

ABSTRACT
Prompt reperfusion after cerebral ischemia is critical for neuronal survival. Any strategies that extend the limited reperfusion window will be of great importance. Acidic postconditioning (APC) is a mild acidosis treatment that involves inhaling CO2 during reperfusion following ischemia. APC attenuates ischemic brain injury although the underlying mechanisms have not been elucidated. Here we report that APC reinforces ischemia-reperfusion-induced mitophagy in middle cortical artery occlusion (MCAO)-treated mice, and in oxygen-glucose deprivation (OGD)-treated brain slices and neurons. Inhibition of mitophagy compromises neuroprotection conferred by APC. Furthermore, mitophagy and neuroprotection are abolished in Park2 knockout mice, indicating that APC-induced mitophagy is facilitated by the recruitment of PARK2 to mitochondria. Importantly, in MCAO mice, APC treatment extended the effective reperfusion window from 2 to 4 h, and this window was further extended to 6 h by exogenously expressing PARK2. Taken together, we found that PARK2-dependent APC-induced mitophagy renders the brain resistant to ischemic injury. APC treatment could be a favorable strategy to extend the thrombolytic time window for stroke therapy.
KEYWORDS: acidic postconditioning, cerebral ischemia, mitophagy, neuroprotection, PARK2, time window
Introduction
Ischemic stroke is one of the leading causes of mortality and disability worldwide. Great effort has focused on the development of potential neuroprotectants; however, few attempts have led to satisfactory results.1 Prompt blood restoration by thrombolysis is vital for neuronal survival and recovery. Unfortunately, the effective therapeutic time window for reperfusion is limited to approximately 4 h after ischemia.2 Therefore, any neuroprotective strategy extending the reperfusion window will be of great importance.
Postconditioning is a process induced by subtoxic stresses following an ischemic episode. Because they trigger endogenous adaptive mechanisms, ischemic, hypoxic, low-glucose and remote ischemic postconditioning treatments have proven to be promising therapies against cerebral ischemia,3-6 and they are proposed as a way to extend the reperfusion window.7 These postconditioning manipulations, however, are either impractical or dangerous. To overcome these disadvantages, we have developed acidic postconditioning (APC) as a therapy for cerebral ischemia. Induced simply by inhaling 20% CO2, APC significantly reduces focal cerebral ischemia and allows a wider time window for reperfusion than ischemic postconditioning (IPC).8 APC can preserve mitochondrial membrane potential, and, similarly, acidic pre-conditioning, where CO2 is inhaled before ischemia, also alleviates mitochondrial dysfunction.9 ASIC1/ASIC1α (acid-sensing [proton-gated] ion channel 1), the primary proton sensor in the nervous system, is localized in mitochondria and decreases mitochondrial membrane potential upon oxidative stress.10 A more recent study showed that acidosis can maintain mitochondrial functions against hypoxia in neurons.11 These studies highlight the possibility that acidosis targets mitochondrial function to protect the brain from ischemic injury.
Although it has not been fully elucidated how acidosis or APC may affect mitochondrial homeostasis, selective mitochondria turnover by the macroautophagy/autophagy machinery (mitophagy) may play a key role.12,13 Several postconditioning strategies have been demonstrated to activate autophagy in the ischemic heart and brain.14,15 However, it is unclear whether mitophagy is involved, even though the neuroprotective effects of mitophagy in cerebral ischemia-reperfusion have been documented.16-18 Moreover, the contributions of autophagy in general to postconditioning are controversial. Autophagy seems to eliminate the neuroprotective effects of IPC,19-21 but some groups have found that autophagy activation mimics the IPC-mediated neuroprotection.15,22 This discrepancy may be attributed to the complex nature of autophagy in selecting cargoes.23 For instance, in ischemic brains, the clearance of damaged mitochondria by mitophagy is beneficial for neuronal survival, whereas nonselective autophagy is detrimental.16,24 Therefore, in the present study we aimed to test the hypothesis that mitochondria are the principal targets of autophagy upon APC treatment and that mitophagy thus protects ischemic brains.
Results
APC activated mitophagy after cerebral ischemia in vivo and in vitro
To explore whether APC activated mitophagy in ischemic brains, mice were subjected to transient middle cerebral artery occlusion (tMCAO) and inhaled 20% CO2 for 5 min after a specified period of reperfusion. The levels of the mitochondrial markers TOMM20 and COX4I1 were used as indicators of mitophagy activity. We found that 5 min of APC initiated at 5 min after the onset of reperfusion (5/5 min) significantly decreased the levels of TOMM20 and COX4I1 (Fig. 1B). This loss of mitochondrial mass may be due to mitophagy, or alternatively caused by decreased mitochondrial biogenesis. We thus determined the mRNA levels of Cox4i1 and Tomm20 and 2 mitochondria biogenesis-related genes, Ppargc1a/Pgc1α and Nrf1, by real-time PCR. None of these mitochondrial markers or biogenesis-related genes was significantly downregulated with APC treatment (Fig. S1A to D). These data suggest that APC treatment (5/5 min) reduces mitochondria mass by activating mitophagy. In contrast, we found that APC failed to activate mitophagy when initiated 50 or 100 min after reperfusion (Fig. 1B and C). In addition to acidosis, inhaling CO2 increases blood flow and respiration rate.25,26 To exclude the possibility that these factors also affect mitophagy activation, NaHCO3 was used during APC to prevent acidosis. NaHCO3 alkalization reversed the APC-induced reduction in TOMM20 and COX4I1 expression levels, indicating that mitophagy was abolished in the absence of acidosis (Fig. 1D). Consistent with our results, NaHCO3 also abolishes the neuroprotective effects of APC,8 suggesting that acidosis is critical for both APC-mediated activation of mitophagy and neuroprotection.
Figure 1.

Acidic postconditioning activates mitophagy after cerebral ischemia in vivo and in vitro. (A) The schematic protocol of surgery for cerebral ischemia with or without acidic postconditioning (APC). For the in vivo model, mice were subjected to 1-h MCAO and then treated by inhaling 20% CO2 for 5 min at 5, 50, or 100 min after reperfusion. X indicates the duration of reperfusion. Mice of sham groups inhaled CO2 at corresponding times. For the in vitro model, OGD-treated corticostriatal slices, slices were subjected to OGD for 15 min and then treated with acidic buffer (pH 6.8) for 1, 3, or 5 min at 5 min after the onset of reperfusion. Y indicates the duration of APC treatment. (B) and C) COX4I1, TOMM20 and LC3 protein levels were determined by western blot analysis at 6 after the onset of reperfusion in tMCAO mice. (D) NaHCO3 was infused at 3 min before APC treatment (inhaling 20% CO2), which was applied for 5 min at 5 min after reperfusion of MCAO. After 6 h of reperfusion, COX4I1 and TOMM20 protein levels were determined by western blot analysis. NaHCO3 administration reversed APC-induced mitochondrial mass reduction. ((E)and F) COX4I1, TOMM20 and LC3 protein levels were determined by western blot analysis at 1 h after the onset of reperfusion in vitro. Data are expressed as mean ± SD n = 3 for each group. *P < 0.05 and **P < 0.01 vs. the indicated groups. N.S., not statistically significant.
