

Abstract
Long-lived plasma cells (PC) in the bone marrow (BM) are a critical source of antibodies after infection or vaccination, but questions remain about the factors that control PCs. We found that systemic infection alters the BM, greatly reducing PCs and regulatory T (Treg) cells, a population that contributes to immune privilege in the BM. The use of intravital imaging revealed that BM Treg cells display a distinct behavior characterized by sustained co-localization with PCs and CD11c-YFP+ cells. Gene expression profiling indicated that BM Treg cells express high levels of Treg effector molecules, and CTLA-4 deletion in these cells resulted in elevated PCs. Furthermore, preservation of Treg cells during systemic infection prevents PC loss, while Treg cell depletion in uninfected mice reduced PC populations. These studies suggest a role for Treg cells in PC biology and provide a potential target for the modulation of PCs during vaccine-induced humoral responses or autoimmunity.
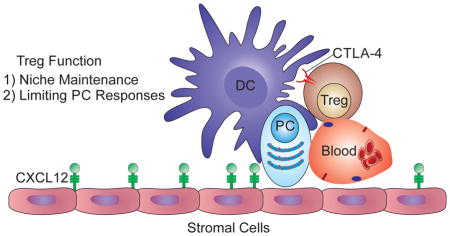
Graphical Abstract
INTRODUCTION
A variety of immune cell precursors reside and develop in the bone marrow (BM), a site that is also home to several populations of mature lymphocytes. There are multiple mechanisms to allow pluripotent or long-lived cells, including hematopoietic and cancer stem cells, plasma cells (PC), and memory T cells, to persist in the BM (Fujisaki et al., 2011; Kawano et al., 2015). However, the spatial relationship and interactions between these disparate cellular populations are still being defined. For example, BM stromal cells provide growth and survival factors necessary for PC and hematopoietic stem cell (HSC) maintenance, but the relationship between these niches is unclear (Sugiyama et al., 2006; Zehentmeier et al., 2014). Moreover, in the BM, regulatory T cells (Treg) are enriched and may contribute to the maintenance of the BM as an immune privileged site, necessary for HSC survival (Fujisaki et al., 2011). However, the behavior of Treg cells in the BM and their interactions with other immune populations have not been visualized and it remains unclear whether their activity is relevant to other hematopoietic cell populations in the BM.
Long-lived PCs present in the BM constitutively produce high levels of antibodies that result in life long serum antibody titers against previously encountered pathogens or vaccines (Manz et al., 1997; Slifka et al., 1998). Consequently, there is interest in understanding the mechanisms that maintain these cells (Chu and Berek, 2013). It is known that stromal cells provide survival signals to PC through the production of CXCL13, BLyS, April, and IL-6 (Roth et al., 2014). Furthermore, eosinophils, basophils, and megakaryocytes are implicated in the maintenance of PCs in the BM (Chu et al., 2011; Rodriguez Gomez et al., 2010; Winter et al., 2010) and there is evidence that perivascular clusters of DCs in the BM provide critical signals for B cells (Rozanski et al., 2011; Sapoznikov et al., 2008). Although these factors promote PC survival, they are not sufficient, and the cellular composition of this niche and requirements for PC maintenance are major questions (Chu and Berek, 2013). However, there is a paucity of intravital imaging studies to describe the behavior of PCs and their interactions with other cell populations. Thus, there remains a need to better define the composition of this niche to understand how PCs are maintained and whether there are regulatory networks that limit PC responses.
Many studies have demonstrated that systemic infection or inflammation results in marked changes in BM populations (Glatman Zaretsky et al., 2012; MacNamara et al., 2009; Ueda et al., 2005). Here, challenge with Toxoplasma gondii, a clinically relevant parasite for which the mouse is a natural host (Montoya and Liesenfeld, 2004) demonstrated that infection-induced changes in the BM lead to a transient loss of PCs and Tregs. Multiphoton imaging revealed that at homeostasis, PCs interact with a CD11c-YFP+ population and Treg cells in the BM. Further studies indicate that Treg cells have a complex interaction with BM PCs, including a CTLA-4-dependent mechanism of PC regulation, while supporting PC populations. Together, these studies suggest an unanticipated role for Treg cells in the maintenance and operation of the PC niche within the BM.
RESULTS
Plasma cells in the BM are disrupted by systemic infection
Changes in the BM, including increased hematopoiesis and reduced lymphopoiesis, are a hallmark of the immune response to numerous pathogens (Glatman Zaretsky et al., 2014; Manz and Boettcher, 2014). Indeed, following infection with the systemic pathogen T. gondii, histological analysis of the BM revealed extensive changes in the cellular composition of this compartment, including increased numbers of hematopoietic progenitors (Figure 1A–B). Unexpectedly, resident PC populations, identified based on their oval shape, purple cytoplasm, and characteristic eccentric nucleus with clock-face chromatin and perinuclear clearing, were diminished (large arrows) (Figure 1A–B). Long-lived PC populations are an independent pool of terminally differentiated B cells maintained through slow homeostatic proliferation with a half life of >120 days (Slifka et al., 1998). PCs express high levels of the transcription factor BLIMP-1 (Minnich et al., 2016; Tellier et al., 2016; Turner et al., 1994) and in uninfected BLIMP-YFP or BLIMP-GFP mice, 70–85% of the CD138+ PCs were positive for the reporter, which indicated sufficient fidelity to visualize PC populations (Wolf et al., 2011). Therefore, intravital imaging of the BM in the skull of BLIMP1-YFP reporter mice was used to visualize the location and motility of PC populations in uninfected and infected mice (Figure 1C). In naïve mice, PCs are localized in clusters adjacent to blood vessels in the endosteal region of the bone and were stationary over the imaging periods used in these studies (Movie S1). However, by 14 days post infection, the PC population was markedly reduced (Figure 1C) and flow cytometry confirmed the infection-induced loss of PCs (Figure 1D–E). This analysis also revealed a decrease of >95% in the total number of pro, pre, immature, and mature recirculating B cells (Figure S1A–C). Although short-term blockade of B cell lymphopoiesis has minimal effects on mature B cell compartments (Hao and Rajewsky, 2001), it was possible that these alterations might impact the development of PC populations. To ascertain whether infection reduced an established PC population, mice were immunized with 4-Hydroxy-3-nitrophenylacetyl (NP)-Ovalbumin. This antigen results in a long-lived NP-specific PC population in the BM, detectable by intracellular staining with NP conjugated to allophycocyanin (Figure 1F). During acute T. gondii infection, the number of NP-specific B cells in the spleen was unchanged (data not shown), but there was a decrease in the number of NP+ PCs in the BM, accompanied by a significant drop in serum NP-specific IgG1 (Figure 1F–G). However, by the chronic phase of infection, the NP+ population was restored to its original levels (data not shown) indicating that infection results in the transient loss of a pre-established PC compartment.
