

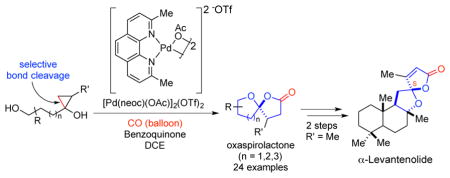
Abstract
A palladium-catalyzed cascade carbonylative spirolactonization of hydroxycyclopropanols has been developed to efficiently synthesize oxaspirolactones common to many complex natural products of important therapeutic value. The mild reaction conditions, high atom economy, broad substrate scope, and scalability of this new method were highlighted in expedient total syntheses of the Turkish tobacco natural products α-levantanolide and α-levantenolide in two and four steps respectively. The hydroxycyclopropanol substrates are readily available in one step via a Kulinkovich reaction of the corresponding lactones. Mechanistic studies utilizing high-resolution electrospray ionization mass spectrometry (ESI-MS) identified several key intermediates in the catalytic cycle as well as those related to catalyst decomposition and competitive pathways.
Graphical Abstract
INTRODUCTION
Our ongoing efforts to develop catalytic carbonylation methodologies to build complex, biologically active molecules,1 and to exploit cyclopropanols in ring opening cross coupling reactions2 inspired endeavors to develop efficient and general oxaspirolactone synthesis by combining carbonylation chemistry3 and cyclopropanol ring opening chemistry.4 The oxaspirolactone motif occurs prominently in many bioactive natural products. Some selected representatives are shown in Figure 1. The triterpenoid phainanoid F (1)5 was found to have potent immunosuppressive activity. Phomoidride B (also known as CP-263,114, 2) and related analogs6 were shown to inhibit squalene synthase as well as Ras farnesyl transferase and have attracted plenty of synthetic attention.7 Massarinolin A (3)8 and its analogs expansolides A (4) and B (5)9 have shown potent antibacterial activity against drug resistant strains. Sequoiamonascins (cf. 6, 7)10 are novel anticancer metabolites isolated from a redwood endophyte. Lancifodilactone G (8),11 a highly oxygenated nortriterpenoid, exhibits potent anti-HIV activity. Abiespirosides (cf. 9),12 stemonines (cf. 10),13 and levantenolides (cf. 11, 12)14 are representatives of their family members with various biological activities. The latter belong to a class of labdane derivaties which were recently found to inhibit LPS-induced nitric oxide (NO) production and LPS-stimulated production of pro-inflammatory cytokines.15
Figure 1.

Selected natural products.
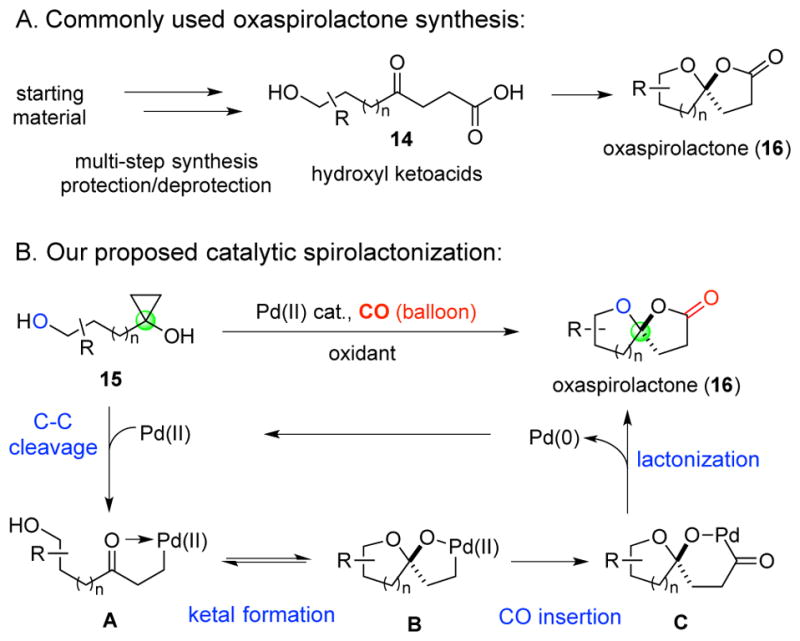
While many synthetic methods have been developed for the synthesis of spiroketal systems,16 less attention has been devoted to the synthesis of oxaspirolactones and few synthetic methods exist. Early methods rely on special reagents,17 stoichiometric transition metals to form reactive intermediates,18 or several protection-deprotection steps to make appropriate hydroxyl ketoacids for acid-promoted cyclizations (cf. 14→16, Figure 2A).19 Recently, Rodriguez et al. reported a gold-catalyzed coupling reaction to access α-amino oxaspirolactones, albeit with poor diastereoselectivity;20 Sridhar et al. reported a Lewis-acid promoted ring opening and cyclization of cyclopropanecarboxylates.21 Notably, White and coworkers have used one substrate to demonstrate that the directed metal (oxo)-promoted C-H hydroxylation with non-heme iron catalysts can be used to synthesize oxaspirolactones.22 New synthetic methods, particularly catalytic methods, to convert readily available starting materials to oxaspirolactones would be an enabling advance to provide access to this class of bioactive natural products. Moreover, oxaspirolactones can be readily converted to spiroketals (cf. 13),23 which are an important and challenging structural motif prevalent in many bioactive natural products.24
Figure 2.
