-6–KO mice.
However, the difference in cartilage thickness between KO and control mice was much less pronounced at 36 weeks than at 12 weeks. Since the superficial zone appeared to be structurally intact (although with decreased Safranin O staining), the relative loss of cartilage in older KO mice might be due to an advance of the tidemark and, ultimately, replacement of cartilage by bone through endochondral ossification. This model is supported by increased TRAP staining in subchondral bone, as well as by earlier studies showing that TGFα/EGFR signaling in cartilage promotes RANKL expression, osteoclast recruitment, and cartilage-to-bone transition (12,13). Moreover, these data suggest a biphasic mode of EGFR/MIG-6 signaling, where it initially promotes articular cartilage growth through chondrocyte proliferation, but ultimately causes events associated with cartilage maturation and replacement by bone.
Loss of Mig6 has been shown to induce the production of abnormal skeletal growths. Whole-body KO of Mig6 leads to the formation of osteochondral nodules in the knee, ankle, and TMJ (25,26). Conditional knockout of Mig6 in the limb mesenchyme also results in growths that have been described as central and lateral osteophytes in the knee, although their presence in other joints was not reported (28). In our study, targeted deletion of Mig6 in chondrocytes resulted in the formation of chondro-osseous nodules primarily in the knee joint. In contrast to previous findings (25,26), we did not identify abnormal calcified tissue in any other appendicular synovial joints or in the TMJ. Histologic analysis also failed to show abnormal tissue in the elbows of 36-week-old MIG-6–KO animals. However, calcified nodular growths were identified in the spine of 36-week-old animals, indicating that the nodule phenotype is not restricted to the knees.
In many of the MIG-6–KO animals used in this study, nodules did not form with the same dynamics in both knees. Differences in size or complete absence of nodular formation in one knee occurred in both sexes at 12 weeks of age. However, by 36 weeks, the nodules had grown substantially and were present bilaterally in nearly every animal. Osteophytes, which are similar to these nodules, commonly form as a result of surgical insult to the joint in many rodent models (5–7), although usually not as exaggerated as in this study. Induction of MIG-6 has been shown in canine impact models of OA (45,46) and induction of TGFα in rat surgical models of OA (18), demonstrating that EGFR signaling is sensitive to joint injury and mechanical stress. This may indicate that an insult to the joint is required to initiate the process of abnormal growth, perhaps in a dysregulated attempt to repair localized damage.
One open question is the cellular origin of the nodules. Since cells in the nodules show high levels of phospho-EGFR, it seems likely that they derive directly from cells in which the Mig6 gene has been inactivated (e.g., in which type II collagen–Cre is or was expressed). This is further supported by the findings of our PCR analyses of genomic recombination. While it is possible that Col2a1-Cre–mediated Mig6 deletion during development of the synovium may also be involved (47), the lack of nodules observed in other synovial joints indicates that a structure specific to the knee is most likely the source of this pathologic change. Indeed, many nodules appear to start from the meniscus, which expresses Col2a1 during its formation (48).
Novel to this study, cartilage-specific MIG-6–KO mice developed erosive lesions surrounding ligament entheses and exhibited increased TRAP staining in the trabeculae, subchondral bone plate, and cartilaginous tissue–ossified tissue junctions that form in the knee nodules of KO animals. The EGFR pathway has previously been identified as a regulator of osteoclast recruitment to developing bone as a normal component in endochondral ossification (12–14,20). Diminished EGFR signaling through deletion of TGFα or pharmacologic inhibition of EGFR results in decreased RANKL expression in cartilage and decreased TRAP staining in the underlying trabecular bone (12,13,49). However, it is still unclear how osteoclasts would be recruited and sustained at the ligament entheses to create the levels of damage observed at such an early age in our MIG-6–KO animals.
Recent studies described by Staal et al (50) produced data in similar animal models that support the results shown here. Taken together, these studies suggest that a fine balance in EGFR signaling is required to maintain joint homeostasis. Under controlled conditions, therapeutic agents that increase EGFR signaling may be a viable option for promoting anabolic activity in articular cartilage. However, the complications of over-stimulation of the EGFR pathway have been demonstrated here, and additional work is required to further elucidate this complex pathway and its effects in cartilage and joint homeostasis.
Acknowledgments
We would like to thank Holly Dupuis for her assistance with the TRAP staining.
Supported by the Canadian Institutes of Health Research (grant MOP 86574 to Dr. Beier). Mr. Pest’s work was supported by the Canadian Institutes of Health Research (Canadian Graduate Scholarship Doctoral Award and the CIHR Joint Motion Program) and by the Canadian Arthritis Network. Dr. Beier is recipient of a Canada Research Chair in Musculoskeletal Health award.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Beier had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Pest, Beier.