Figure 1.

Acute Toxoplasma gondii infection results in a loss of PC in the BM. (A) Naïve mouse. The marrow cavity contains vascular sinuses (V) surrounded by mature neutrophils (arrowheads) admixed with predominantly myelopoietic precursors and few mature PC (arrows). Bone cortex (C). (B) Day 14 T. gondii infected mouse. Medullary vascular sinuses (V) are surrounded by increased numbers of hematopoietic progenitors characterized by hyperchromatic nuclei. Few mature neutrophils (arrowheads) and immature band neutrophils (arrows) are observed. Mature PC are not identified. Bone cortex (C). (C) Naïve or infected BLIMP1-YFP reporter mice were imaged using intravital 2-photon microscopy of the skull BM. The BLIMP1-YFP-expressing cells are yellow and quantum dots were injected intravenously to label the vasculature red. At least 3 mice were imaged for each time point. (D–E) BM from naïve or infected WT mice was evaluated by flow cytometry (using a “dump” gate to eliminate CD3+, F4/80+, and/or Gr1+ potential contaminating cells) for the presence of PC. (F–G) WT mice were immunized with NP-OVA. 6 weeks later, these mice were challenged with T. gondii and evaluated for NP-specific PC (F) and NP-specific antibody titers (G).
Plasma cells interact with T regulatory cells and CD11c+ cells in the BM
The infection-induced loss of PCs in the BM raises questions about which of the regulatory or cellular elements that support the PC niche are disrupted. Because Foxp3-GFP+ cells are preferentially localized in the endosteal region of naive BM and contribute to the status of the BM as an immune privileged site (Fujisaki et al., 2011), we examined the impact of infection on this population. Intravital imaging of uninfected mice showed Foxp3-GFP+ Treg cells throughout the BM, predominantly in extravascular sites, but by day 14 post-infection, these cells were largely absent (Figure 2A). Flow cytometric analysis confirmed that infection resulted in an ~80% decrease in Treg cells in the BM (Figure 2B–C) that mirrored the changes in Treg cell numbers at peripheral sites during acute toxoplasmosis (Benson et al., 2012; Oldenhove et al., 2009). The generation of mice that express both BLIMP1-YFP and Foxp3-GFP allowed for simultaneous imaging of PC and Treg cell populations in the BM. While Treg cells exhibited a greater motility than the PCs, these cells have a range of mobility with a large proportion of the Treg cell population displaying a more sessile phenotype (Movie S2). Indeed, the majority of the Foxp3-GFP+ cells were found closely associated with BLIMP1-YFP+ PCs (Figure 2D–F), as ~10% of PCs interact with Treg cells for 4 minutes or less, while more than 60% of interactions between Foxp3-GFP+ and BLIMP1-YFP+ cells were >10 minutes, with 30% exceeding the 20 minute imaging window (Figure 2F, Movies S3–S4). In contrast, Foxp3-GFP+ cells in the spleen exhibited a higher velocity and shorter interaction times with BLIMP1-YFP+ cells (Figure 2F, Movie S5). It should be noted that the transfer of polyclonally activated CD8+ T cells into Foxp3-GFP reporter mice resulted in a small population of CD8+ T cells in the BM that could be imaged simultaneously with Treg cells. These effector T cells exhibited a higher velocity than that of the resident Treg cell population (Figure S2A, Movie S6). This internal control indicates that the sessile Treg cell phenotype in the BM was not an artifact associated with sample preparation. Further analysis of the cellular localization of PCs and Treg cells using fixed long bones showed that they were present in areas rich in HSCs (CD150+) and that PCs were intimately associated with the network of stromal cells that are a source of CXCL12 in the BM (Figure S2B–C, Movie S7). Thus, Treg cells in the BM display a behavior distinct from that observed in the spleen and many are spatially associated with PCs within local niches in the BM.
Figure 2.

Treg cells and PC interact in the BM. (A) Naïve or infected Foxp3-GFP reporter mice were imaged using intravital 2-photon microscopy of the skull BM. The Foxp3-GFP-expressing cells are green (demarcated by the *) and quantum dots were injected intravenously to label the vasculature red. At least 3 mice were imaged for each time point. (B–C) BM from naïve or infected WT mice was evaluated by flow cytometry for the presence of Treg cells. (D–F) BLIMP1-YFP/Foxp3-GFP dual reporter mice were imaged using intravital 2-photon microscopy of the skull BM. (D–E) BLIMP1-YFP (white), Foxp3-GFP (green), and vasculature (red) expression in BLIMP1-YFP/Foxp3-GFP dual reporter mice reveals short and long term interactions, highlighted in the close up (E). 3 mice were imaged for these experiments, with 53 Foxp3-GFP cells recorded in the BM and 104 imaged in the spleen. (F) Quantification of the duration of PC-Treg cell interactions in the spleen and BM.
