

ABSTRACT
Recent discoveries on the role of commensal microbiota have significantly changed our understanding of human physiology. The host-microbiota interplay is now an important aspect to take into account to understand immune responses and immunological diseases. Autoimmune uveitis is a sight-threatening disease that arises without a known infectious etiology. It is unknown where and how autoreactive T cells become primed to trigger disease in the eye, which is an immune privileged site. We recently reported data supporting the notion that retina-specific T cells receive a signal in the gut from commensal microbiota-derived cross-reactive antigen(s) and trigger autoimmune uveitis in the R161H mouse model. Here we discuss our published findings, as well as our recent attempts to identify the responsible microbe(s) by using different antibiotic treatments, 16S rDNA sequencing and homology searches for candidate antigenic mimic(s) of the retinal antigen.
KEYWORDS: microbiota, retina, autoimmune uveitis, immune privilege, T cell, interphotoreceptor retinoid-binding protein (IRBP)
Introduction
As is the case for many autoimmune diseases, the knowledge on pathogenesis of autoimmune uveitis has been helped by studies of its animal model, known as experimental autoimmune uveitis (EAU).1-4 In the rat and mouse models of EAU, disease is driven by autoreactive T cells that recognize tissue-specific antigens that are unique to the eye. Examples include retinal arrestin (S-Ag) and the interphotoreceptor retinoid-binding protein (IRBP).5-7 Interestingly, uveitis patients exhibit immunological responses to these antigens.8 Activated retina-specific T cells infiltrate the eye, secrete effector cytokines, such as IFN-γ and IL-17, and cause destruction of the retina.6,7
Although similar pathogenic processes are shared by other tissue-specific autoimmune diseases, unique features of the eye make uveitis particularly interesting. The retina is separated from the immune system by a tight blood-retinal-barrier (BRB) that is difficult to traverse by non-activated T cells. Within the eye, the retina is surrounded by intraocular fluids, a milieu that is rich in anti-inflammatory substances, such as retinoic acid and TGF-β, which tend to inhibit effector T cells and convert naive T cells into regulatory cells.9 Given this protected status of the eye, uveitis represents something of a paradox. Retina-specific T cells must be activated to be able to cross the BRB to cause uveitis, but the unique retinal antigens are sequestered inside the eye and are not available in the periphery to prime these T cells. Therefore, a fundamental question is how and where are retina-specific T cells first activated and acquire the ability to elicit uveitis.
Recently, we demonstrated that retina-specific autoreactive T cells receive an activation signal through their T cell receptor (TCR) in the lamina propria (LP) of the intestine.10 This signal involves retina-specific TCR and requires gut microbiota.10 This was the first report that links gut commensals to disruption of the immune privileged status of the eye and to uveitis, and provides implications that such a scenario may be generalizable to other autoimmune diseases.11
In these studies, we took advantage of the genetically engineered R161H mouse model of uveitis developed in our lab. These mice develop spontaneous uveitis due to having a high number of autoreactive T cells that are specific for the retinal antigen IRBP.12 We examined various organs of these mice to look for activated T cells and found that large numbers of retina-specific T cells were being activated in the lamina propria (LP) of the intestine, even before disease became apparent in the eye. Notably, depletion of the gut microbiota by broad-spectrum antibiotics treatment (ampicillin, metronidazole, neomycin and vancomycin = AMNV13), or by rearing the mice in germ-free conditions, resulted in attenuation of uveitis, and in parallel, in reduction of Th17 cells in the intestinal LP. This activation signal in the intestine was transduced through the clonotypic TCR of R161H T cells, but the endogenous antigen IRBP was not required for this activation, suggesting that it came from the microbiota. Indeed, microbiota-rich protein extracts from intestinal contents14 activated retina-specific T cells, making them pathogenic enough to transfer disease in naïve wild-type recipients.10
This addendum aims to extend our discussion to additional aspects of our published findings in the context of recent reports in other autoimmune diseases and gut immune physiology, which are influenced by the microbiota.