Acquisition of data. Pest, Russell.
Analysis and interpretation of data. Pest, Zhang, Jeong, Beier.
References
- 1.Losina E, Weinstein AM, Reichmann WM, Burbine SA, Solomon DH, Daigle ME, et al. Lifetime risk and age at diagnosis of symptomatic knee osteoarthritis in the US. Arthritis Care Res (Hoboken) 2013;65:703–11. doi: 10.1002/acr.21898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aigner T, Sachse A, Gebhard PM, Roach HI. Osteoarthritis: pathobiology—targets and ways for therapeutic intervention. Adv Drug Deliv Rev. 2006;58:128–49. doi: 10.1016/j.addr.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Goldring SR, Goldring MB. Bone and cartilage in osteoarthritis: is what’s best for one good or bad for the other? Arthritis Res Ther. 2010;12:143. doi: 10.1186/ar3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooke D, Scudamore A, Li J, Wyss U, Bryant T, Costigan P. Axial lower-limb alignment: comparison of knee geometry in normal volunteers and osteoarthritis patients. Osteoarthritis Cartilage. 1997;5:39–47. doi: 10.1016/s1063-4584(97)80030-1. [DOI] [PubMed] [Google Scholar]
- 5.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061–9. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Appleton CT, McErlain DD, Pitelka V, Schwartz N, Bernier SM, Henry JL, et al. Forced mobilization accelerates pathogenesis: characterization of a preclinical surgical model of osteoarthritis [published erratum appears in Arthritis Res Ther 2008;10:407] Arthritis Res Ther. 2007;9:R13. doi: 10.1186/ar2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welch ID, Cowan MF, Beier F, Underhill TM. The retinoic acid binding protein CRABP2 is increased in murine models of degenerative joint disease. Arthritis Res Ther. 2009;11:R14. doi: 10.1186/ar2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joiner DM, Less KD, Van Wieren EM, Zhang YW, Hess D, Williams BO. Accelerated and increased joint damage in young mice with global inactivation of mitogen-inducible gene 6 (MIG-6) after ligament and meniscus injury. Arthritis Res Ther. 2014;16:R81. doi: 10.1186/ar4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider MR, Sibilia M, Erben RG. The EGFR network in bone biology and pathology. Trends Endocrinol Metab. 2009;20:517–24. doi: 10.1016/j.tem.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Humtsoe JO, Kramer RH. Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene. 2010;29:1214–26. doi: 10.1038/onc.2009.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. 2009;218:460–6. doi: 10.1002/jcp.21635. [DOI] [PubMed] [Google Scholar]
- 12.Usmani SE, Pest MA, Kim G, Ohora SN, Qin L, Beier F. Transforming growth factor α controls the transition from hypertrophic cartilage to bone during endochondral bone growth. Bone. 2012;51:131–41. doi: 10.1016/j.bone.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Siclari VA, Lan S, Zhu J, Koyama E, Dupuis HL, et al. The critical role of the epidermal growth factor receptor in endochondral ossification. J Bone Miner Res. 2011;26:2622–33. doi: 10.1002/jbmr.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito K, Horiuchi K, Kimura T, Mizuno S, Yoda M, Morioka H, et al. Conditional inactivation of -converting enzyme in chondrocytes results in an elongated growth plate and shorter long bones. PLoS One. 2013;8:e54853. doi: 10.1371/journal.pone.0054853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Appleton CT, Usmani SE, Mort JS, Beier F. Rho/ROCK and MEK/ERK activation by transforming growth factor-α induces articular cartilage degradation. Lab Invest. 2010;90:20–30. doi: 10.1038/labinvest.2009.111. [DOI] [PubMed] [Google Scholar]
- 16.Long DL, Willey JS, Loeser RF. Rac1 is required for matrix metalloproteinase 13 production by chondrocytes in response to fibronectin fragments. Arthritis Rheum. 2013;65:1561–8. doi: 10.1002/art.37922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Appleton CT, McErlain DD, Henry JL, Holdsworth DW, Beier F. Molecular and histological analysis of a new rat model of experimental knee osteoarthritis. Ann N Y Acad Sci. 2007;1117:165–74. doi: 10.1196/annals.1402.022. [DOI] [PubMed] [Google Scholar]