Previous studies have described a population of mature recirculating B cells in the BM that are closely associated with DCs (Sapoznikov et al., 2008), but it is unclear whether DCs are a component of the PC niche. Therefore, mice that express CD11c-YFP/BLIMP1-GFP or CD11c-YFP/Foxp3-GFP were generated to determine whether DCs are localized near to or interact with PCs or Treg cells in the BM. In the cavarial BM of naïve CD11c-YFP/BLIMP1-GFP mice, there were numerous large stationary CD11c-YFP+ populations that displayed irregularly shaped dendrites (Figure 3A–B) and ~70% of PCs interacted with a population of CD11c-YFP+ cells, while 40–50% of Treg cells interacted with CD11c-YFP+ cells (Figure 3C, Movie S8). The use of mixed BM chimeras demonstrated that the CD11c-YFP+ cells present in the BM are of hematopoietic origin, as WT recipients that were reconstituted with BM from CD11c-YFP mice contained an extensive network of YFP+ cells in the BM, while YFP+ cells were rare in irradiated reporter mice that were recipients of WT BM, associated with a 40 fold decrease in the YFP volume (Figure S3). These data support a model in which CD11c-YFP+ cells, implicated in providing co-stimulation required for PC survival and antibody production (Rozanski et al., 2011), are a component of the PC niche and form close associations with PCs and Treg cells.
Figure 3.

CD11c-YFP+ cells interact with PC and Treg cells in the BM. (A–C) Intravital imaging of skull BM in BLIMP-GFP/CD11c-YFP dual reporter mice with quantum dot-labeled vasculature (red), with interacting cells highlighted in close up (B). 4 mice were imaged, for a total of 83 BLIMP-GFP cells counted for quantification. (C) Quantification of the percentage of cells observed in interactions between CD11c-YFP+ cells and BLIMP1-GFP+ or Foxp3-GFP+ cells in respective dual reporter mice. (D–H) Imaging and quantification of Treg cell velocity (D) and CD11c+ cell-Treg cell location (E–F) in the spleen and BM of Foxp3-GFP (53 BM Foxp3-GFP cells and 150 splenic Foxp3-GFP cells) (D) or CD11c-YFP/Foxp3-GFP dual (E–G) reporter mice, as well as interaction times between these cells (68 BM Foxp3-GFP cells and 94 splenic Foxp3-GFP cells) (G) and Treg cell movement shown by tracks (H).
Few studies have used multi-photon imaging to visualize Treg cell behavior and interactions with DCs in situ, and these were performed in LNs in the context of immunization and diabetes (Matheu et al., 2015; Tadokoro et al., 2006; Tang et al., 2006). Therefore, we decided to compare how Treg cells in the BM or spleen interact with resident DC populations at homeostasis. Analysis of reporter mice revealed that splenic Treg cells were highly motile based on their velocity and displacement rate, and that in the spleens of Foxp3-GFP/CD11c-YFP dual reporter mice, less than 10% of the Treg cells had sustained interactions with CD11c-YFP+ cells (Figure 3D, E, G H, and Movie S9). In contrast, Treg cells in the BM had reduced velocity, low displacement, and >50% of Treg cells had sustained interactions with CD11c-YFP+ cells (Figure 3C, D, F–H, Movie S10). Therefore, the Treg cell populations in the spleen and BM are characterized by fundamental differences in their behavior and those in the BM display unanticipated interactions with PCs, as well as with a CD11c-YFP+ population.
Treg cells in the BM exhibit an effector phenotype with regulatory effects on PCs
Treg cells in non-lymphoid tissues have unique transcriptional profiles and functions associated with individual tissues (Burzyn et al., 2013a; Burzyn et al., 2013b; Feuerer et al., 2010). However, despite the enrichment of Treg cells in the BM, these cells have not been transcriptionally characterized. Therefore, to determine whether BM Treg cells have a transcriptional signature distinct from that of peripheral Treg cells, gene expression profiles of BM or splenic Treg cells were compared (Figure 4A, Table S1). Differentially expressed transcripts are dominated by chemokine/cytokine receptors (Ccr2, Ccr3, Cxcr3, Il9r, Il1rl1) and effector molecules (Ctla4, Il10, Fgl2, Gzmb), suggestive of an activated population with increased Treg suppressive capabilities (Figure 4A–B). These transcripts are also over-expressed in other tissue-Treg cells relative to those isolated from lymphoid organs, with those from injured muscle being most similar to BM Treg cells (Figure 4B). Indeed, Principal Component Analysis encompassing all differentially represented transcripts showed that BM Treg cells reside with tissue Treg cells, not lymphoid Treg cells (Figure 4C). These findings highlight the effector capacity of BM Treg cells, consistent with the idea that these cells have a role in the maintenance of an immune privileged niche.
Figure 4.

BM Treg cells exhibit a more effector-like phenotype than splenic Treg cells, with a higher expression of suppressor genes. (A–C) Treg cells sorted from spleen and BM of naïve Foxp3-GFP+ mice were evaluated by microarray. Cells were assessed for differentially regulated genes (A), which were then compared to Treg cells from other sites by specific gene expression (B) or as total populations by Principle Componant Analysis (C). (D) CTLA-4 expression was determined by flow cytometry on BM or splenic Treg cells, which was concatenated from 5 mice (left) and MFI was quantified (right). (E) PC and Treg cell populations in the BM, Spl, and LN of WT or Foxp3Cre/CTLA4flox/flox mice were assessed by flow cytometry.