Proposed method.
We investigated whether a palladium-catalyzed carbonylative spirolactonization of readily available hydroxycyclopropanols would provide an expedient synthesis of oxaspirolactones (cf. 15→16, Figure 2B). A palladium(II)-catalyzed C-C bond cleavage25 of the strained three-membered ring would provide palladium-homoenolate A from cyclopropanol 15 with a tethered alcohol. The tethered alcohol would then react with the newly generated ketone to form the corresponding ketal B. Carbon monoxide migratory insertion26 followed by lactonization would provide the desired product 16 and a Pd(0) complex. The latter will be oxidized to the Pd(II) catalyst by an external oxidant.
RESULTS
Methodology Development
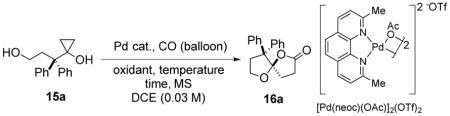
To test our hypothesis, we investigated the model substrate 15a (Table 1), which is conveniently prepared in one step from the corresponding commercially available lactone using the Kulinkovich reaction.27 Treatment of 15a with a catalytic amount of Pd(OAc)2 and benzoquinone (BQ) at 60 °C in 1,2-dichloroethane (DCE) in presence of molecular sieves afforded the desired oxaspirolactone 16a in 13% isolated yield (entry 1). Variation of the terminal oxidant did little to improve the yield. Palladium catalysts such as Pd(PPh3)2Cl2 and Pd(MeCN)2Cl2 were less effective than Pd(OAc)2. Higher yields were obtained when Pd(TFA)2 or [(Cinnamyl)PdCl]2 were used. These results suggested that electron deficient palladium centers might facilitate coordination of the cyclopropanol and suppress competitive reactions. We therefore investigated [Pd(neoc)(OAc)]2(OTf)2, a cationic dimeric palladium catalyst developed in the Waymouth laboratory.28 The use of this complex resulted in significantly higher yields: the spirolactone 16a was obtained in 85% yield with 0.1 equiv. of [Pd(neoc)(OAc)]2(OTf)2 after 60 h at room temperature. The reaction is faster at an elevated temperature and carrying out the reaction in the absence of molecular sieves was beneficial. Notably, oxygen can be used as oxidant instead of BQ, but the yields were lower and other oxidized byproducts were produced in the presence of oxygen (entry 6). A simple combination of Pd(OAc)2 and neocuproine ligand was not effective (entry 12), which indicates the importance of the cationic nature of the [Pd(neoc)(OAc)]2(OTf)2 catalyst. Though palladium black is produced during the course of the reaction, palladium black is not a competent heterogeneous catalyst for this system (entry 13). After optimization, it was found that carrying out the reaction with 0.05 equiv. of [Pd(neoc)(OAc)]2(OTf)2 at 50 °C for 18 h afforded an 89% isolated yield of the spirolactone 16a.
Table 1.
Reaction condition optimization.
| ||||
|---|---|---|---|---|
| entry | Pd catalyst (equiv) | oxidant (equiv) | T (°C)/time (h) | yielda |
| 1 | Pd(OAc)2 (0.1) | BQ (2.0) | 60/14 | 13% |
| 2 | Pd(TFA)2 (0.1) | BQ (2.0) | RT/14 | 30% |
| 3 | Pd(TFA)2 (0.1)/PPh3 (0.2) | BQ (2.0) | RT/14 | LC |
| 4 | [(Cinnamyl)PdCl]2 (0.1) | BQ (2.0) | RT/60 | 66% |
| 5 | [Pd(neoc)(OAc)]2(OTf)2 (0.1) | BQ (2.0) | RT/60 | 85% |
| 6 | [Pd(neoc)(OAc)]2(OTf)2 (0.1) | O2 (balloon) | RT/60 | 60% |
| 7b | [Pd(neoc)(OAc)]2(OTf)2 (0.05) | BQ (2.0) | RT/60 | 57% |
| 8b | [Pd(neoc)(OAc)]2(OTf)2 (0.05) | BQ (2.0) | RT/60 | 77% |
| 9b | [Pd(neoc)(OAc)]2(OTf)2 (0.01) | BQ (2.0) | RT/60 | 63% |
| 10b | [Pd(neoc)(OAc)]2(OTf)2 (0.1) | BQ (2.0) | 40/36 | 79% |
| 11b | [Pd(neoc)(OAc)]2(OTf)2 (0.05) | BQ (2.0) | 50/18 | 89% |
| 12b | Pd(OAc)2 (0.1)/neocuproine (0.1) | BQ (2.0) | RT/60 | LC |
| 13b | Pd black recycled from reaction | BQ (2.0) | 50/18 | 0% |
Isolated reaction yields with 0.1 mmol of 15a;
Without MS; BQ: Benzoquinone; LC: Low Conversion; RT: Room Temperature; MS: 4Å Molecular Sieves.