- 18.Appleton CT, Usmani SE, Bernier SM, Aigner T, Beier F. Transforming growth factorα suppression of articular chondrocyte phenotype and Sox9 expression in a rat model of osteoarthritis. Arthritis Rheum. 2007;56:3693–705. doi: 10.1002/art.22968. [DOI] [PubMed] [Google Scholar]
- 19.Hallbeck AL, Walz TM, Briheim K, Wasteson A, Wasteson A. TGF-α and ErbB2 production in synovial joint tissue: increased expression in arthritic joints. Scand J Rheumatol. 2005;34:204–11. doi: 10.1080/03009740510017715. [DOI] [PubMed] [Google Scholar]
- 20.Yi T, Lee HL, Cha JH, Ko SI, Kim HJ, Shin HI, et al. Epidermal growth factor receptor regulates osteoclast differentiation and survival through cross-talking with RANK signaling. J Cell Physiol. 2008;217:409–22. doi: 10.1002/jcp.21511. [DOI] [PubMed] [Google Scholar]
- 21.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–17. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 22.Frosi Y, Anastasi S, Ballaro C, Varsano G, Castellani L, Maspero E, et al. A two-tiered mechanism of EGFR inhibition by RALT/MIG6 via kinase suppression and receptor degradation. J Cell Biol. 2010;189:557–71. doi: 10.1083/jcb.201002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Descot A, Hoffmann R, Shaposhnikov D, Reschke M, Ullrich A, Posern G. Negative regulation of the EGFR-MAPK cascade by actin-MAL-mediated Mig6/Errfi-1 induction. Mol Cell. 2009;35:291–304. doi: 10.1016/j.molcel.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 24.Makkinje A, Quinn DA, Chen A, Cadilla CL, Force T, Bonventre JV, et al. Gene 33/Mig-6, a transcriptionally inducible adapter protein that binds GTP-Cdc42 and activates SAPK/JNK: a potential marker transcript for chronic pathologic conditions, such as diabetic nephropathy. Possible role in the response to persistent stress. J Biol Chem. 2000;275:17838–47. doi: 10.1074/jbc.M909735199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang YW, Su Y, Lanning N, Swiatek PJ, Bronson RT, Sigler R, et al. Targeted disruption of Mig-6 in the mouse genome leads to early onset degenerative joint disease. Proc Natl Acad Sci U S A. 2005;102:11740–5. doi: 10.1073/pnas.0505171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin N, Gilbert JL, Broaddus RR, Demayo FJ, Jeong J. Generation of a Mig-6 conditional null allele. Genesis. 2007;45:716–21. doi: 10.1002/dvg.20348. [DOI] [PubMed] [Google Scholar]
- 27.Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, et al. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006;12:568–73. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 28.Shepard JB, Jeong JW, Maihle NJ, O’Brien S, Dealy CN. Transient anabolic effects accompany epidermal growth factor receptor signal activation in articular cartilage in vivo. Arthritis Res Ther. 2013;15:R60. doi: 10.1186/ar4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terpstra L, Prud’homme J, Arabian A, Takeda S, Karsenty G, Dedhar S, et al. Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol. 2003;162:139–48. doi: 10.1083/jcb.200302066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solomon LA, Li JR, Berube NG, Beier F. Loss of ATRX in chondrocytes has minimal effects on skeletal development. PLoS One. 2009;4:e7106. doi: 10.1371/journal.pone.0007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillespie JR, Bush JR, Bell GI, Aubrey L, Dupuis H, Ferron M, et al. GSK-3β function in bone regulates skeletal development, whole-body metabolism, and male life span. Endocrinology. 2013;154:3702–18. doi: 10.1210/en.2013-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang G, Woods A, Agoston H, Ulici V, Glogauer M, Beier F. Genetic ablation of Rac1 in cartilage results in chondrodysplasia. Dev Biol. 2007;306:612–23. doi: 10.1016/j.ydbio.2007.03.520. [DOI] [PubMed] [Google Scholar]