Since PCs in the BM co-localize with Treg cells, it was relevant to determine whether the reduction in PCs during infection is a consequence of the loss of the BM Treg cell population or a secondary consequence of inflammation due to the loss of the suppressive niche. Based on the transcriptional profiling, we focused on CTLA-4 as being highly expressed by BM Treg cells and having a prominent role in Treg suppressive activity (Wing et al., 2008). Analysis of CTLA-4 expression revealed that splenic Treg cells express negligible surface levels of CTLA-4, whereas BM Treg cells have elevated levels (Figure 4D). Next, we analyzed mice with a Treg cell-specific loss of CTLA-4 expression (Foxp3Cre/CTLA4flox/flox), which develop systemic inflammation (Wing et al., 2008). Despite the presence of an ongoing inflammatory response, there was no decrease in the numbers of Foxp3+ T cells in the BM and these mice had a ~3-fold increase in the percentage of Treg cells (Figure 4E). However, in contrast to the loss of PCs observed when Treg cells are reduced during infection-induced inflammation, compromising Treg cell function in this system without decreasing Treg cell number resulted in a 3-fold increase in the number of PCs (Figure 4E). However, CTLA-4 expression on Treg cells negatively regulates PC development in the periphery (Sage et al., 2014; Wing et al., 2014) and PCs in the BM express high levels of CD28, an important regulator of PC function (Rozanski et al., 2011). Thus, although CTLA4-deficient Treg cells are unable to prevent fatal inflammation (Wing et al., 2008), these experiments suggest that Treg cell expression of CTLA-4 acts to limit the size of the PC pool in the BM.
Treg cells and PC maintenance
Next, complementary approaches were used to understand whether Treg cells are directly involved in the maintenance of PCs. First, in gain-of-function experiments, we asked whether treatment with an IL-2-anti-IL-2 antibody complex that promotes Treg cell survival (Boyman et al., 2006) could prevent the loss of BM PCs during infection. Treatment of naïve mice with IL-2 complex resulted in elevated Treg cell numbers by percentage, but not absolute number in the BM (Figure 5A–B), and did not alter PC numbers (Figure 5C–D). IL-2 complex treatment of infected mice did result in a modest increase in parasite burden (data not shown), but mitigated the loss of the BM Treg cells and preserved the PCs in the BM (Figure 5A–D). As PCs do not express the IL-2 receptor component CD25 (Jego et al., 2001) (data not shown), this rescue is unlikely due to direct effects of the IL-2-anti-IL-2 complex on PCs. Of note, because of the experimental variation we observed in the levels of PC reduction, the overall relationship between the frequency of Treg cells and PCs was assessed by comparing Treg cell numbers with the numbers of PCs in the BM from these experiments. When the number of Treg cells was plotted versus the number of PCs in the BM from these IL-2C treatment experiments, a highly significant correlation was observed between these populations (Figure 5E). Of note, it has been proposed that eosinophils promote PC survival (Chu et al., 2011) and these cells are transiently lost from the BM along with PCs during acute infection (Figure 5F). However, as previously reported (Van Gool et al., 2014), treatment of naïve mice with the IL-2 complex resulted in an eosinophilia, but when infected mice were treated with IL-2 complex, the eosinophil population was not rescued, although the PC population was maintained (Figure 5F). These data suggest that the role for Treg cells in supporting PC populations is distinct from that of the eosinophils present in the BM.
Figure 5.

BM Treg cells support PC maintenance in the BM. (A–F) WT mice were infected with T. gondii and treated with IL-2-anti-IL-2 complex every 3 days for 14 days. BM was evaluated for Treg cells (A–B), PC (C–D), and representative flow plots are shown, with a concatenation of mice within each group for PC plots (D). The numbers of Treg cells and PC were plotted to identify correlations between the cell numbers of these two populations (E). Eosinophils were evaluated and quantified by flow cytometry (F).
Secondly, to assess the impact of Treg cell loss on PC populations in the absence of infection, Treg cells were depleted in naïve Foxp3-DTR mice using diphtheria toxin (DT) and the PC compartment was assessed. In these mice, DT treatment resulted in a >90% decrease in the percentage and number of Foxp3+ Treg cells in the BM (Figure 6A–B) and a >50% decrease in PCs (Figure 6C–D). Of note, when Treg cells were eliminated and PCs were reduced, the numbers of CD11c+MHCIIhi cells remained constant, though these cells did express elevated levels of CD80 (data not shown). Thus, despite recent findings that Treg cell depletion enhances PC formation in the periphery (Wing et al., 2014), the depletion of Treg cells resulted in a reduction in the BM PC population, similar to the phenomenon observed in infected mice. Although Treg cell depletion using the Foxp3-DTR system results in widespread inflammation (Christiaansen et al., 2014), the studies described in Figure 5 are performed in the context of systemic infection-induced inflammation. Thus, the ability of IL-2C treatment to preserve Treg cells and mitigate the PC loss suggests that inflammation alone would not result in the PC reduction observed in the Foxp3-DTR experiments. In these mice, Treg cell depletion did not result in significant changes in the eosinophil population, further emphasizing that eosinophils are not sufficient for PC maintenance (Figure 6E–F). These independent approaches implicate Treg cells as part of a network that maintains and regulates PC populations in the BM.
Figure 6.

(A–F) Naïve Foxp3-DTR mice were treated with DT on days 0, 1, 4, and 6, and evaluated on day 7. Populations of Treg cells (A–B), PC (C–D), and eosinophils (E–F) were evaluated and enumerated in the BM.
DISCUSSION
There have been many advances in defining the events that lead to the generation of PCs and the idea that BM PCs require multiple survival factors for their maintenance is established, but the cellular interactions that promote PC survival are poorly characterized (Goodnow et al., 2010). Stromal cells are part of this niche (Tokoyoda et al., 2004; Zehentmeier et al., 2014), and diverse migratory cells in the BM influence PC survival (Tangye, 2011; Winter et al., 2012) and data presented here highlight the association of BM PCs and Treg cells with CD11c-YFP+ cells. Together with evidence that Treg cells contribute to HSC survival (Fujisaki et al., 2011), our studies connecting Treg cells with maintenance of BM PCs suggest that Treg cell activities in the BM may affect the homeostasis of many cell types. Indeed, Treg cells appear to be in excess of the BM PCs, which may prevent small changes in Treg cell numbers from resulting in major changes in PC homeostasis. Alternatively, “excess” Treg cells may reflect their role in other niches (Fujisaki et al., 2011) and the principles that govern distinct micro-environments may offer insight into the mechanisms that preserve memory cells or which help maintain PC tumors or cancer stem cells in the BM (Winter et al., 2012).