With the optimized conditions in hand, the scope and generality of this catalytic oxidative spirolactonization was evaluated (Table 2). The reaction was found to give generally high yields for substrates bearing primary and secondary alcohols, while tertiary alcohols gave moderate yields. Ring size could be varied to give spiro(4.4) (cf. 16a–i), (5.4) (cf. 16j–u), and (6.4) (cf. 16v–w) ring systems. The reaction conditions tolerate a broad range of functional groups such as halogens including fluorine and bromine (16b, 16d, 16f, 16v), N-Boc carbamate (16m), alkenes (16o and 16t), benzoate (16q) and TBS ether (16r). More complex polycyclic systems such as 16h, 16i, 16o, 16q, 16r, 16t, and 16u could be accessed easily and in good yield. Notably, the tricyclic ring system of 16h and 16i resembles the core structure of ascospiroketals22 (cf. 13, Figure 1). In general, the stereoselectivity for the spiro(4.4) ring systems is poor (cf. 16g, 16h, and 16i), which was also reflected in the expansolides (cf. 4, 5), levantenolides (cf. 11, 12), and other natural products containing such a spiro(4.4) lactone systems. However, the stereoselectivity for the spiro(5.4) ring systems are excellent, presumably due to the anomeric effect. The structures of 16r, 16s, 16t, and 16u were determined unambiguously by single crystal X-ray diffraction.
Table 2.
Substrate scope for the carbonylative spirolactonization.

|
Total Syntheses of α-Levantanolide and α-Levantenolide
The C12 oxygenated labdanolic diterpenes α- and β-levantenolides (11 and 12, respectively, Figure 1),29 isolated from extracts of Turkish tobacco, were selected to test the utility of this protocol. The commercially available (3aR)-(+)-sclareolide (17) provided a convenient optically active starting point, which was converted to unsymmetrical cyclopropanol 18 as a 4:1 mixture of diastereomers in 57% yield (Scheme 1). The catalytic oxidative spirolactonization of cyclopropanol 18 occurred with high regioselectivity to afford a mixture of three diastereomeric oxaspirolactone products in 53% combined yield with 2 mol% of the catalyst. The major diastereomer was shown to be the natural product α-levantanolide (19),30 which could be isolated in 30% yield.
Scheme 1.

Synthesis of α-Levantenolide and α-Levantanolide.
For cyclopropanol 18, there are two cyclopropane C-C bonds that could potentially be cleaved during the β-carbon elimination process. NMR analysis of the three diastereomeric oxaspirolactone products indicated that only the less-substituted cyclopropane C-C bond was cleaved in the ring-opening step. In addition to 19, an inseparable mixture of minor products 20 (23% yield) was obtained which corresponds to two oxaspirolactones of undetermined configuration at C12 and C13 in a 4:1 ratio. The mixture of 19 and 20 was subjected to a α-selenylation/oxidative elimination process to provide α-levantenolide 11 as a single product in 46% yield (two steps). None of the corresponding isomer β-levantenolide 12 was observed. The structure of 11 was confirmed by single crystal X-ray diffraction. In summary, the new catalytic carbonylative spirolactonization method has enabled expedient total syntheses of the Turkish tobacco natural products α-levantanolide and α-levantenolide in two and four steps respectively from readily available and cheap starting materials. These syntheses are significantly shorter and efficient than the previous ones29,30 and set the stage to access other natural and unnatural analogs for further biological evaluations.
Mechanistic Studies using High Resolution Electrospray Ionization Mass Spectrometry
One possible mechanism for the catalytic oxidative spirolactonization of 15a is illustrated in Figure 3. Key steps in this proposed cycle involve β-carbon elimination from the Pd-alkoxide intermediate E to generate the palladium-homoenolate H. Intramolecular cyclization to the lactol I followed by CO insertion would afford the hydroxyacyl intermediate K. Elimination of the spirolactone 16a and reoxidation of intermediate L would regenerate the [(neoc)PdOAc]+ D. To provide support for this mechanism, the catalytic reactions were monitored by high resolution electrospray ionization mass spectrometry (ESI-MS)31 in the presence of both O2 and BQ using methods described previously.32 ESI-MS was also performed on the reaction of partially deuterated substrate 15a-d4 (Figure 4) to provide further evidence for structures of side products and reactive intermediates. Collision induced dissociation (CID) coupled tandem mass spectrometry (MS/MS) was applied to key intermediates to gain structural information through fragmentation of trapped ions.
