

ABSTRACT
Macroautophagy/autophagy is a well-established process involved in maintaining cellular homeostasis, but its role in cancer is complex and even controversial. Many studies have reported a correlative relationship between increased autophagy and evolving cancer cells under stress conditions such as nutrient or oxygen deprivation; however, there has been a lack of a plausible mechanistic link to properly target the autophagy process in the context of this microenvironment. We recently unveiled a positive regulatory loop involving TGM2 (transglutaminase 2)-NFKB/NF-κB signaling, IL6 and autophagy in cancer using mantle cell lymphoma (MCL) as a model system. These pathways are functionally connected to each other, thereby promoting malignant B cell survival and leading to enhanced lymphoma progression both in mice and in patients. Disruption of this network could provide an opportunity to increase the efficacies of current therapies and to reduce MCL drug resistance.
KEYWORDS: autophagy, cancer, IL-6, lymphoma, NF-κB, survival, TG2, therapeutic target
The physiological roles of autophagy in different stages of cancer development are quite distinct and paradoxical. At a very early stage, autophagy maintains homeostasis and prevents cancer initiation by eliminating oncogenic protein substrates. Once a tumor forms and develops, autophagy becomes a cytoprotective mechanism that facilitates the adaptation of cancer cells to multiple stress stimuli ranging from nutrient shortage and chronic hypoxia to oxidative stress from chemotherapies. This stress-induced autophagy restores the intracellular energy balance and provides cellular building blocks during cancer progression. In this context, the inhibition of autophagy has been explored to stop tumor progression in multiple clinical studies, although its anticancer efficacy is cell type- and treatment-dependent. Nevertheless, the mechanistic underpinnings that influence autophagy in evolving cancer cells remain elusive. To validate autophagy targeting as a potential therapeutic option, it is important to dissect the multifaceted influence of increased autophagy in cancer.
One such interconnected network that modulates autophagy is TGM2-NFKB signaling and cytokine IL6, which facilitates malignant B cell survival under stressed conditions and confers their survival upon dispersal to organs. TGM2 is a Ca2+-dependent crosslinking enzyme that regulates multiple cellular processes during normal cell development. Our recent report has demonstrated that TGM2 is closely associated with the constitutive expression of NFKB in cancer cells. In MCL, the TGM2-mediated polymerization of nonphosphorylated NFKBIA/IκBα leads to NFKBIA proteasomal degradation. As a consequence, TGM2 forms complexes with NFKB components, which leads to the constitutive expression of NFKB by driving the translocation of NFKB to the nucleus.
Intriguingly, TGM2 is a stress-responsive gene. TGM2 activity is upregulated by various stressors, including tissue injury and inflammation, inflammatory cytokines, and reactive oxygen species. Given that both autophagy and TGM2 activity can be induced under cellular stress, we asked whether autophagy induction is connected to TGM2-NFKB signaling. Using multiple genetic approaches, including CRISPR-Cas9-mediated knockout technology, an shRNA-mediated knockdown system, and ectopic overexpression, we discovered that the inhibition of the autophagy pathway leads to undetectable TGM2 levels and further reduced NFKB expression, whereas TGM2 in turn regulates autophagosome and autolysosome formation.
We next consolidated these connections using xenograft models and patient samples. Xenografts bearing TGM2-overexpressing lymphomas show increased autophagy formation and autophagic flux. Compared with normal B cells, MCL patient cells exhibit upregulated autophagy formation, which can be repressed by BAPTA/AM, an intracellular calcium chelator that inhibits TGM2 enzymatic activities. To gain more insight into the reciprocal correlation between autophagy and TGM2, we performed additional rescue experiments. First, inhibiting autophagy in TGM2-overexpressing cells reduces cell proliferation and decreases TGM2-induced chemoresistance. The protective effects of TGM2 overexpression in MCL are reversed by autophagy inhibitors, revealing a novel role of TGM2-mediated autophagy in MCL drug resistance. Second, the forced overexpression of TGM2 partially rescues the phenotype of autophagy-inhibited cells. These data support the idea that decreased TGM2 levels could be a causative factor for the reduced survival of autophagy-inhibited MCL cells. In addition, these results demonstrate that TGM2-NFKB signaling confers MCL survival via enhanced autophagy, which in turn regulates TGM2-NFKB signaling, indicating a positive feedback loop.