Many acute infections lead to depletion of peripheral Treg cells (Benson et al., 2012; Oldenhove et al., 2009), and this reduction in Treg populations in the BM correlates with the loss of pre-existing long-lived PCs. While evidence is presented that supports the idea that Treg cells maintain PCs, the underlying mechanism is unclear. The close association of Treg cells and PCs suggests there may be communication between these populations through cell contact or via soluble factors. Alternatively, the extended contacts between Treg cells and CD11c-YFP+ cells in the BM may be relevant to PC maintenance. Thus, the close association of BM PCs with CD11c-YFP+ cells is consistent with the idea that PC survival is dependent on constitutive signals through CD80/CD86 (Rozanski et al., 2011). Although DCs are required for Treg homeostasis (Darrasse-Jeze et al., 2009; Hanig and Lutz, 2008; Salomon et al., 2000), Treg cells regulate the activation of DC (Liu et al., 2009; Veldhoen et al., 2006) and transcriptional profiling indicated that BM Treg cells express an effector phenotype and pathways that are relevant to the modulation of DC function (Burzyn et al., 2013a; Burzyn et al., 2013b; Feuerer et al., 2010). Indeed, when Treg cells did not express CTLA-4, there were increased BM PCs suggesting that this pathway can limit local PC activity. However, these data have to be interpreted with care as the increased plasmablasts and PCs in the periphery of Foxp3Cre/CTLA4flox/flox mice may contribute to the increased PC number in the BM (Matheu et al., 2015; Wing et al., 2014; Wing et al., 2008). In the absence of a specific means to deplete or inactivate BM Treg cells, it is difficult to exclude the possibility that systemic effects of Treg cell deficiencies impact on BM PC numbers. An alternative explanation may be that inflammation leads to a simultaneous, but non-causal loss of both PCs and Treg cells, while delivery of IL-2 complexes may promote the survival or proliferation of other cell types required for the support of these populations. Regardless, we favor the possibility that the ability of Treg cells to interact with DCs may have a dual role: allowing Treg cells to contribute to the maintenance of the PC population, but also to limit PC activities.
The alterations observed in the BM during acute infection have been characterized in response to numerous pathogens, (Glatman Zaretsky et al., 2014) and the work presented here highlights effects on pre-existing PC populations, that appears to be common to infection or vaccination (Ng et al., 2014; Slocombe et al., 2013; Xiang et al., 2007). While the alterations in BM PCs are transient, the benefit of this disruption is unclear. It has been suggested that because the PC niche is of limited size, this process may allow newly created PCs to displace pre-existing PCs in order to expand the repertoire of the BM PC population to ensure that recent systemic pathogens are represented (Radbruch et al., 2006; Xiang et al., 2007). The observation that PCs removed from the BM undergo rapid apoptosis indicates that PCs are not intrinsically long lived and require constant signals for maintenance, however our studies do not distinguish whether disruption of the niche leads to PC death or evacuation as a consequence of competition with plasmablasts (Odendahl et al., 2005). While the mechanisms by which PCs contract and subsequently are restored in the BM are uncertain, they may include cell death, a selection mechanism for the repopulation of the niche, and/or expansion of memory B cell populations (Bernasconi et al., 2002; DiLillo et al., 2008; Ng et al., 2014).
The success of rituximab highlights the clinical benefits of B cell depletion for many disease states, although this approach does not affect PC populations (Edwards and Cambridge, 2006). There are antibody-mediated disorders (allergy, lupus, and rheumatoid arthritis) where eliminating pre-existing PCs could have value (Edwards and Cambridge, 2006; Radbruch et al., 2006). However, as PCs express few traditional B cell markers, downregulate most surface receptors, including the BCR and MHC class II, and are radio-resistant there are few treatments to deplete established PCs (Clatworthy, 2011; Lu et al., 2011). The studies presented here offer a model to understand the factors that govern homeostasis and maintenance of PCs and suggest an unexpected role for Treg cells in the processes that support PC populations, in addition to perhaps limiting PC activity. Understanding the PC-Treg cell interaction could provide novel strategies to treat immune-mediated diseases in which long lived PCs are involved.
EXPERIMENTAL PROCEDURES
Animals
C57BL/6J mice were obtained from Taconic or Sage at 6–8 weeks of age. BAC transgenic BLIMP1-YFP mice were originally from E. Meffre (Yale University). CD11c-YFP mice were provided by Michel Nussenzweig (Rockefeller University). Foxp3-GFP mice were from Vijay Kuchroo (Harvard University). Foxp3-DTR mice were obtained from Alexander Rudensky (Memorial Sloan Kettering). BLIMP1-GFP mice were originally from Stephen Nutt (WEHI). Foxp3-IRES-Cre Balb/c CTLA4flox/flox mice were previously described (Wing et al., 2008). All mouse strains were housed, maintained, and bred under specific pathogen–free conditions at the University of Pennsylvania, Harvard University, Osaka University, or UCSD animal facilities. All experimental procedures with mice were approved by the Institutional Animal Care and Use Committee of the respective institution.
Infections, immunizations, and irradiation
T. gondii tachyzoites of the Prugniaud strain were maintained in vitro by serial passage in human fibroblasts monolayers and parasites were isolated via serial needle passage and filtration through 5μm filters. Mice were infected i.p. with 104 parasites in 100μl PBS. NP-Ovalbumin (Biosearch Technologies) with a conjugation ratio of 16–18 was emulsified in alum and 50μg/mouse was injected i.p. To make IL-2 complexes, 1.5ug carrier-free rmIL-2 (eBioscience) was incubated with 25ug anti-IL-2 (JES6-1A12, eBioscience) for 5 minutes at room temperature before injection. Control mice received 25ug Rat IgG (BioXcell). Foxp3-DTR mice were treated with DT on days 0, 1, 4, and 6 for Treg cell depletion. For mixed BM chimeras, WT or CD11c-YFP recipient mice were irradiated with 1000Rd. After 6 hours, 2×106 cells isolated from BM of Foxp3-GFP and CD11c-YFP (1:1) or only Foxp3-GFP (100%) donor mice were transferred iv to recipient mice. Mice were provided sulfamethoxazole in the drinking water for 2 weeks after irradiation. After 8–12 weeks of reconstitution, recipient mice were imaged as described.