Figure 3.

Proposed catalytic cycle and observed intermediates and byproducts with ESI-MS analysis.
Figure 4.

Observed organic products, byproducts and inorganic intermediates unique to deuterated substrate 15a-d4 by ESI-MS analysis.
The catalytic reaction of 15a carried out under either one balloon of O2 or 2 equivalents of BQ in DCE for 4–24 hours were analyzed by ESI-MS (Figure S3, S4). The major organic ion observed corresponds to the spirolactone 16a (m/z 295.1349). The doubly oxidized byproduct 23 (m/z 265.1223) and the singly oxidized 21 or 22 (m/z 267.1370) were also observed, though these three ions were observed in lower abundance than that corresponding to 16a. Analysis of reactions carried out under analogous conditions with the labeled substrate 15a-d4 yielded ions at m/z 299.1584, 270.1558 and 271.1634, corresponding to 16a-d4, 21-d3, and 22-d4, respectively (Figure S5). When the reaction was carried out with 15a-d4 in the absence of CO, both 21-d3 and 22-d4, are formed in a 1:1 ratio (assuming equal ionization efficiencies). Thus, while the overoxidized products corresponding to 21, 22 and 23 were not isolated from the preparative experiments, the high sensitivity of mass spectrometry reveals that competitive primary alcohol oxidation to afford 22 or β-H elimination (from an intermediate such as E) to give 21 are competitive reactions that occur during the catalytic cycle.
ESI-MS also provided information on the speciation of Pd during the catalytic cycle. Analysis of reactions carried out under one balloon of CO and one balloon of O2 in DCE revealed ions (Figure S1, Supporting Information) at m/z 373.0166, corresponding to intermediate D, and at m/z 581.1415, corresponding to intermediates E, H, or I (which cannot be distinguished as they are isobaric). The CID spectra of this ion shows fragmentation to m/z 331.0065, (neoc)PdOH+, and m/z 424.0429, (neoc)PdCH2CH3+, (Figure S10). Analysis of an analogous reaction using the labeled substrate 15a-d4 revealed ions at m/z 585.1675, corresponding to intermediates E-d4, H-d4, I-d4 (Figure S2). The CID spectra of the ions at m/z 585.1675 (Figure S11) reveal fragmented ions at m/z 331.0065, corresponding to (neoc)PdOH+ and m/z 347.0669, corresponding to (neoc)PdCD2CD2H+. No perprotio (neoc)PdCH2CH3+ was observed. These labeling studies indicate that the most reasonable structure for the ion at m/z 581.1415 (substrate 15a) or m/z 585.1675 (substrate 15a-d4) are those corresponding to intermediate E where the tertiary alkoxide is bound to the Pd center.
Several additional ions were identified that provide some insight on competitive processes and catalyst decomposition mechanisms. An ion at m/z 817.2364 was observed (Figure S1), corresponding to species G, bearing two neocuproine ligands, the substrate 15a, and CO. The CID spectrum of G (Figure S8) yields daughter ions of G that have lost one neocuproine, or CO and neocuproine (to form E). Similar to ions E/H/I, further fragmentation yields (neoc)PdOH+ and (neoc)PdCH2CH3+ (Figure S12) analogous to results observed in the CID spectra of the m/z 581.1415 (assigned as intermediate E). When substrate 15a-d4 is used, an ion at m/z 821.2645, corresponding to intermediate G-d4 is observed (Figure S2). The CID spectrum of G-d4 reveals a fragment ion at m/z 347.0669 corresponding to (neoc)PdCD2CD2H+ (Figure S13). These data suggest that CID of the ion corresponding to G generates ion E by loss of neocuproine and CO. Overall, these data support the assignment of the ion at m/z 817.2364 as the structure G given in Figure 2 with two κ1-bound neocuproines.
To confirm if the assigned structure G was reasonable, analogous ESI-MS studies were performed using isopropanol and t-butanol as alcohol substrates (Figure 5 and Figures S8–S9). Similar to using the cyclopropyl diol substrate, Pd black formation was observed after several minutes of reaction. The dominant ions observed in both reactions were those at m/z 609.1482, G-iPr, and m/z 623.1638, G-tBu, corresponding to Pd alkoxides bearing two neocuproine ligands and one CO ligand, analogous to that of ion G. The CID spectra of these ions (Figure S17–S18) showed similar fragmentation patterns where CO and neocuproine were lost, consistent with ions derived from a Pd alkoxide bearing two κ1-bound neocuproines, and one CO. These results further validate the proposed structure of G and support the conclusion that in the presence of excess ligand generated from the formation of Pd black, two κ1-bound neocuproines instead of one κ2-bound neocuproine occurs. Furthermore, Casati et al. have also observed similar bis-neocuproine palladium halide complexes.33
Figure 5.