We next investigated APC-mediated activation of mitophagy in the in vitro ischemic models, oxygen-glucose deprivation (OGD)-treated corticostriatal slices and OGD-treated primary cultured neurons. A 3-min APC treatment of corticostriatal slices applied after 5 min of reperfusion (5/3 min), which is one of the most prominent neuroprotection strategies,8 reduced TOMM20 and COX4I1 protein levels to a greater extent compared with other APC treatments (Fig. 1E and F). In OGD-treated neurons, APC decreased TOMM20 and COX4I1 levels (Fig. 2B). The mt-Atp6:Rpl13 ratio, an indicator of relative mitochondria amount,27 decreased as well (Fig. 2C). These reductions can be reversed by the lysosome inhibitor chloroquine (CQ), confirming that autophagic mitochondria lysis occurred28 (Fig. 2B and C). To monitor mitophagy in neurons, mitochondria were visualized with fluorescent MitoTracker Red, and autophagosomes were visualized based on the expression of green fluorescent protein-tagged microtubule-associated protein 1 light chain 3B (GFP-LC3).28 APC increased the overlap between mitochondria and autophagosomes in OGD-Rep neurons compared with OGD-Rep neurons that did not receive APC treatment (Fig. 2D). We also determined the levels of LC3-II after APC treatment in both in vivo and in vitro models and found that APC did not significantly reinforce LC3-II expression in these models (Fig. 1B, E). These results indicate that in ischemic brains APC selectively enhances mitophagy rather than generally enhancing autophagy. Taken together, these data suggest that APC-induced acidosis activates mitophagy after cerebral ischemia.
Figure 2.
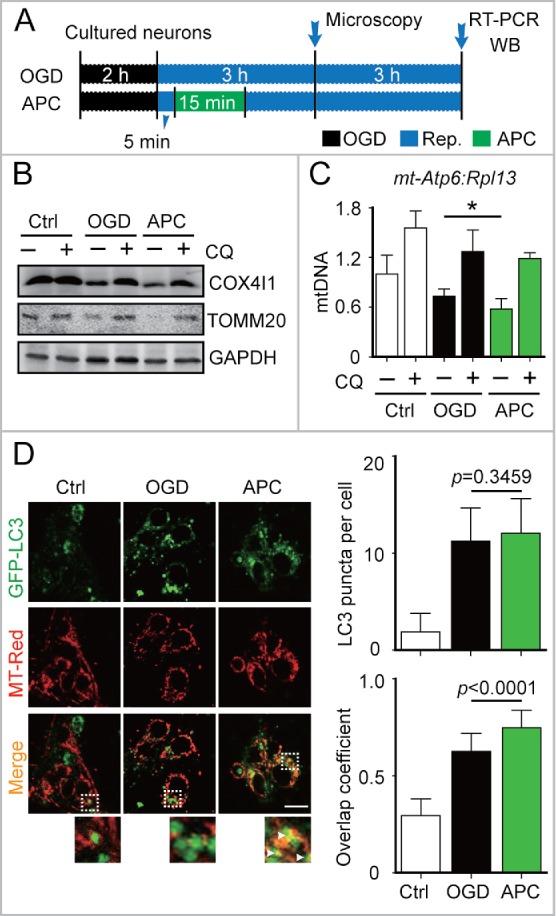
APC-activated mitophagy in primary cultured neurons. (A) OGD and APC protocols in primary cultured neurons. Primary neuronal cultures were subjected to OGD for 2 h and treated with acidosis for 15 min at 5 min after the onset of reperfusion. (B) After 6 h of reperfusion, TOMM20 and COX4I1 protein levels were determined by western blot analysis in the presence or absence of chloroquine (CQ), and (C) relative mitochondrial DNA levels as indicated by the ratio of mt-Atp6 (mitochondria-encoded DNA) to Rpl13 (nucleus encoded DNA) were assessed by real-time PCR. (D) Cells transfected with AAV-GFP-LC3 were loaded with MitoTracker Red (MT-Red; 100 nmol/L). Images were captured at 3 h after the onset of reperfusion by confocal microscopy. Areas in the white boxes are enlarged at the bottom. Right upper panel columns represent the numbers of GFP-LC3-positive puncta per cell, and the lower panel columns represent Manders' overlap coefficient. At least 30 cells from 3 independent experiments for each group were included. Scale bar: 20 µm. Data are expressed as mean ± SD n = 3 for each group. *P < 0.05 vs. the indicated groups.
Mitophagy inhibition blocks APC-mediated neuroprotection
To clarify the involvement of activated mitophagy in APC-mediated neuroprotection, mitophagy was blocked with the autophagy inhibitor 3-methyladenine (3-MA). In tMCAO mice, infarct volumes and neurologic deficit scores were assessed 24 h after the ischemic insult. Based on these measures, APC treatment significantly reduced ischemic brain injury. However, 3-MA injection (intracerebroventricular) before APC treatment abolished its neuroprotective effects as reflected by the reversed infarct volumes and neurologic scores. Moreover, neuroprotection was also diminished by the mitophagy inhibitor mitochondrial division inhibitor-1 (mdivi-1) (Fig. 3A and B). The mdivi-1 treatment blocks mitochondrial fission by inhibiting DNM1L (dynamin 1-like) and makes it difficult for the mitochondria that have not undergone fission to be engulfed by phagophores, the precursors to autophagosomes.16,29,30 The in vitro ischemic models, primary cultured neurons and corticostriatal slices, were subjected to 2 h and 15 min of OGD, respectively. Then the neurons and slices were exposed to 3-MA or mdivi-1 at the onset of reperfusion. The inhibititory effects of 3-MA and mdivi-1 on mitophagy were confirmed by western blot detection of TOMM20 and COX4I1 (Fig. 3C and D). We found that both 3-MA and mdivi-1 cancelled the neuroprotective effects of APC (Fig. 3D). We further blocked mitophagy by silencing Atg7, a gene that is essential for autophagy induction. APC did not reverse the cell viability with knockdown of Atg7 expression (Fig. 3E). Overall, these data offer support for the hypothesis that mitophagy is involved in APC-mediated neuroprotection against cerebral ischemia.
Figure 3.
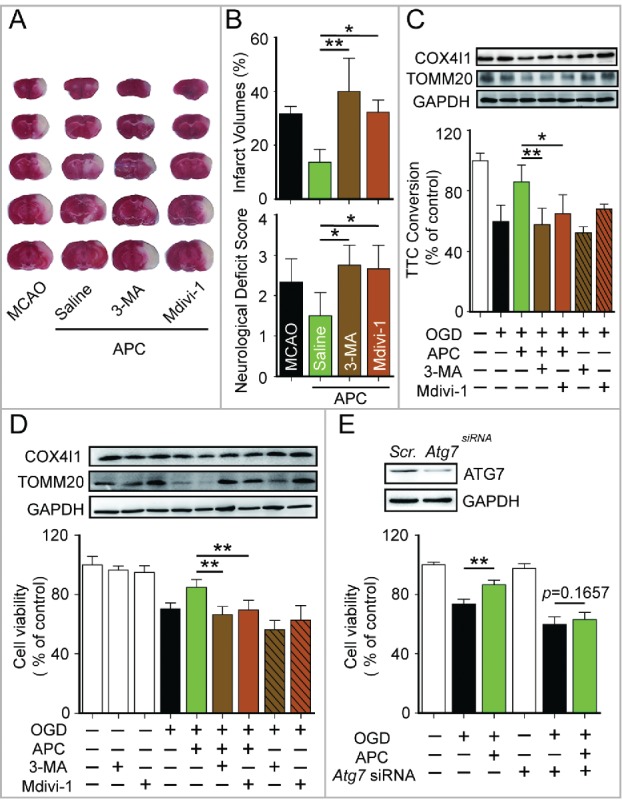
Mitophagy inhibition blocks APC-mediated neuroprotection. (A) and B) Mice were subjected to 1-h MCAO and then treated by inhaling 20% CO2 for 5 min at 5 min after reperfusion. (A) Representative brain slices after TTC staining are shown. (B) Infarct volumes and neurological deficit scores were measured 24 h after surgery. 7.5 µg of 3-methyladenine (3-MA, intracerebroventricular) or 3 mg/kg mdivi-1 (intraperitoneal) was injected at the onset of reperfusion. n = 4 to 6 for each group. Corticostriatal slices (C) and cultured neurons (D) were subjected to OGD and then treated with acidic buffer (pH 6.8) for 5 or 15 min at 5 min after the onset of reperfusion. The inhibitors, 1.25 mmol/L 3-MA or 25 µmol/L mdivi-1, were applied at the onset of reperfusion. COX4I1 and TOMM20 levels were examined by western blot at 1 or 6 h after the onset of reperfusion respectively (upper panel). Corticostriatal slice viability was quantified by TTC assay at 1 h after reperfusion, and cell viability was assessed by MTT assay at 24 h after reperfusion. (E) Atg7 was knocked down by transfection with microRNA 72 h before OGD. The knockdown of Atg7 expression was confirmed by western blot (upper panel). A scrambled siRNA sequence was used as a negative control (scr.). Cell viability was assessed by MTT assay at 24 h after reperfusion. APC failed to improve cell viability in Atg7 knockdown neurons. n = 3 for each group. Data are expressed as mean ± SD. *P < 0.05 and **P < 0.01 vs. the indicated groups.