Cell purification and flow cytometry
Briefly, mouse spleens and BM were collected and dissociated through a 40μm strainer to produce a single cell suspension. Red blood cells were lysed using 0.86% ammonium chloride (Sigma). Cells were counted, washed in flow cytometry buffer (1% BSA (Sigma), 2mM EDTA (Invitrogen) in PBS. FcR were blocked with 2.4G2 (BioXCell) and Rat IgG (Invitrogen) while staining with McAb at 40 for 30 minutes. Intracellular staining was carried out using the eBioscience kit. Samples were acquired using a FACSCanto II or LSRFortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star, Inc.). The following mAb against mouse antigens were used as FITC, PE, PerCP-Cy5.5, PE-Cy7, allophycocyanin (APC), APC-ef780, Pacific blue, AF450, AF700, PE Texas Red, or biotin conjugates: FcεRI, AA4.1, CD23, CD43, IgM, FcγRII/III, ckit, Sca-1, CD34, CD138, CD45R (B220; RA3-6B2), CD278 (ICOS; C398.4A), IgD (11–26) (eBioscience), CD4 (RM4–5), CD8a (53–6.7), CD95 (Fas; Jo2), CXCR5 (2G8) (BD Biosciences), CD3 (17A2), CD19 (6D5), Foxp3, CTLA-4, Siglec F, CD11b, F4/80, TCRβ, Gr-1, FcεRI, and CD279 (PD-1; J43) (Biolegend). Biotinylated antibodies were detected using PerCP-Cy5.5– (BD Biosciences) or APC-conjugated streptavidin (eBioscience). FITC or biotin-conjugated PNA was obtained from Vector Laboratories. Plots shown are on a Logicle scale.
Microarray profiling and analysis
C57Bl/6Foxp3-IRES-gfp mice were bred in specific-pathogen-free facilities at Harvard Medical School. BM Treg cells (GFP+CD25hiCD4+TCRβ+CD19−CD8α−TCRγδ−CD11b−CD11c−NK1.1−Gr-1−Ter-119−) were triple-sorted (necessary to eliminate contamination from dominant myeloid populations) into Trizol from pools of 2–3 mice. Splenic Tregs were double-sorted from the same mice. Profiling was performed on Affymetrix MoGene 1.0 ST microarray as described [Sitrin PMID 23650440]. Data were analyzed with the Multiplot Studio and Gene-E modules from GenePattern. Principal component analysis-based 3D plotting was generated with the Population PCA module [http://cbdm.hms.harvard.edu/LabMembersPges/SD.html].
Histology
Mouse femurs were isolated and placed in 10% buffered formalin (Sigma). Tissues were paraffin embedded, sectioned, and stained with hematoxylin and eosin (H&E) and slides were reviewed by a board-certified veterinary anatomic pathologist. Identification of PCs was based on characteristic morphologic features, including eccentrically located round to oval nuclei with coarsely clumped chromatin peripheral to the nuclear envelope, moderate amounts of deep purple cytoplasm with a distinct perinuclear clearing, indicative of a well-developed Golgi apparatus. Immature (e.g. band) neutrophils were identified as round cells with amphiphilic cytoplasm and horseshoe-shaped nuclei. Mature neutrophils were identified as round cells with pale eosinophilic cytoplasm and nuclei containing hyper-condensed chromatin with multiple segmentations.
Microscopy
Mice were anaesthetized and maintained at core temperature of 37°C. Thinned-skull and open-skull surgery were performed as described previously (Nimmerjahn et al., 2005; Yang et al., 2010). For preparation of the spleen for imaging, mice were euthanized by CO2 asphyxiation, the spleen was removed and immediately transferred into an imaging chamber (Warner Instruments). Tissue was perfused with cRPMI (RPMI plus 10% fetal-calf serum, 1% pen/strep, 1% glutamine, 1% nonessential amino acids, and 0.1%β-mercaptoethanol), bubbled with 95% oxygen and 5% carbon dioxide, and maintained at 37°C. Imaging was performed using a Leica SP5 2-photon microscope system (Leica Microsystems) equipped with a picosecond or femtosecond laser (Coherent). GFP, YFP, RFP and tdTomato were excited using laser light of 900 nm or as indicated. Images were obtained using a 20x water-dipping lens. Fluorescence emission was separated by high efficiency custom dichroic mirrors and collected with 4 external non-descanned detectors. The use of all 4 detectors allowed the separation of GFP and YFP expressing cells and from autofluorescent cells, which appear in “orange” due to their detection in several channels simultaneously. The resulting images were analyzed with Volocity software (PerkinElmer). In these images, the frequency of interaction and the duration of interaction between cells were assessed manually using 3 dimensional reconstructions of the acquired movies (Supplemental Movies 4 and 8). 3–4 mice from each of the BLIMP1-YFP, Foxp3-GFP BLIMP1-YFP dual reporter, Foxp3-GFP CD11c-YFP dual reporter, and BLIMP1-GFP CD11c-YFP dual reporter strains were imaged and analyzed at each of the indicated time points. Within the Foxp3-GFP BLIMP-YFP dual reporter mice, 53 BM and 104 splenic Foxp3-GFP+ cells were imaged and analyzed. 83 BLIMP1-GFP cells were imaged and quantified in BLIMP1-GFP CD11c-YFP dual reporter mice. Within the Foxp3-GFP CD11c-YFP dual reporter mice, 68 Foxp3-GFP+ cells were imaged and evaluated in the BM and 94 splenic Foxp3-GFP+ cells were imaged and analyzed. 53 Foxp3-GFP+ cells in the BM and 150 splenic Foxp3-GFP+ cells were analyzed in BM chimeras that were made as described in the text. For transfer of total splenic T cells, splenocytes were isolated from naïve mice and enriched for CD8+ T cells by negative selection (Miltenyi). These cells were then activated and expanded in vitro for 3 days with plate bound αCD3(1μg/ml)/αCD28(1μg/ml) and IL-2 (200U/ml−1) before transfer of 1 million cells to recipient Foxp3-GFP mice.