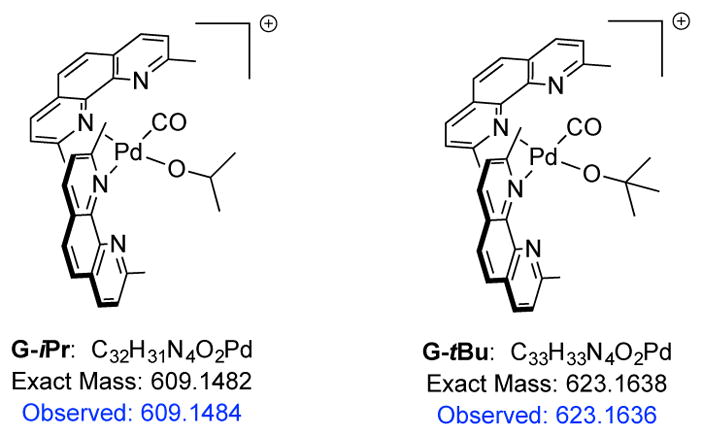
Key ions observed in a reaction with iPrOH and tBuOH as the substrate, respectively.
The observation of G is independent of the oxidant used and of the reaction time, though it is in higher relative abundance in longer reactions. It is likely that this complex is a dormant state of the catalyst which inhibits the progress of the reaction.
Before Pd black formed, several dimeric Pd intermediates m/z 951.1366, 657.0087, and 690.9705 were observed—corresponding to O, P, and Q (Figure S1). Casati et al.34 have observed similar Pd(I) dimers with bridging COs, leading us to assign O, P, and Q as ions with dimeric structures bearing two Pd(I)s and bridging COs. The CID spectrum of an intermediate analogous to O shows that neither of the COs are inserted into the substrate as both COs are readily displaced (Figure S16). Of these intermediates, only Q is observed after Pd black forms—albeit in low abundance—indicating that O, P, and Q are not catalytically active and are on a decomposition pathway to form Pd black.
Additionally, at reaction times of 4–24 hours, an ion at m/z 595.1208, formulated as the overoxidized species N was detected under O2. Under BQ, N was present but in much smaller abundance, this could be caused by O2 exposure prior to and during ESI-MS analysis. The atomic composition of this ion indicates that the primary alcohol of the substrate has been oxidized to a carboxylic acid. The CID spectrum of ion N (Figure S15) yields daughter ions corresponding to a palladium hydride and a palladium methyl, indicating that the substrate is C-bound. Loss of CO and CH2O2 fragments indicates that there is a carboxylic acid moiety which cannot be on the ligand. Therefore the most reasonable structure, N, has the substrate ring opened and C-bound with primary alcohol oxidized to the corresponding carboxylic acid.
The ion observed at m/z 551.0663 (Figure S6, S7) corresponds to the Pd carboxylate R,32a,c which was previously shown to be an unproductive species derived from oxidative degradation of the ligand under O2. The identification of these overoxidized products under O2 provides a possible explanation for the lower yields observed when O2 is used as the terminal oxidant (Table 1).
CONCLUSIONS
In summary, we report a novel Pd-catalyzed cascade reaction of hydroxycyclopropanols for the synthesis of oxaspirolactones of varying substitution and ring size. The reaction features mild conditions and excellent functional group tolerability. Its potential application in complex natural product synthesis was demonstrated by short syntheses of α-levantanolide and α-levantenolide. Mechanistic studies with ESI-MS have detected several key intermediates in the catalytic cycle as well as the potential side reaction pathways.
EXPERIMENTAL
Representative oxaspirolactone synthesis procedure
A stirred solution of 15a (27 mg, 0.1 mmol) and benzoquinone (22 mg, 0.2 mmol) in DCE (3 ml) was evacuated and backfilled three times using a carbon monoxide balloon. [Pd(neoc)(OAc)]2(OTf)2 (5.4 mg, 0.005 mmol) was added in one portion and the resulting solution was stirred at 50 °C for 18 hours. The reaction mixture was filtered through a pad of celite and concentrated under reduced pressure. The resulting black residue was dissolved in methylene chloride and purified by flash chromatography (hexane/ethyl acetate = 20/1 to 4/1) to obtain 16a as a white solid (27 mg, 89%).
Mass Spectrometry Details
Mass spectrometry experiments were conducted on an LTQ-Orbitrap XL instrument, with a resolution 100,000 at m/z 400 and mass accuracy of less than 5 ppm, manufactured by Thermo Fisher Scientific (San Jose, CA). Collision induced dissociation (CID) spectra were taken in tandem MS mode by colliding desired ions with helium. Data were preliminarily analyzed in Thermo Fisher’s Qual Browser tool, and then exported as text files for further processing in MatLab. Additional compound identification was performed using Cerno MassWorks.