PARK2 is required for APC-induced mitophagy activation and neuroprotection
PARK2 plays a key role in the mitophagy process. In ischemic neurons, PARK2 senses mitochondrial transmembrane potential loss and accumulates in the damaged mitochondria that are targeted for degradation.17,31 However, it is unclear whether PARK2 is required for APC-induced mitophagy. To determine whether APC induces PARK2 accumulation on mitochondria, we co-immunostained brain slices from wild-type mice with antibodies against PARK2 and the mitochondrial marker TOMM20. Our data show that APC enhances the recruitment of PARK2 to mitochondria in tMCAO mice that receive APC treatment compared with those that do not (Fig. 4A). In addition, we determined the intracellular distribution of PARK2 by separating the mitochondrial fraction from the cytosolic fraction of ischemic brains. APC treatment increased mitochondrial PARK2 levels in parallel with decreased cytosolic levels, suggesting that APC increased PARK2 translocation onto the mitochondria (Fig. 4B).
Figure 4.

APC facilitates PARK2 translocation onto mitochondria. Wild-type or park2−/− mice were subjected to 1-h MCAO and then treated by inhaling 20% CO2 for 5 min at 5 min after reperfusion. (A) APC-induced PARK2 translocation onto mitochondria can be seen at 2 h after the onset of reperfusion (green, upper panels). Mitochondria are labeled with the mitochondrial marker TOMM20 (red, lower panels). Nuclei were stained with DAPI. (B) PARK2 levels in both mitochondrial (mito.) and cytosolic (cyto.) fractions were determined by western blot analysis at 2 h after the onset of reperfusion. n = 3 for each group. (C) and D) Primary cultured neurons were transfected with AAV-GFP-PARK2 in advance of treatment. They were then subjected to 2-h OGD and treated with acidic buffer for 15 min at 5 min after the onset of reperfusion. Cells were loaded with MitoTracker Red (MT-Red), and images were captured by confocal microscopy. (C) Images show representative examples from 3 independent experiments. Scale bar: 20 µm. (D) Columns represent the percentage of neurons with PARK2 translocation onto mitochondria. At least 100 cells from 3 independent experiments for each group were included. Data are expressed as mean ± SD. ** P < 0.01 vs. the indicated groups.
To further confirm that APC facilitates PARK2 translocation to mitochondria, cultured neurons were infected with adeno-associated viruses (AAV) expressing GFP-PARK2. The ratio of neurons with PARK2 recruitment to mitochondria to those with no PARK2 recruitment was quantified. We found that APC increased the recruitment of PARK2 to mitochondria compared with the OGD-Rep group (70.31 ± 15.09% vs. 51.35 ± 10.97%, respectively, P < 0.01) (Fig. 4C and D). We next used Park2 knockout (park2−/−) mice to investigate the function of PARK2 in APC-induced mitophagy. We found that APC increased the overlap between TOMM20 and LC3 in tMCAO wild-type mice compared with untreated tMCAO mice, but not in park2−/− tMCAO mice (Fig. 5A). Similarly, we found that the APC-induced mitochondria clearance was largely impaired in park2−/− mice, as reflected by the lack of reduction in TOMM20 and COX4I1 protein levels (Fig. 5B). These results indicate that PARK2 is required for APC-induced mitophagy activation.
Figure 5.

PARK2 is required for APC-induced activation of mitophagy and neuroprotection. Wild-type or park2−/− mice were subjected to 1-h MCAO and then treated by inhaling 20% CO2 for 5 min at 5 min after reperfusion. (A) APC-activated mitophagy at 3 h after the onset of reperfusion was visualized using the autophagosome marker LC3 (green, upper panels) and the mitochondrial marker TOMM20 (red, lower panels). Nuclei were stained with DAPI. (B) COX4I1 and TOMM20 levels were determined by western blot at 6 h after the onset of reperfusion. n = 3 for each group. (C) Infarct volumes were quantified by TTC staining at 24 h after surgery. n = 6 for each group. (D) Primary cultured neurons were subjected to 2-h OGD, and treated with acidosis for 15 min at 5 min after the onset of reperfusion. ATP levels in cellular supernatants were determined at 24 h after the onset of reperfusion. n = 3 for each group. (E) Primary cultured neurons were transfected with AAV-GFP-PARK2 in advance. The neurons were then treated as described in (D). The PARK2 level in wild-type, park2−/− and park2−/− with GFP-PARK2 neurons was measured by western blot (left panel). Cell viability was examined by MTT assay after 24 h of reperfusion (right panel). n = 3 for each group. Data are expressed as mean ± SD. * P < 0.05 and ** P < 0.01 vs. the indicated groups. N.S., not statistically significant.
To further explore the role of PARK2-dependent mitophagy in APC-conferred neuroprotection, park2−/− mice were subjected to tMCAO. Park2 deletion significantly enhanced the brain infarct volume (Fig. 5C), and APC treatment did not exhibit any rescuing effect, indicating that the neuroprotective effect of APC was almost completely blocked in park2−/− mice. Therefore PARK2 is required for APC-conferred benefits. Mitochondrial health in neurons is reflected by the recovery of ATP supply after OGD.32 We therefore examined the intracellular ATP level of primary cultured neurons 24 h after OGD insult. We found significant ATP loss in park2−/− neurons after OGD, which confirmed the increased mitochondrial vulnerability caused by Park2 deletion.33 Moreover, APC treatment significantly reversed the OGD-induced decrease in intracellular ATP in wild-type neurons, but not in park2−/− neurons, suggesting the involvement of PARK2 in APC-conferred mitochondrial quality control (Fig. 5D). To further support this notion, GFP-PARK2 was exogenously overexpressed in park2−/− neurons. Based on the cell viability 24 h after OGD-Rep. treatment, we found that PARK2 overexpression in park2−/− neurons restored the neuroprotective effects of APC (Fig. 5E). These results indicate that PARK2-induced mitophagy is required for the APC-mediated neuroprotection.