- 33.Schmitz N, Laverty S, Kraus VB, Aigner T. Basic methods in histopathology of joint tissues. Osteoarthritis Cartilage. 2010;18(Suppl 3):S113–6. doi: 10.1016/j.joca.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 34.McErlain DD, Ulici V, Darling M, Gati JS, Pitelka V, Beier F, et al. An in vivo investigation of the initiation and progression of subchondral cysts in a rodent model of secondary osteoarthritis. Arthritis Res Ther. 2012;14:R26. doi: 10.1186/ar3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulici V, Hoenselaar KD, Agoston H, McErlain DD, Umoh J, Chakrabarti S, et al. The role of Akt1 in terminal stages of endochondral bone formation: angiogenesis and ossification. Bone. 2009;45:1133–45. doi: 10.1016/j.bone.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Lin CI, Du J, Shen WT, Whang EE, Donner DB, Griff N, et al. Mitogen-inducible gene-6 is a multifunctional adaptor protein with tumor suppressor-like activity in papillary thyroid cancer. J Clin Endocrinol Metab. 2011;96:E554–65. doi: 10.1210/jc.2010-1800. [DOI] [PubMed] [Google Scholar]
- 37.Zhang YW, Staal B, Su Y, Swiatek P, Zhao P, Cao B, et al. Evidence that MIG-6 is a tumor-suppressor gene. Oncogene. 2007;26:269–76. doi: 10.1038/sj.onc.1209790. [DOI] [PubMed] [Google Scholar]
- 38.Zhang YW, Vande Woude GF. Mig-6, signal transduction, stress response and cancer. Cell Cycle. 2007;6:507–13. doi: 10.4161/cc.6.5.3928. [DOI] [PubMed] [Google Scholar]
- 39.Kim TH, Lee DK, Cho SN, Orvis GD, Behringer RR, Lydon JP, et al. Critical tumor suppressor function mediated by epithelial Mig-6 in endometrial cancer. Cancer Res. 2013;73:5090–9. doi: 10.1158/0008-5472.CAN-13-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Z, Dong Q, Wang Y, Qu L, Qiu X, Wang E. Downregulation of Mig-6 in nonsmall-cell lung cancer is associated with EGFR signaling. Mol Carcinog. 2012;51:522–34. doi: 10.1002/mc.20815. [DOI] [PubMed] [Google Scholar]
- 41.Ying H, Zheng H, Scott K, Wiedemeyer R, Yan H, Lim C, et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc Natl Acad Sci U S A. 2010;107:6912–7. doi: 10.1073/pnas.0914930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ling S, Chang X, Schultz L, Lee TK, Chaux A, Marchionni L, et al. An EGFR-ERK-SOX9 signaling cascade links urothelial development and regeneration to cancer. Cancer Res. 2011;71:3812–21. doi: 10.1158/0008-5472.CAN-10-3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aigner T, Stove J. Collagens—major component of the physiological cartilage matrix, major target of cartilage degeneration, major tool in cartilage repair. Adv Drug Deliv Rev. 2003;55:1569–93. doi: 10.1016/j.addr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 44.Radin EL, Rose RM. Role of subchondral bone in the initiation and progression of cartilage damage. Clin Orthop Relat Res. 1986;213:34–40. [PubMed] [Google Scholar]
- 45.Mateescu RG, Todhunter RJ, Lust G, Burton-Wurster N. Increased MIG-6 mRNA transcripts in osteoarthritic cartilage. Biochem Biophys Res Commun. 2005;332:482–6. doi: 10.1016/j.bbrc.2005.04.144. [DOI] [PubMed] [Google Scholar]
- 46.Burton-Wurster N, Mateescu RG, Todhunter RJ, Clements KM, Sun Q, Scarpino V, et al. Genes in canine articular cartilage that respond to mechanical injury: gene expression studies with Affymetrix canine GeneChip. J Hered. 2005;96:821–8. doi: 10.1093/jhered/esi105. [DOI] [PubMed] [Google Scholar]
- 47.Fosang AJ, Golub SB, East CJ, Rogerson FM. Abundant LacZ activity in the absence of Cre expression in the normal and inflamed synovium of adult Col2a1-Cre; ROSA26RLacZ reporter mice. Osteoarthritis Cartilage. 2013;21:401–4. doi: 10.1016/j.joca.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 48.Kambic HE, McDevitt CA. Spatial organization of types I and II collagen in the canine meniscus. J Orthop Res. 2005;23:142–9. doi: 10.1016/j.orthres.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Zhu J, Li Y, Lin T, Siclari VA, Chandra A, et al. Epidermal growth factor receptor (EGFR) signaling regulates epiphyseal cartilage development through β-catenin-dependent and -independent pathways. J Biol Chem. 2013;288:32229–40. doi: 10.1074/jbc.M113.463554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Staal B, Williams BO, Beier F, Vande Woude GF, Zhang YW. Cartilage-specific deletion of Mig-6 results in osteoarthritis-like disorder with excessive articular chondrocyte proliferation. Proc Natl Acad Sci. 2014;111:2590–5. doi: 10.1073/pnas.1400744111. [DOI] [PMC free article] [PubMed] [Google Scholar]