General Procedures for ESI Experiments
Procedure Using Benzoquinone as Oxidant: To a 1 dram vial with septum cap was added substrate (0.03 mmol) and benzoquinone (6.4 mg, 0.06 mmol). 1 mL of DCE (dried over molecular sieves) was added and the reaction mixture was stirred until all solids dissolved. A balloon of CO was bubbled through the solution for 10 minutes, then [Pd(neoc)(OAc)]2(OTf)2 (1.6 mg, 0.003 mmol) was added as a solid while stirring vigorously. The reaction mixture was stirred at room temperature. Aliquots were removed via syringe and filtered through a 22 μm PVDF syringe filter. The resultant solution was diluted 100x into either dichloromethane or acetonitrile and immediately used to collect ESI-MS data. Procedure Using O2 as Oxidant: Same as above but omits BQ and a separate balloon of O2 is added at the same time as the balloon of CO is added.
Supplementary Material
Acknowledgments
MD thanks NSF (CAREER 1553820) and the ACS petroleum research foundation (PRF# 54896-DNI1) for financial support and the NIH P30CA023168 for supporting shared NMR resources to Purdue Center for Cancer Research. KLW thanks the Center for Molecular Analysis and Design for a fellowship, and RMW acknowledges support from the Department of Energy (DE-SC0005430). RNZ thanks the Air Force Office of Scientific Research for support (FA-9550-16-1-0113).
Footnotes
The authors declare no competing financial interests.
This paper is dedicated to Professor Samuel J. Danishefsky and Professor Stuart Schreiber on the occasion of their 80th and 60th birthdays, respectively.
ASSOCIATED CONTENT
Experimental procedures, compound characterization, and ESI-MS spectra are available in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bai Y, Davis DC, Dai MJ. Angew Chem, Int Ed. 2014;53:6519. doi: 10.1002/anie.201403006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Li Y, Ye Z, Bellman TM, Chi T, Dai MJ. Org Lett. 2015;17:2186. doi: 10.1021/acs.orglett.5b00782. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye Z, Dai MJ. Org Lett. 2015;17:2190. doi: 10.1021/acs.orglett.5b00828. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ye Z, Gettys KE, Shen X, Dai MJ. Org Lett. 2015;17:6074. doi: 10.1021/acs.orglett.5b03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu XF, Neumann H, Beller M. Chem Rev. 2013;113:1. doi: 10.1021/cr300100s. [DOI] [PubMed] [Google Scholar]
- 4.For reviews: Kulinkovich OG, de Meijere A. Chem Rev. 2000;100:2789. doi: 10.1021/cr980046z.Rosa D, Nikolaev A, Nithiy N, Orellana A. Synlett. 2015;26:441.
- 5.Fan YY, Zhang H, Zhou Y, Liu HB, Tang W, Zhou B, Zuo JP, Yue JM. J Am Chem Soc. 2015;137:138. doi: 10.1021/ja511813g. [DOI] [PubMed] [Google Scholar]
- 6.(a) Dabrah TT, Harwood HJ, Jr, Huang LH, Jankovich ND, Kaneko T, Li J-C, Lindsey S, Moshier PM, Subashi TA, Therrien M, Watts PC. J Antibiot. 1997;50:1. doi: 10.7164/antibiotics.50.1. [DOI] [PubMed] [Google Scholar]; (b) Spencer P, Agnelli F, Sulikowski GA. Org Lett. 2001;3:1443. doi: 10.1021/ol015707k. [DOI] [PubMed] [Google Scholar]
- 7.For a review: Spiegel DA, Njardarson JT, McDonald IM, Wood JL. Chem Rev. 2003;103:2691. doi: 10.1021/cr020408+.For selected examples: Chen C, Layton ME, Sheehan SM, Shair MD. J Am Chem Soc. 2000;122:7424.Nicolaou KC, Baran PS. Angew Chem, Int Ed. 2002;41:2678. doi: 10.1002/1521-3773(20020802)41:15<2678::AID-ANIE2678>3.0.CO;2-V.Waizumi N, Itoh T, Fukuyama T. J Am Chem Soc. 2000;122:7825.Meng DF, Tan Q, Danishefsky SJ. Angew Chem, Int Ed. 1999;38:3197. doi: 10.1002/(sici)1521-3773(19991102)38:21<3197::aid-anie3197>3.0.co;2-6.Bio MM, Leighton JL. J Org Chem. 2003;68:1693. doi: 10.1021/jo026478y.Murphy GK, Shirahata T, Hama N, Bedermann A, Dong P, McMahon TC, Twenter BM, Spiegel DA, McDonald IM, Taniguchi N, Inoue M, Wood JL. J Org Chem. 2013;78:477. doi: 10.1021/jo302353g.