APC-activated mitophagy extends the reperfusion window of cerebral ischemia
Circulation restoration to the ischemic territory remains the most effective strategy to rescue ischemic brains. However, the effective reperfusion time window is limited to approximately 4 h after ischemia. We previously reported that after 1 h of MCAO in mice, APC treatment within 50 min of reperfusion is protective.8 This postconditioning manipulation time-window is wider than that of IPC. Nevertheless, it is still unclear whether APC is able to extend the reperfusion window for cerebral ischemia. To address this issue, cerebral blood reperfusion was established at 2, 4 and 6 h after MCAO in mice, and these mice were not killed until 24 h after the onset of ischemia. Another group of mice that received no reperfusion and were also killed 24 h after MCAO was used as the permanent ischemia control (pMCAO). As shown in Fig. 6A, compared with the pMCAO mice, reperfusion reduced infarct volumes at 2 h, but not at 4 h or longer periods of ischemia, suggesting that the reperfusion window was approximately 2 h in our model of ischemia.
Figure 6.

APC-activated mitophagy extends the reperfusion window for cerebral ischemia. (A)and B) Mice were subjected to permanent middle cerebral artery occlusion (pMCAO) for 24 h or transient middle cerebral artery occlusion (tMCAO) for the indicated durations of reperfusion. Mice with acidosis treatment were treated by inhaling 20% CO2 for 5 min at 1 h after the onset of occlusion (pMCAO) or at 5 min after reperfusion (tMCAO), respectively. (A) A representative TTC-stained brain slice from each group is shown. (B) Infarct volumes were determined 24 h after surgery by TTC staining. n = 6 for each group. (C) and D) The impact of APC on the expression of COX4I1 and TOMM20 in ipsilateral cortical tissue after (C) the indicated tMCAO procedures; (D) tMCAO treatment with 1 h of ischemia + 5 h of reperfusion compared with pMCAO treatment with 6 h of ischemia. (E) Mice were injected with AAV-GFP-PARK2 one month in advance of treatment. They were then subjected to 6-h MCAO and then treated by inhaling 20% CO2 for 5 min at 5 min after reperfusion. Infarct volumes were determined by TTC staining 24 h after surgery, and a representative TTC-stained brain slice from each group is shown. n = 6 for each group. Data are expressed as mean ± SD. * P < 0.05 and ** P < 0.01 vs. the indicated groups. N.S., not statistically significant.
Surprisingly, we found that an APC treatment of 5-min CO2 inhalation after 5 min of reperfusion significantly reduced infarct volumes even after 4 h of ischemia (Fig. 6A and B). These results indicate that the APC procedure extends the reperfusion window to at least to 4 h after ischemia. We next examined the activation of mitophagy by APC after different durations of ischemia. APC significantly reduced the levels of mitochondrial markers after 2 or 4 h, but not after 6 h of ischemia (Fig. 6C) or in the pMCAO control (Fig. 6D). This time scale is consistent with the extended reperfusion window conferred by APC, indicating that APC-activated mitophagy may be connected to reperfusion window extension. Therefore, we hypothesized that enhanced mitophagy may provide additional extension of the reperfusion window. To test this, GFP-PARK2 was exogenously expressed in mice. As expected even after 6 h of MCAO, the APC treatment remained effective in mice overexpressing PARK2 (Fig. 6E). This result indicates that reinforced PARK2-dependent mitophagy further extends the reperfusion window up to at least 6 h after ischemia. Taken together, these data suggest that APC could be a promising strategy to extend the reperfusion window.
Discussion
Pre- and postconditioning strategies are considered promising therapies for cerebral ischemia.34,35 The mechanisms by which these approaches provide neuroprotection are not fully elucidated, and there is growing interest in studying the role of autophagy in these processes.36 Ischemic postconditioning inhibits lethal ischemia-induced autophagy.19 However, another investigation found a further increase in autophagy upon remote ischemic postconditioning in the limb.15 These discrepancies may be due to differences in models and postconditioning procedures, but they may also suggest bidirectional regulation of autophagy by postconditioning.
In the present study we found that APC did not significantly upregulate the level of LC3-II, suggesting that autophagic flux was not robustly reinforced (Fig. 1B; Fig. 2D). However, interestingly we observed increased mitochondria loss with APC treatment in distinct ischemic models (Fig. 1C, E; Fig. 2C, D). Mitochondrial loss was autophagic because it was reversed by lysosome inhibition (Fig. 2B). PARK2 plays a key role in the mitophagy process. The loss of mitochondrial membrane potential (ΔΨm) triggers PARK2 translocation and thereby recruits autophagy machinery to mitochondria in ischemic neurons.17 Consistent with this, we also observed PARK2 translocation after ischemia treatments (Fig. 4A to C) and the abolishment of mitophagy in park2−/− mice (Fig. 5A and B), further supporting mitophagy activation by APC. These results show for the first time that mitophagy can be selectively activated by APC.
Nevertheless, it is not clear how APC enhances selective recognition of mitochondria by phagophores. In the very first minutes of reperfusion, there is a rapid reversal of intracellular acidosis to alkalosis,37 which facilitates H+ pumping to the mitochondrial intermembrane space by mitochondrial oxidative phosphorylation (OxPhos) complexes.38 The resulting increased H+ gradient transiently hyperpolarizes the ΔΨm.39,40 Given that mitochondrial PARK2 recruitment is reversible with ΔΨm regulation,41 it is plausible that PARK2 may temporally detach from mitochondria due to the ΔΨm increase. APC treatment may prevent PARK2 detachment by attenuating the alkalosis of reperfusion and thus promote mitophagy. Although direct evidence is lacking, we found that NaHCO3 treatment reduces the amount of mitophagy induced by APC (Fig. 1D), suggesting that there is an association between pH and mitophagy. This relationship may also explain why APC promotes mitophagy only in transient but not permanent ischemia (Fig. 6D); reperfusion provides an opportunity for APC to reverse alkalosis and ΔΨm hyperpolarization.
Autophagy selectively targets a variety of cellular components, and the indiscriminant experimental manipulation of autophagy may lead to incorrect interpretation of the contributions of autophagy to acute brain injury.23 Here, we used mdivi-1, a known inhibitor of mitochondrial fission, to inhibit mitophagy. Although mdivi-1 was shown to have other effects in addition to inhibiting mitophagy,42,43 recent studies have suggested that mdivi-1 acts as an inhibitor of mitophagy by blocking autophagosome/mitophagosome formation.30,44 We found that mdivi-1 abolishes the neuroprotective effects of APC (Fig. 3B–D), suggesting the involvement of mitophagy. Therefore, APC-induced autophagy may target defective mitochondria and thereby render neuronal cells resistant to reperfusional injury. A recent study showed that low pH modulates mitochondrial shapes and sustains mitochondrial function and cell survival in hypoxia.11 Together with our results, it appears that acidosis not only helps to remodel the healthy mitochondria but also to remove the defective ones by mitophagy.
PARK2 is involved in ischemia-induced mitophagy,16,17 and deletion of the corresponding gene has no impact on autophagic flux,45 indicating that Park2 deletion is an ideal approach to selectively block mitophagy. The park2−/− mice have larger infarct volumes and reduced ATP recovery (Fig. 5C and D). Conversely, GFP-PARK2 expression reduces ischemic injury (Fig. 5E). These data provide direct evidence that PARK2-dependent mitophagy is required for the neuroprotective effects of APC. In our study, PARK2 was exogenously expressed in neurons before the ischemic event. Although it is unlikely that PARK2 promotes mitophagy in intact neurons, we cannot exclude the possibility that overexpressed PARK2 may act before APC. Prompt induction of PARK2 expression upon APC treatment may be needed to address this issue. Taken together, these data strongly suggest that mitochondria are the target of APC-induced autophagy and that mitophagy confers the protective effects of APC against ischemic neuronal injury.