For clearing and imaging bones were fixed in 4% PFA for 6 h and immersed in 30% sucrose O/N before cryopreservation in OCT (Fisher Scientific) as previously described (Acar et al., 2015). Bones were bisected longitudinally on a cryostat, washed in PBS and blocked overnight in staining solution (10% DMSO, 0.5% IgePal630 and 5% donkey serum) plus FcR block. Tissues were incubated with labeled antibodies against GFP (Life Technologies), CD150 (Clone TC15-12F12.2, Biolegend) and Laminin 1+2 (polyclonal rabbit anti-mouse, Abcam) in staining solution for 3 days and then washed with PBS for 1 day. For clearing, half bones were dehydrated by incubation in increasing ethanol concentrations and overnight incubation in BABB solution (1:2, benzyl alcohol: benzyl benzoate). Images were obtained using a Zeiss LSM710 confocal microscope and analyzed using Volocity 6.3 (Perkin Elmer) software. These experiments were performed 2–10 times on a total of >10mice per image and representative images were selected.
ELISA
Levels of NP-specific serum IgG1 were determined by ELISA. Immulon 4HBX plates (Thermo Fisher Scientific) were coated overnight at 4°C with 5μg NP-BSA/mL (Biosearch Technologies), in 50μl/well. Plates were then blocked with 10% FBS, and incubated with serial dilutions of sera, followed by a peroxidase-coupled anti-IgG1 conjugate (Southern Biotech) and SureBlue TMB or ABTS substrate (KPL).
Statistical analysis
Statistical significance was assessed using a Student’s T test. A p value of p ≤ 0.05 was considered significant. Statistical analysis was performed using Prism software (GraphPad software, Inc.).
Supplementary Material
Acknowledgments
The authors thank Gordon Ruthel for technical assistance. Imaging experiments were performed in the PennVet ImagingCore Facility on instrumentation supported by NIH S10RR027128, the School of Veterinary Medicine, the University of Pennsylvania, and the Commonwealth of Pennsylvania.
This work was supported by NIH grant AI-110201 (C.A.H.), AI 41158 (C.A.H.), T32 AI-055400 (A.G.Z.), AI-51530 (C.O.B.), AI-123782 (L.L), R01-AI-097590 (D.A.) and R01-AI-097590 (D.A.), the JSPS Grant-in-Aid for Scientific Research (A grant no. 26253030 to S.S.), the JSPS Grant-in-Aid for Young Scientists (B grant no.15K19129 to J.B.W), and the Lupus Foundation of America, Philadelphia Tri-State Chapter (A.G.Z).
Footnotes
AUTHOR CONTRIBUTIONS
A.G.Z, C.K., F.D., J.B.W. R.G., D.G.A, J.S.S., S.C., A.I.W., and W.J.Q. performed experiments and analysis. J.B.E. provided pathology expertise. D.C.B. offered statistical analysis. D.B., J.E., D.A., M.P.C., S.S., L.L., C.O.B. provided technical and intellectual guidance and commented on the manuscript. A.G.Z. and C.A.H. wrote the manuscript.
ACCESSION NUMBERS: GSE76264
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, Jaiyeola C, Zhao Z, Luby-Phelps K, Morrison SJ. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–130. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson A, Murray S, Divakar P, Burnaevskiy N, Pifer R, Forman J, Yarovinsky F. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J Immunol. 2012;188:800–810. doi: 10.4049/jimmunol.1100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013a;14:1007–1013. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D. A special population of regulatory T cells potentiates muscle repair. Cell. 2013b;155:1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiaansen AF, Boggiatto PM, Varga SM. Limitations of Foxp3(+) Treg depletion following viral infection in DEREG mice. J Immunol Methods. 2014;406:58–65. doi: 10.1016/j.jim.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Berek C. The establishment of the plasma cell survival niche in the bone marrow. Immunol Rev. 2013;251:177–188. doi: 10.1111/imr.12011. [DOI] [PubMed] [Google Scholar]
- Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, Lee JJ, Lohning M, Berek C. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. 2011;12:151–159. doi: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- Clatworthy MR. Targeting B cells and antibody in transplantation. Am J Transplant. 2011;11:1359–1367. doi: 10.1111/j.1600-6143.2011.03554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, Masilamani RF, Dustin ML, Rudensky A, Liu K, Nussenzweig MC. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. 2009;206:1853–1862. doi: 10.1084/jem.20090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLillo DJ, Hamaguchi Y, Ueda Y, Yang K, Uchida J, Haas KM, Kelsoe G, Tedder TF. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. 2008;180:361–371. doi: 10.4049/jimmunol.180.1.361. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc Natl Acad Sci U S A. 2010;107:5919–5924. doi: 10.1073/pnas.1002006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo Celso C, Tsuyuzaki H, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474:216–219. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatman Zaretsky A, Engiles JB, Hunter CA. Infection-induced changes in hematopoiesis. J Immunol. 2014;192:27–33. doi: 10.4049/jimmunol.1302061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatman Zaretsky A, Silver JS, Siwicki M, Durham A, Ware CF, Hunter CA. Infection with Toxoplasma gondii alters lymphotoxin expression associated with changes in splenic architecture. Infect Immun. 2012;80:3602–3610. doi: 10.1128/IAI.00333-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11:681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- Hanig J, Lutz MB. Suppression of mature dendritic cell function by regulatory T cells in vivo is abrogated by CD40 licensing. J Immunol. 2008;180:1405–1413. doi: 10.4049/jimmunol.180.3.1405. [DOI] [PubMed] [Google Scholar]
- Hao Z, Rajewsky K. Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J Exp Med. 2001;194:1151–1164. doi: 10.1084/jem.194.8.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. 2001;97:1817–1822. doi: 10.1182/blood.v97.6.1817. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Moschetta M, Manier S, Glavey S, Gorgun GT, Roccaro AM, Anderson KC, Ghobrial IM. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263:160–172. doi: 10.1111/imr.12233. [DOI] [PubMed] [Google Scholar]
- Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, Nussenzweig M. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–397. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, Dugan BJ, Angeles TS, Albom MS, Mason JL, et al. Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J Immunol. 2011;187:3840–3853. doi: 10.4049/jimmunol.1101228. [DOI] [PubMed] [Google Scholar]
- MacNamara KC, Racine R, Chatterjee M, Borjesson D, Winslow GM. Diminished hematopoietic activity associated with alterations in innate and adaptive immunity in a mouse model of human monocytic ehrlichiosis. Infect Immun. 2009;77:4061–4069. doi: 10.1128/IAI.01550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388:133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- Matheu MP, Othy S, Greenberg ML, Dong TX, Schuijs M, Deswarte K, Hammad H, Lambrecht BN, Parker I, Cahalan MD. Imaging regulatory T cell dynamics and CTLA4-mediated suppression of T cell priming. Nat Commun. 2015;6:6219. doi: 10.1038/ncomms7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, Busslinger M. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol. 2016 doi: 10.1038/ni.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- Ng DH, Skehel JJ, Kassiotis G, Langhorne J. Recovery of an antiviral antibody response following attrition caused by unrelated infection. PLoS Pathog. 2014;10:e1003843. doi: 10.1371/journal.ppat.1003843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, Berek C, Hiepe F, Manz R, Radbruch A, Dorner T. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;105:1614–1621. doi: 10.1182/blood-2004-07-2507. [DOI] [PubMed] [Google Scholar]
- Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O’Brien S, Blank R, Lamb E, Natarajan S, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, Hiepe F. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–750. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, Mack M. Basophils support the survival of plasma cells in mice. J Immunol. 2010;185:7180–7185. doi: 10.4049/jimmunol.1002319. [DOI] [PubMed] [Google Scholar]
- Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85:15–24. doi: 10.1002/cyto.a.22355. [DOI] [PubMed] [Google Scholar]
- Rozanski CH, Arens R, Carlson LM, Nair J, Boise LH, Chanan-Khan AA, Schoenberger SP, Lee KP. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med. 2011;208:1435–1446. doi: 10.1084/jem.20110040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41:1026–1039. doi: 10.1016/j.immuni.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Sapoznikov A, Pewzner-Jung Y, Kalchenko V, Krauthgamer R, Shachar I, Jung S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat Immunol. 2008;9:388–395. doi: 10.1038/ni1571. [DOI] [PubMed] [Google Scholar]
- Slifka M, Antia R, Whitmire J, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- Slocombe T, Brown S, Miles K, Gray M, Barr TA, Gray D. Plasma cell homeostasis: the effects of chronic antigen stimulation and inflammation. J Immunol. 2013;191:3128–3138. doi: 10.4049/jimmunol.1301163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, Dustin ML. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med. 2006;203:505–511. doi: 10.1084/jem.20050783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye SG. Staying alive: regulation of plasma cell survival. Trends Immunol. 2011;32:595–602. doi: 10.1016/j.it.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, Kallies A, Busslinger M, Nutt SL. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol. 2016 doi: 10.1038/ni.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707–718. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Turner CA, Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell. 1994;77:297–306. doi: 10.1016/0092-8674(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gool F, Molofsky AB, Morar MM, Rosenzwajg M, Liang HE, Klatzmann D, Locksley RM, Bluestone JA. Interleukin-5-producing group 2 innate lymphoid cells control eosinophilia induced by interleukin-2 therapy. Blood. 2014;124:3572–3576. doi: 10.1182/blood-2014-07-587493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Moncrieffe H, Hocking RJ, Atkins CJ, Stockinger B. Modulation of dendritic cell function by naive and regulatory CD4+ T cells. J Immunol. 2006;176:6202–6210. doi: 10.4049/jimmunol.176.10.6202. [DOI] [PubMed] [Google Scholar]
- Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity. 2014;41:1013–1025. doi: 10.1016/j.immuni.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- Winter O, Dame C, Jundt F, Hiepe F. Pathogenic long-lived plasma cells and their survival niches in autoimmunity, malignancy, and allergy. J Immunol. 2012;189:5105–5111. doi: 10.4049/jimmunol.1202317. [DOI] [PubMed] [Google Scholar]
- Winter O, Moser K, Mohr E, Zotos D, Kaminski H, Szyska M, Roth K, Wong DM, Dame C, Tarlinton DM, et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood. 2010;116:1867–1875. doi: 10.1182/blood-2009-12-259457. [DOI] [PubMed] [Google Scholar]
- Wolf AI, Mozdzanowska K, Quinn WJ, 3rd, Metzgar M, Williams KL, Caton AJ, Meffre E, Bram RJ, Erickson LD, Allman D, et al. Protective antiviral antibody responses in a mouse model of influenza virus infection require TACI. J Clin Invest. 2011;121:3954–3964. doi: 10.1172/JCI57362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, Walker EU, Manz RA, Tarlinton DM, Smith KG. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol. 2007;8:419–429. doi: 10.1038/ni1440. [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc. 2010;5:201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehentmeier S, Roth K, Cseresnyes Z, Sercan O, Horn K, Niesner RA, Chang HD, Radbruch A, Hauser AE. Static and dynamic components synergize to form a stable survival niche for bone marrow plasma cells. Eur J Immunol. 2014;44:2306–2317. doi: 10.1002/eji.201344313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.