MTOR (mechanistic target of rapamycin) is a nutrient-sensitive kinase that has been identified as a promising target in cancers. However, MTOR inhibition induces autophagy that can counteract the effect of MTOR inhibitors in MCL. Therefore, we investigated whether there is a correlation between differential TGM2 expression and differential responses to MTOR inhibitors. Indeed, TGM2 overexpression results in a significant resistance to MTOR inhibitors, which is reversed after autophagy inhibitor treatments. This finding highlights the potential of inhibiting autophagy to modulate therapeutic resistance. The simultaneous blocking of TGM2 and autophagy is a potential strategy for overcoming chemoresistance in MCL.
Similar to other cancers, patients with advanced MCL with bone marrow involvement exhibit increased drug resistance; one of the mechanisms of acquired resistance is the accessory cells, which reside in regions near cancer cells and which provide protection from therapies. IL6 is one of the cytokines that is secreted by bone marrow stromal cells and that has been considered as a microenvironment-dependent survival agent for cancer cells. In addition to its affects on diverse environmental stresses, autophagy also plays an important role in innate and adaptive immunity and is regulated by different cytokines. Therefore, we further delineated whether the downstream target of TGM2-NFKB signaling, IL6, is autophagy dependent.
Both autocrine IL6 and paracrine IL6 enhance autophagy induction and autophagic flux. Most strikingly, autophagy, in addition to being regulated by IL6, can augment IL6 secretion, suggesting that intact autophagy is required for the efficient production of cytokines under stress. Combined with our results that demonstrated IL6 enrichment in MCL patient samples and bone marrow, our findings indicate that IL6-mediated autophagy is likely an important way to modify MCL cell behavior to enable malignant B cell dispersal and progression.
Together, our data unveil a new cytoprotective response of autophagy in MCL and add a new layer in the understanding of how autophagy supports cancer progression. The mechanisms of this positive effect are connected to the TGM2-NFKB and IL6-STAT3 signaling pathways (Fig. 1). Disruption of the autophagy, TGM2-NFKB signaling and IL6 secretion loop may represent a novel therapeutic tool for the treatment of patients with MCL and provide alternative strategies to overcome chemoresistance in clinical medicine. Future work exploring other inducers involved in this regulatory network will likely reveal more connections between autophagy and the pathogenesis of MCL.
Figure 1.
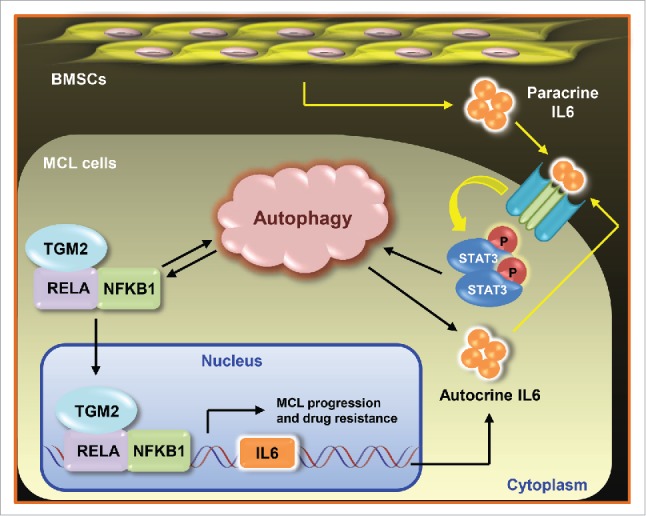
Interconnected network of TGM2-NFKB and IL6-STAT3 signaling and autophagy regulation in MCL. MCL cells exploit both TGM2-NFKB signaling and the cytokine IL6 to trigger a cytoprotective autophagy response, which in turn enhances TGM2-NFKB signaling and facilitates IL6 secretion, suggesting a positive feedback loop underlying the survival and drug resistance mechanisms in MCL cells. As an upstream activator of STAT3, IL6 is stimulated by either TGM2-NFKB signaling or paracrine signals from BMSCs. MCL cells elicit IL6-mediated autophagy as an important way to favor MCL progression. MCL, mantle cell lymphoma; BMSCs, bone marrow stromal cells.
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Funding
N.M. is supported by R01CA181319 (NCI) and R21CA202212 (NCI).