Thrombolysis and thrombectomy are common stroke therapy procedures used to achieve recanalization. However, these procedures are clinically appropriate only in a small portion of patients due to the limited reperfusion window. Unfortunately, few therapies address this issue in clinical practice. Hence, there is a consensus that any strategy that extends the reperfusion window will be valuable. Here we recapitulated the reperfusion window in mice by retrieval of occlusions after distinct periods of ischemia (Fig. 6B, dark columns). We found that the reperfusion window in our model is approximately 2 h. Surprisingly, APC treatment extended the reperfusion window to at least 4 h, which is approximately a 2-h extension (Fig. 6B, green columns). This increase could be of great importance because the mortality rate increases approximately 1% with each 1-h delay in reperfusion.46 A variety of mechanisms may be involved in determining the reperfusion window.47 Delayed reperfusion causes excessive free radical accumulation, which disrupts the blood-brain barrier and results in deteriorated neurovascular functions.48 Oxidative stress may play a key role in regulating the reperfusion window. Mitochondria are the principal source of reactive oxygen species upon cerebral ischemia reperfusion. APC-activated mitophagy may remove these stressed mitochondria and subsequently extend the reperfusion window. PARK2 is also likely involved in the APC-mediated extension of the reperfusion window because APC did not potentiate the protection of reperfusion in park2−/− mice even after only 1 h of ischemia (Fig. 5C). Moreover, it is noteworthy that APC had neuroprotective effects even after 6 h of ischemia in wild-type mice that exogenously express PARK2 (Fig. 6E). This finding raises the possibility of extending the reperfusion window by reinforcing PARK2-dependent mitophagy.
In summary, the present study has provided strong evidence that PARK2-dependent mitophagy is responsible for APC-induced neuroprotection and reperfusion window extension. These findings bring forth the notion that mitochondria are critical targets of autophagy in APC and possibly other postconditioning procedures. Therefore, APC could be a powerful strategy to overcome the reperfusion window limitation in stroke therapy.
Materials and methods
Animals
Male adult C57BL/6 mice weighing 22–25 g were used. The park2−/− mice (C57BL/6 strain background) were kindly provided by Prof. Zhuohua Zhang (Central South University). For primary cortical neuronal culture, pregnant Sprague-Dawley rats, C57BL/6 mice or park2−/− mice with embryonic (E18) fetuses were used. All experimental protocols and animal handling procedures were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the experimental protocols were approved by the Zhejiang University Animal Experimentation Committee. Every effort was made to minimize any pain or discomfort, and the minimum number of animals was used.
MCAO mouse models, acidic postconditioning and drug administration
Mice were anesthetized for surgery by inhalation of isoflurane. Cerebral blood flow (CBF) was determined in the area of the middle cerebral artery (MCA) by laser Doppler flowmetry (Model Moor VMS-LDF2, Moor Instruments Ltd, UK). A flexible fiber-optic probe was affixed to the skull over the cortex supplied by the proximal part of the right MCA (2-mm caudal to bregma and 6-mm lateral to midline). Focal cerebral ischemia was induced by MCA occlusion (MCAO) as described previously.49 Briefly, a 6–0 nylon monofilament suture, blunted at the tip and coated with 1% poly-l-lysine, was inserted 10 mm into the internal carotid to occlude the origin of the MCA. Animals that had less than 80% reduction in CBF in the core of the MCA area were excluded from the study. Body temperature was maintained at 37°C by a heat lamp (FHC, Bowdoinham, ME, USA) during surgery. For transient MCAO, reperfusion was allowed after a specified duration by monofilament removal. Acidic postconditioning (APC) was performed as we described previously.8 Briefly, mice inhaled 20 % CO2 for 5 min after 5, 50, or 100 min of reperfusion. For permanent MCAO, acidosis treatment was performed for 5 min at 1 h after the onset of occlusion.
Mice were given an intracerebroventricular injection of 7.5 µg 3-MA (Sigma, M9281) at the onset of reperfusion in tMCAO as we described previously.16 Control mice were injected with the same volume of saline. Mdivi-1 (Abcam, ab144589) was injected intraperitoneally at the onset of reperfusion (3 mg/kg body weight). NaHCO3 (2 mL/animal, 78 mg/mL) was intracerebroventricularly injected at 3 min after reperfusion.
Infarct volumes were determined at 24 h after surgery by 2,3,5-triphenyltetrazolium hydrochloride (TTC; 0.25%; Sigma, T8877) staining, and the extent of the normal and infarct areas were measured with ImageJ software and determined by an indirect method, which corrects for edema. The percentage of the corrected infarct volume was calculated by dividing the infarct volume by the total contralateral hemispheric volume, and this ratio was then multiplied by 100.
Neurological deficit scores were evaluated 24 h after surgery as follows: 0, no deficit; 1, flexion of contralateral forelimb upon lifting of the whole animal by the tail; 2, circling to the contralateral side; 3, falling to contralateral side and 4, no spontaneous motor activity.
Virus delivery in vivo
Mice were anesthetized by inhalation of isoflurane and mounted in a stereotaxic apparatus (512600, Stoelting, Wood Dale, Il, USA), AAV-GFP-PARK2 (1 μl) was injected into the corpus striatum (AP +0.5 mm; L −2.0 mm; V −3.0 mm ) with a 10-μl syringe and a 34 gauge needle at 100 nl/min using an injection pump (Micro 4, WPI, Sarasota, Fl, USA). After each injection, the needle was left in place for an additional 5 min and then slowly withdrawn. The virus was allowed to express target proteins for a minimum of 1 mo.
Preparation of brain slices, oxygen-glucose deprivation and APC procedures, drug administration and slice viability determination
Acute brain slices were prepared from adult male C57BL/6 mice, and oxygen-glucose deprivation (OGD) procedures were performed as we described previously.5 Briefly, corticostriatal slices (400-mm thick) were cut in ice-cold artificial cerebrospinal fluid (ACSF) bubbled with 5% CO2+95% O2 (pH 7.4). The slices were immersed in oxygenated ACSF for 1 h and then at 37°C for 15 min before experiments. For OGD, slices were transferred into glucose-free ACSF bubbled with 5% CO2+95% N2 for 15 min, then returned to oxygenated ACSF for 1 h. Control slices were kept in oxygenated ACSF. For APC, after 5 min of reperfusion by returning the slices to oxygenated ACSF, slices were transferred to acidic buffer equilibrated with 80% O2+20% CO2 (pH 6.8) for 1, 3, or 5 min. Inhibitors, 1.25 mmol/L 3-MA or 25 µmol/L mdivi-1, were applied at the onset of reperfusion. Slice viability was determined by 0.25% TTC staining. Formazan extracted with ethanol:dimethylsulfoxide (1:1) was measured at 490 nm. Viability was determined by absorbance normalized to the dry weight of the slice and expressed as the percentage of control slices included in each experiment.
Cell culture, OGD and APC procedures, drug administration and cell viability determination
For primary cortical neuronal culture, E18 mice were used. Briefly, the dissected cortex was treated with 0.125% trypsin (Invitrogen, 25200–056) in Hank's buffer (in mmol/L: 137 NaCl, 5.4 KCl, 0.4 KH2PO4, 0.34 Na2HPO4·7H2O, 10 glucose and 10 HEPES) for 10 min at 37°C and dissociated by repeated passage through a series of fire-polished Pasteur pipettes. Approximately 2 × 105 cells/cm2 were seeded onto poly-L-lysine (10 μg/ml)-coated plates and dishes. The neurons were cultured in Neurobasal medium (Invitrogen, 21103–049) supplemented with 2% B27 (Invitrogen, 17504–044), 10 U/ml penicillin, 10 U/ml streptomycin and 0.5 mmol/L glutamine at 37°C in a humidified atmosphere with 5% CO2. Cultures were maintained for 8 d before further treatment and were routinely observed under a phase-contrast inverted microscope. To quantify the neuron percentage in our cultures, we immunostained the neurons against RBFOX3/NeuN. We found that the neuron percentage was approximately 85–90%.