- 8.Oh H, Gloer JB, Shearer CA. J Nat Prod. 1999;62:497. doi: 10.1021/np980447+. [DOI] [PubMed] [Google Scholar]
- 9.Massias M, Rebuffat S, Molho L, Chiaroni A, Riche C, Bodo B. J Am Chem Soc. 1990;112:8112. [Google Scholar]
- 10.Stierle DB, Stierle AA, Bugni T. J Org Chem. 2003;68:4966. doi: 10.1021/jo0340253. [DOI] [PubMed] [Google Scholar]
- 11.Xiao WL, Zhu HJ, Shen YH, Li RT, Li SH, Sun HD, Zheng YT, Wang RR, Lu Y, Wang C, Zheng QT. Org Lett. 2005;7:2145. doi: 10.1021/ol050502n. [DOI] [PubMed] [Google Scholar]
- 12.Yang X-W, Li S-M, Li Y-L, Xia J-H, Wu L, Shen Y-H, Tian J-M, Wang N, Liu Y, Zhang W-D. Eur J Org Chem. 2010:6531. [Google Scholar]
- 13.Wang P, Liu AL, Li ZH, Du GH, Qin HL. J Asian Nat Prod Res. 2008;10:311. doi: 10.1080/10286020701833511. [DOI] [PubMed] [Google Scholar]
- 14.For isolation, see: Giles JA, Schumacher JN. Tetrahedron. 1961;14:246.For structural revision, see: González AG, Francisco CG, Freire R, Hernández R, Salazar JA, Suárez E. Tetrahedron Lett. 1976;17:1897.
- 15.(a) Girón N, Través PG, Rodríguez B, López-Fontal R, Boscá L, Hortelano S, Heras BL. Toxicology and Applied Pharmacology. 2008;228:179. doi: 10.1016/j.taap.2007.12.006. [DOI] [PubMed] [Google Scholar]; (b) Girón N, Pérez-Sacau E, López-Fontal R, Amaro-Luis JM, Hortelano S, Estevez-Braun A, Heras BL. Eur J Med Chem. 2010:3155. doi: 10.1016/j.ejmech.2010.04.007. [DOI] [PubMed] [Google Scholar]; (c) Kiem PV, Anh HLT, Nhiem NX, Minh CV, Thuy NTK, Yen PH, Hang DT, Tai BH, Mathema VB, Koh YS, Kim YH. Chem Pharm Bull. 2012;60:246. doi: 10.1248/cpb.60.246. [DOI] [PubMed] [Google Scholar]
- 16.(a) Vaillancourt V, Pratt NE, Perron F, Albizati KF. Total Synthesis of Natural Products. John Wiley & Sons, Inc; 2007. The Total Synthesis of Spiroketal-Containing Natural Products; p. 533. [Google Scholar]; (b) Sperry J, Wilson ZE, Rathwell DCK, Brimble MA. Nat Prod Rep. 2010;27:1117. doi: 10.1039/b911514p. [DOI] [PubMed] [Google Scholar]; (c) Cala L, Fananas FJ, Rodriguez F. Org Biomol Chem. 2014;12:5324. doi: 10.1039/c4ob00737a. [DOI] [PubMed] [Google Scholar]
- 17.Bundy GL, Lin CH, Sih JC. Tetrahedron. 1981;37:4419. [Google Scholar]
- 18.(a) DeShong P, Rybczynski PJ. J Org Chem. 1991;56:3207. [Google Scholar]; (b) Bueno AB, Hegedus LS. J Org Chem. 1998;63:684. doi: 10.1021/jo971665v. [DOI] [PubMed] [Google Scholar]
- 19.(a) Brimble MA, Fares FA, Turner P. Aust J Chem. 2000;53:845. [Google Scholar]; (e) Mellor JM, Mohammed S. Tetrahedron. 1993;49:7547. [Google Scholar]; (c) Robertson J, Chovatia PT, Fowler TG, Withey JM, Woollaston DJ. Org Biomol Chem. 2010;8:226. doi: 10.1039/b918091e. [DOI] [PubMed] [Google Scholar]
- 20.Cala L, Mendoza A, Fananas FJ, Rodriguez F. Chem Commun. 2013;49:2715. doi: 10.1039/c3cc00118k. [DOI] [PubMed] [Google Scholar]
- 21.Ramakrishna B, Sridhar PR. RSC Advances. 2015;5:8142. [Google Scholar]
- 22.Bigi MA, Reed SA, White MC. J Am Chem Soc. 2012;134:9721. doi: 10.1021/ja301685r. [DOI] [PubMed] [Google Scholar]
- 23.(a) Seibert SF, Krick A, Eguereva E, Kehraus S, König GM. Org Lett. 2007;9:239. doi: 10.1021/ol0626802. [DOI] [PubMed] [Google Scholar]; (b) Chang S, Hur S, Britton R. Angew Chem, Int Ed. 2015;54:211. doi: 10.1002/anie.201408905. [DOI] [PubMed] [Google Scholar]