For OGD treatment, cells were rinsed once with warm glucose-free DMEM (Invitrogen, 12800–017), and then refreshed with O2- and glucose-free DMEM (pre-balanced in an O2-free chamber at 37°C). Cells were immediately placed in a sealed chamber (Billups Rothenburg, MIC-101) loaded with mixed gas containing 20% CO2+80% N2 for 6 min at 25 L/min. The sealed chambers were then incubated at 37°C for 2 h. Reperfusion was performed by refreshing the culture with normal medium. For APC, cells were exposed to an acidic culture medium (pH 6.8) equilibrated with normoxic mixed gas containing 20% CO2 for 15 min at 5 min after reperfusion. Inhibitors, 10 μmol/L CQ (Sigma, C6628), 1.25 mmol/L 3-MA or 25 µmol/L mdivi-1, were applied at the onset of reperfusion. Cell viability was determined by the 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide assay (MTT, Sigma, M2128) after 24-h reperfusion. Cells were incubated with 0.5mg/ml MTT at 37°C for 4 h. The reaction product formazan was dissolved in DMSO. Absorbance at 570nm was determined using a microplate reader (DTX880, Beckman Coulter, CA, USA). Cell viability of vehicle-treated controls was set as 100%.
Transfection and RNA silencing in primary cultured neurons
To achieve a high efficiency of transfection in primary cultured neurons, AAVs containing GFP-LC3 and GFP-PARK2 were constructed (Obio Technology Corp., Ltd. Shanghai, China). AAVs were incubated with neurons at 6 d in vitro.
A small interfering RNA (siRNA) targeting mice Atg7 (5′-GCAUCAUCUU UGAAGUGAA-3′) and a scrambled control siRNA (5′-AUGAAGTGA AUUGCUCAA-3′) were synthesized by GenePharm (Shanghai). Primary neurons were transfected on 5 d in vitro with 20 nmol Atg7 siRNA or scrambled siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778100). After transfection in antibiotic-free medium for 8 h, cells were refreshed with normal medium.
Confocal microscopy
For microscopy examination, primary neurons were seeded onto a poly-l-lysine-treated glass bottom dish (In Vitro Scientific, D35–20–0-N) and transfected with GFP-LC3 or GFP-PARK2 as aforementioned. After OGD, cells were reperfused for the indicated duration of time. Cells were incubated with MitoTracker Red (100 nmol/L; Life Technologies, M7512) for 15 min before observation. Dishes were observed on a confocal microscope (Fluoview FV1000, Olympus, Tokyo, Japan). The Manders overlap coefficiency was measured and analyzed by Image Pro-Plus 7.0 software. Five randomly selected fields from one coverslip were included to get an average, and experiments were repeated independently at least 3 times.
Real-time PCR
mt-Atp6:Rpl13 assay: After 6 h of OGD-reperfusion, total cellular DNA was extracted with a DNeasy Blood and Tissue kit (Tiangen, DP304–03). Aliquots of 20 ng total DNA were used for PCR detection of the mitochondrial gene mt-Atp6 and the nuclear gene Rpl13, as we described previously.16 The primer sequences were as follows: rat mt-Atp6 (Fw: 5′-ATT ACG GCT CCT GCT CAT A-3′; Rev: 5′-TGG CTC AAC CAA CCT TCT A-3′) and rat Rpl13 (Fw: 5′-CAC AAG AAA ATG GCA CGC AC-3′; Rev: 5′-GAG CAG AAG GCT TCC TGG G-3′). The amount of PCR product was calculated and normalized using the standard curve. Relative expression was presented as the mt-Atp6:Rpl13 ratio.
For the mitochondria biogenesis gene assay, after 6 h of tMCAO-reperfusion, total RNA was isolated from ischemic mice cerebral cortex using Trizol (Sigma, T9424) according to the manufacturer's instructions. Total RNA was reverse-transcribed into cDNA using the Prime Script One-Step RT-PCR Kit (TakaRa, RR037A) according to the manufacturer's instructions. RNA concentrations were measured using a NanoDrop (Thermo Scientific). The following primer sequences were used: Tomm20 (Fw: 5′-TGGGCTTTCCAAGTTACCTGATT-3′; Rev: 5′- CACCCTTCTCGTAGTCACCTTGT-3′), Cox4i1 (Fw: 5′-GAATGTTGGCTTCCAGAGCG-3′; Rev: 5′-TCACAACACTCCCATGTGCT-3′), Ppargc1a/Pgc1α (Fw: 5′-ACTGACGGCCTAACTCCACCCA-3′; Rev: 5′-ACTCGGATTGCTCCGGCCCT-3′) and Nrf1 (Fw: 5′-GCGCAGCCGCTCTGAGAACTTAT-3′; Rev: 5′-ATGGGCGGCAGCTTGACTGT-3′).
Determination of ATP Levels
The level of ATP in cultured neurons was determined using the ATP Assay Kit (Beyotime Institute of Biotechnology, S0026), according to the manufacturer's instructions. Briefly, cultured cells were harvested after 24 h reperfusion, were lysed with a lysis buffer, and were centrifuged at 12,000 × g for 10 min at 4°C. The level of ATP was determined by mixing 50 μl of the supernatant with 50 μl of luciferase reagent. Luciferase uses ATP to catalyze the oxidation of luciferin to produce light. The emitted light, which is linearly related to the ATP concentration, was measured using a microplate luminometer (Varioskan Flash, 5250040, Thermo).
Immunohistochemistry
Mice were killed and were perfused with saline solution, followed by 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS, Corning, 21–040-CVR). The brain was then removed, fixed in 4% paraformaldehyde overnight and equilibrated in 30% sucrose (Vetec, V900116) at 4°C. Coronal sections (2 μm) were cut on a freezing microtome (Thermo, CRYOSTAR NX50). After rinsing with 0.5 % Triton X-100 (vol/vol; Sangon, T0694) in 0.1 M PBS (30 min) and blocking with 10% (wt/vol) normal bovine serum (Jackson ImmonoResearch, 017–000–121) for 4 h at room temperature, sections were incubated with the following primary antibodies diluted in PBS with 0.15% Triton X-100 overnight at 4°C: anti-TOMM20 (1:400; Abcam, ab56783), anti-PARK2 (1:400; Cell Signaling Technology, 2132S) and anti-LC3 (1:400; Sigma, L7543). After rinsing with PBS, sections were incubated with Alexa Fluor 594- or Alexa Fluor 488-conjugated secondary fluorescent antibody (1:400; Jackson ImmunoResearch, 715–585–150, 711–545–152) for 2 h at room temperature. After rinsing, the sections were mounted on slides using Vectashield Mounting Media (Sigma, F6057). Confocal images were captured using a Fluoview FV1000 confocal microscope (Olympus, Tokyo, Japan).
Immunoblotting
Brain tissues and cells were homogenized in RIPA buffer (20 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton X-100, 0.5% sodium deoxycholate (Sigma, 30970), and 2% Protease Inhibitor Cocktail Tablets (Roche, 04693132001). For cytosolic and mitochondrial protein extraction, an isolation kit (Thermo, 89801) was used according to the manufacturer's instructions. An aliquot of 40 µg protein from each sample was separated using SDS-PAGE and transferred to a nitrocellulose membrane, which was then blocked with 5% nonfat milk in PBS (pH 7.4).
The membrane was incubated with primary antibodies against LC3 (1:1,000), COX4I1 (1:1,000; Cell Signaling Technology, 4844S), TOMM20 (1:800; Anbo Biotechnology, C16678), PARK2 (1:1,000), ATG7 (1:1,000; Epitomics, 5146–1) and GAPDH (1:3,000; KangChen, KC-5G4) at 4°C overnight. Secondary antibodies against rabbit (IRDye 800-coupled, 1:10,000; LI-COR, 926–32211) or mouse IgG (IRDye 700-coupled, 1:5,000; LI-COR, 926–68070) were incubated for 2 h at room temperature. Blots were visualized using an Odyssey infrared imaging system (LI-COR Biosciences, 9120) and analyzed using the Odyssey software. The relative optical density was obtained by comparing the measured values with the mean values from the control group.
Statistical analysis
All data were collected and analyzed in a blind manner. Data are presented as mean ± SD. One-way ANOVA (analysis of variance) with Dunnett T3 post-hoc tests was applied for multiple comparisons. P < 0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- AAV
adeno-associated virus
- ACSF
artificial cerebrospinal fluid
- APC
acidic postconditioning
- COX4I1
cytochrome c oxidase IV isoform 1
- CQ
chloroquine
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- IPC
ischemic postconditioning
- Isc.-Rep
ischemia-reperfusion
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- mdivi-1
mitochondrial division inhibitor-1
- mt-Atp6
mitochondrially encoded ATP synthase 6
- mtDNA
mitochondrial DNA
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- Nrf1
nuclear respiratory factor 1
- OGD-Rep.
oxygen and glucose deprivation-reperfusion
- OxPhos
oxidative phosphorylation
- PARK2
Parkinson disease (autosomal recessive
- juvenile) 2
parkin
- Ppargc1a
peroxisome proliferative activated receptor, gamma, coactivator 1 α
- pMCAO
permanent middle cerebral artery occlusion
- Rpl13
ribosomal protein L13
- tMCAO
transient middle cerebral artery occlusion
- TOMM20
translocase of outer mitochondrial membrane 20 homolog (yeast)
- TTC
2,3,5-triphenyltetrazolium hydrochloride.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful to Dr. Zhuohua Zhang for offering the Park2 gene knockout mice. We are grateful to the Imaging Facilities, Zhejiang University School of Medicine for the help in confocal microscopy.
Funding
This work was funded by the National Natural Science Foundation of China (81573406, 81373393, 81273506, 81221003, 81473186 and 81402907), Zhejiang Provincial Natural Science Foundation (LR15H310001) and the Program for Zhejiang Leading Team of S&T Innovation Team (2011R50014).
References
- [1].Moskowitz MA. Brain protection: maybe yes, maybe no. Stroke 2010; 41:S85-6; PMID:20876513; http://dx.doi.org/ 10.1161/STROKEAHA.110.598458 [DOI] [PubMed] [Google Scholar]
- [2].Del Zoppo GJ, Saver JL, Jauch EC, Adams HP Jr, American Heart Association Stroke C . Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke 2009; 40:2945-8; PMID:19478221; http://dx.doi.org/ 10.1161/STROKEAHA.109.192535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab 2006; 26:1114-21; PMID:16736038; http://dx.doi.org/ 10.1038/sj.jcbfm.9600233 [DOI] [PubMed] [Google Scholar]
- [4].Leconte C, Tixier E, Freret T, Toutain J, Saulnier R, Boulouard M, Roussel S, Schumann-Bard P, Bernaudin M. Delayed hypoxic postconditioning protects against cerebral ischemia in the mouse. Stroke 2009; 40:3349-55; PMID:19628803; http://dx.doi.org/ 10.1161/STROKEAHA.109.557314 [DOI] [PubMed] [Google Scholar]
- [5].Fan YY, Zhang XN, He P, Shen Z, Shen Y, Wang XF, Hu WW, Chen Z. Transient lack of glucose but not O2 is involved in ischemic postconditioning-induced neuroprotection. CNS Neuroscience Therapeutics 2013; 19:30-7; http://dx.doi.org/ 10.1111/cns.12033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hess DC, Hoda MN, Bhatia K. Remote limb perconditioning [corrected] and postconditioning: will it translate into a promising treatment for acute stroke? Stroke 2013; 44:1191-7; PMID:23339961; http://dx.doi.org/ 10.1161/STROKEAHA.112.678482 [DOI] [PubMed] [Google Scholar]
- [7].Esmaeeli-Nadimi A, Kennedy D, Allahtavakoli M. Opening the window: Ischemic postconditioning reduces the hyperemic response of delayed tissue plasminogen activator and extends its therapeutic time window in an embolic stroke model. Eur J Pharmacol 2015; 764:55-62; PMID:26123846; http://dx.doi.org/ 10.1016/j.ejphar.2015.06.043 [DOI] [PubMed] [Google Scholar]
- [8].Fan YY, Shen Z, He P, Jiang L, Hou WW, Shen Y, Zhang XN, Hu WW, Chen Z. A novel neuroprotective strategy for ischemic stroke: transient mild acidosis treatment by CO2 inhalation at reperfusion. J Cereb Blood Flow Metab 2014; 34:275-83; PMID:24192637; http://dx.doi.org/ 10.1038/jcbfm.2013.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang CH, Fan YY, Wang XF, Xiong JY, Tang YY, Gao JQ, Shen Z, Song XH, Zhang JY, Shen Y, et al.. Acidic preconditioning protects against ischemia-induced brain injury. Neurosci Lett 2012; 523:3-8; PMID:22583767; http://dx.doi.org/ 10.1016/j.neulet.2012.05.015 [DOI] [PubMed] [Google Scholar]
- [10].Wang YZ, Zeng WZ, Xiao X, Huang Y, Song XL, Yu Z, Tang D, Dong XP, Zhu MX, Xu TL. Intracellular ASIC1a regulates mitochondrial permeability transition-dependent neuronal death. Cell Death Differ 2013; 20:1359-69; PMID:23852371; http://dx.doi.org/ 10.1038/cdd.2013.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, Bergeron R, Cregan SP, Harper ME, Park DS, et al.. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat Commun 2014; 5:3550; PMID:24686499; http://dx.doi.org/ 10.1038/ncomms4550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 2012; 148:1145-59; PMID:22424226; http://dx.doi.org/ 10.1016/j.cell.2012.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012; 337:1062-5; PMID:22936770; http://dx.doi.org/ 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wei C, Li H, Han L, Zhang L, Yang X. Activation of autophagy in ischemic postconditioning contributes to cardioprotective effects against ischemia/reperfusion injury in rat hearts. J Cardiovascular Pharmacol 2013; 61:416-22; PMID:23364609; http://dx.doi.org/ 10.1097/FJC.0b013e318287d501 [DOI] [PubMed] [Google Scholar]
- [15].Su J, Zhang T, Wang K, Zhu T, Li X. Autophagy activation contributes to the neuroprotection of remote ischemic perconditioning against focal cerebral ischemia in rats. Neurochem Res 2014; 39:2068-77; PMID:25082119; http://dx.doi.org/ 10.1007/s11064-014-1396-x [DOI] [PubMed] [Google Scholar]
- [16].Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al.. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013; 9:1321-33; PMID:23800795; http://dx.doi.org/ 10.4161/auto.25132 [DOI] [PubMed] [Google Scholar]