- 24.Aho JE, Pihko PM, Rissa TK. Chem Rev. 2005;105:4406. doi: 10.1021/cr050559n. [DOI] [PubMed] [Google Scholar]
- 25.(a) Aoki S, Fujimura T, Nakamura E, Kuwajima I. J Am Chem Soc. 1988;110:3296. [Google Scholar]; (b) Fujimura T, Aoki S, Nakamura E. J Org Chem. 1991;56:2809. [Google Scholar]; (c) Parida BB, Das PP, Niocel M, Cha JK. Org Lett. 2013;15:1780. doi: 10.1021/ol400666x. [DOI] [PubMed] [Google Scholar]; (d) Cheng K, Walsh PJ. Org Lett. 2013;15:2298. doi: 10.1021/ol4008876. [DOI] [PubMed] [Google Scholar]; (e) Rosa D, Orellana A. Chem Commun. 2013;49:5420. doi: 10.1039/c3cc42080a. [DOI] [PubMed] [Google Scholar]; (f) Nithiy N, Orellana A. Org Lett. 2014;16:5854. doi: 10.1021/ol5027188. [DOI] [PubMed] [Google Scholar]
- 26.(a) Aoki S, Nakamura E, Kuwajima I. Tetrahedron Lett. 1988;29:1541. [Google Scholar]; (b) Aoki S, Nakamura E. Synlett. 1990:741. [Google Scholar]; (c) Kang SK, Yamaguchi T, Ho PS, Kim WY, Yoon SK. Tetrahedron Lett. 1997;38:1947. [Google Scholar]
- 27.Esposito A, Taddei M. J Org Chem. 2000;65:9245. doi: 10.1021/jo0011944. [DOI] [PubMed] [Google Scholar]
- 28.(a) Conley NR, Labios LA, Pearson DM, McCrory CCL, Waymouth RM. Organometallics. 2007;26:5447. [Google Scholar]; (b) Pearson DM, Conley NR, Waymouth RM. Adv Syn Cat. 2011;353:3007. [Google Scholar]; (c) Pearson DM, Conley NR, Waymouth RM. Organometallics. 2010;30:1445. [Google Scholar]; (d) Painter RM, Pearson DM, Waymouth RM. Angew Chem, Int Ed. 2010;49:9456. doi: 10.1002/anie.201004063. [DOI] [PubMed] [Google Scholar]
- 29.For previsous syntheses, see Kato T, Tanemura M, Suzuki T, Kitahara Y. Chem Commun. 1970:28.Tanemura M, Suzuki T, Kato T, Kitahara Y. Tetrahedron Lett. 1970;11:1463.Kato T, Tanemura M, Kanno S, Suzuki T, Kitahara Y. Bioorg Chem. 1971;1:84.González AG, Francisco CG, Freire R, Hernández R, Salazar JA, Suárez E. Tetrahedron Lett. 1976;17:2725.Urones JG, Basabe P, Marcos IS, Díez Martín D, Sexmero MJ, Peral MH. Tetrahedron. 1992;48:10389.
- 30.(a) Giles JA, Schumacher JN, Young GW. Tetrahedron. 1963;19:107. [Google Scholar]; (b) de Pascual Terasa J, Urones JG, Pedrero AM, Barcala PB. Tetrahedron Lett. 1985;26:5717. [Google Scholar]
- 31.(a) Yan X, Sokol E, Li X, Li G, Xu S, Cooks RG. Angew Chem, Int Ed. 2014;53:5648. doi: 10.1002/anie.201310493. [DOI] [PubMed] [Google Scholar]; (c) Vikse KL, Ahmadi Z, McIndoe JS. Coord Chem Rev. 2014;279:96. [Google Scholar]; (d) Ingram AJ, Boeser CL, Zare RN. Chem Sci. 2016;7:39. doi: 10.1039/c5sc02740c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Chung K, Banik SM, De Crisci AG, Pearson DM, Blake TR, Olsson JV, Ingram AJ, Zare RN, Waymouth RM. J Am Chem Soc. 2013;135:7593. doi: 10.1021/ja4008694. [DOI] [PubMed] [Google Scholar]; (b) Ingram A, Solis-Ibarra D, Zare RN, Waymouth RM. Angew Chem, Int Ed. 2014;53:5648. doi: 10.1002/anie.201400134. [DOI] [PubMed] [Google Scholar]; (c) Ingram AJ, Walker KL, Zare RN, Waymouth RM. J Am Chem Soc. 2015;137:13632. doi: 10.1021/jacs.5b08719. [DOI] [PubMed] [Google Scholar]
- 33.Rimoldi M, Ragaini F, Gallo E, Ferretti F, Macchi P, Casti A. Dalton Trans. 2012;41:3648. doi: 10.1039/c2dt11979j. [DOI] [PubMed] [Google Scholar]
- 34.Ragaini F, Larici H, Rimoldi M, Caselli A, Ferretti F, Macchi P, Casati N. Organometallics. 2011;30:2385. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.