- [17].Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z, Zheng Y, Deng T, Yan H, Li W, et al.. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014; 10:1801-13; PMID:25126734; http://dx.doi.org/ 10.4161/auto.32136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yan H, Zhang X, Hu W, Ma J, Hou W, Wang X, Gao J, Shen Y, Lv J, Ohtsu H, et al.. Histamine H3 receptors aggravate cerebral ischaemic injury by histamine-independent mechanisms. Nat Commun 2014; 5:3334; PMID:24566390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One 2012; 7:e46092; PMID:23029398; http://dx.doi.org/ 10.1371/journal.pone.0046092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xie R, Wang P, Cheng M, Sapolsky R, Ji X, Zhao H. Mammalian target of rapamycin cell signaling pathway contributes to the protective effects of ischemic postconditioning against stroke. Stroke 2014; 45:2769-76; PMID:25013017; http://dx.doi.org/ 10.1161/STROKEAHA.114.005406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang J, Han D, Sun M, Feng J. A combination of remote ischemic perconditioning and cerebral ischemic postconditioning inhibits autophagy to attenuate plasma HMGB1 and induce neuroprotection against stroke in rat. J Mol Neurosci 2016; 58:424-31; PMID:26852332; http://dx.doi.org/ 10.1007/s12031-016-0724-9 [DOI] [PubMed] [Google Scholar]
- [22].Qi ZF, Luo YM, Liu XR, Wang RL, Zhao HP, Yan F, Song ZJ, Luo M, Ji X. AKT/GSK3beta-dependent autophagy contributes to the neuroprotection of limb remote ischemic postconditioning in the transient cerebral ischemic rat model. CNS Neurosci Therapeutics 2012; 18:965-73; http://dx.doi.org/ 10.1111/cns.12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci 2016; 17:467-84; PMID:27256553; http://dx.doi.org/ 10.1038/nrn.2016.51 [DOI] [PubMed] [Google Scholar]
- [24].Baek SH, Noh AR, Kim KA, Akram M, Shin YJ, Kim ES, Yu SW, Majid A, Bae ON. Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke 2014; 45:2438-43; PMID:24938837; http://dx.doi.org/ 10.1161/STROKEAHA.114.005183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Simon RP, Niro M, Gwinn R. Brain acidosis induced by hypercarbic ventilation attenuates focal ischemic injury. J Pharmacol Exp Ther 1993; 267:1428-31; PMID:8263804 [PubMed] [Google Scholar]
- [26].Thompson SW. Reactivity of cerebral blood flow to CO 2 in patients with transient cerebral ischemic attacks. Stroke 1971; 2:273-8; PMID:5111576; http://dx.doi.org/ 10.1161/01.STR.2.3.273 [DOI] [PubMed] [Google Scholar]
- [27].Schips TG, Wietelmann A, Hohn K, Schimanski S, Walther P, Braun T, Wirth T, Maier HJ. FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc Res 2011; 91:587-97; PMID:21628326; http://dx.doi.org/ 10.1093/cvr/cvr144 [DOI] [PubMed] [Google Scholar]
- [28].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al.. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008; 14:193-204; PMID:18267088; http://dx.doi.org/ 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, et al.. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 2014; 124:3987-4003; PMID:25083992; http://dx.doi.org/ 10.1172/JCI74985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 2012; 22:545-52; PMID:22342752; http://dx.doi.org/ 10.1016/j.cub.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Connolly NM, Dussmann H, Anilkumar U, Huber HJ, Prehn JH. Single-cell imaging of bioenergetic responses to neuronal excitotoxicity and oxygen and glucose deprivation. J Neurosci 2014; 34:10192-205; PMID:25080581; http://dx.doi.org/ 10.1523/JNEUROSCI.3127-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 2015; 87:371-81; PMID:26182419; http://dx.doi.org/ 10.1016/j.neuron.2015.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 2006; 7:437-48; PMID:16715053; http://dx.doi.org/ 10.1038/nrn1927 [DOI] [PubMed] [Google Scholar]
- [35].Dezfulian C, Garrett M, Gonzalez NR. Clinical application of preconditioning and postconditioning to achieve neuroprotection. Translational Stroke Res 2013; 4:19-24; PMID:24323188; http://dx.doi.org/ 10.1007/s12975-012-0224-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab 2009; 29:873-85; PMID:19240739; http://dx.doi.org/ 10.1038/jcbfm.2009.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Regli L, Anderson RE, Meyer FB. Effects of intermittent reperfusion on brain pHi, rCBF, and NADH during rabbit focal cerebral ischemia. Stroke 1995; 26:1444-51; discussion 51-2; PMID:7631351; http://dx.doi.org/ 10.1161/01.STR.26.8.1444 [DOI] [PubMed] [Google Scholar]
- [38].Cutaia M, Kroczynski J, Tollefson K. pH-dependent oxidant production following inhibition of the mitochondrial electron transport chain in pulmonary endothelial cells. Endothelium: J Endothelial Cell Res 2002; 9:109-21; PMID:12200958; http://dx.doi.org/ 10.1080/10623320212007 [DOI] [PubMed] [Google Scholar]
- [39].Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Huttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 2013; 47:9-23; PMID:23011809; http://dx.doi.org/ 10.1007/s12035-012-8344-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, et al.. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014; 515:431-5; PMID:25383517; http://dx.doi.org/ 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012; 22:320-33; PMID:22280891; http://dx.doi.org/ 10.1016/j.devcel.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol 2013; 1:542-51; PMID:24273737; http://dx.doi.org/ 10.1016/j.redox.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu JM, Yi Z, Liu SZ, Chang JH, Dang XB, Li QY, Zhang YL. The mitochondrial division inhibitor mdivi-1 attenuates spinal cord ischemia-reperfusion injury both in vitro and in vivo: Involvement of BK channels. Brain Res 2015; 1619:155-65; PMID:25818100; http://dx.doi.org/ 10.1016/j.brainres.2015.03.033 [DOI] [PubMed] [Google Scholar]
- [44].Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming QL, Huang C, Li P, Gao N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015; 11:1259-79; PMID:26114658; http://dx.doi.org/ 10.1080/15548627.2015.1056970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, Dorn GW 2nd. Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circulation Res 2015; 117:346-51; PMID:26038571; http://dx.doi.org/ 10.1161/CIRCRESAHA.117.306859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fonarow GC, Smith EE, Saver JL, Reeves MJ, Bhatt DL, Grau-Sepulveda MV, Olson DM, Hernandez AF, Peterson ED, Schwamm LH. Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation 2011; 123:750-8; PMID:21311083; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.110.974675 [DOI] [PubMed] [Google Scholar]
- [47].Zhang L, Zhang ZG, Chopp M. The neurovascular unit and combination treatment strategies for stroke. Trends Pharmacol Sci 2012; 33:415-22; PMID:22595494; http://dx.doi.org/ 10.1016/j.tips.2012.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res 2009; 43:348-64; PMID:19241241; http://dx.doi.org/ 10.1080/10715760902751902 [DOI] [PubMed] [Google Scholar]
- [49].Fan YY, Hu WW, Dai HB, Zhang JX, Zhang LY, He P, Shen Y, Ohtsu H, Wei EQ, Chen Z. Activation of the central histaminergic system is involved in hypoxia-induced stroke tolerance in adult mice. J Cereb Blood Flow Metab 2011; 31:305-14; PMID:20588322; http://dx.doi.org/ 10.1038/jcbfm.2010.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.