Adaptive vs. innate stimuli: Depletion of gut microbiota dampens spontaneous uveitis, but appears less effective in the immunization-induced EAU model
Our study, which revealed the dependence of uveitis on gut microbiota, utilized the R161H transgenic mouse strain expressing a TCR specific for peptide IRBP161-180, the major pathogenic epitope of IRBP, in approximately 25% of peripheral CD4+ T cells. All R161H mice develop spontaneous uveitis that starts soon after weaning and reaches its peak by 3 months of age.12 In the classical model of uveitis, induced by immunization with IRBP161-180 in complete Freund's adjuvant (CFA), the adjuvant provides necessary innate signals via receptors for pathogen-associated molecular patterns that promote antigen presenting cell maturation, which then present the IRBP161-180 antigen to autoreactive T cells in the host in the context of “danger” signals that drive them to a pathogenic, tissue destructive phenotype.
Although in the spontaneous R161H uveitis model the IRBP-specific T cells were seen to signal through their autoreactive TCRs in the gut environment in a microbiota-dependent fashion, this does not negate the possibility that innate adjuvant effects are needed together with the TCR signal to activate these cells for pathogenicity. We hypothesized that the gut microbiota may serve as the source of innate “adjuvant” signals as well, serving as a source of both the antigen and the adjuvant. The findings that treatment by broad-spectrum antibiotic attenuated disease, and that microbiota-rich extracts of intestinal contents could activate and trigger pathogenicity in R161H lymphocytes are compatible with this interpretation,13 but do not allow to resolve the antigen from adjuvant activities. LPS and M. tuberculosis extract as a source of innate stimuli alone were unable to activate R161H cells,10 speaking for the need for a TCR driven signal, but again not contradicting the need for an adjuvant driven one. This issue still remains unresolved.
Another question which still remains open is whether the immunization-induced, acute EAU model can be affected by commensal microbiota. In our studies, we set out to use the EAU model to address the possibility that attenuation of spontaneous uveitis by long-term antibiotic treatment (treatment is given to the pregnant dam and continues indefinitely10) could stem from nonspecific immunosuppressive effects, rather than elimination of the resident gut microbiome. Since R161H mice are bred heterozygously, we had WT littermates treated long-term with antibiotics at our disposal. Upon immunization with the IRBP peptide the antibiotic treated WT mice developed full blown disease, indistinguishable in severity from non-treated WT mice immunized as controls. This eliminated the concern about immunosuppression, but raised a different question. Namely, using a short-term course of the same antibiotic mix, others had reported attenuated disease in induced models of autoimmune diseases including experimental autoimmune encephalomyelitis, the model for multiple sclerosis,15,16 experimental arthritis17 and more recently EAU induced by immunization with IRBP in CFA.18 Furthermore, Nakamura et al.18 reported increased numbers of T regulatory cells as a result of the antibiotic treatment in the induced EAU model, which we did not see in the spontaneous uveitis model.10 It remains to be determined whether the length of antibiotic treatment, or the specific microbiota present in the various animal facilities, might underlie these differences.
The search for the inciting microbe(s): Single antibiotic treatment and candidate organism analysis
The microbe(s) providing the putative crossreactive antigen that was seen to signal through the TCR of the retina-specific T cells in the gut environment remains elusive. The community composition of the microbiota was shown to be important in directing immune responses in the gut. For example, segmented filamentous bacteria (SFB) was shown to promote Th17 responses and Clostridia were able to expand Foxp3+ regulatory T cells in mice,19,20 and later confirmed by the same group that human-derived Clostridia species have a critical role.21 Not surprisingly, 16S rDNA sequencing meta analysis of the microbiota composition in R161H mice treated or untreated with the antibiotic cocktail revealed multiple groups of bacteria that were present at the phylum level. The meta-analysis of the microbiota composition in R161H mice treated or untreated with the antibiotic cocktail revealed differences in bacterial phyla. Bacteroidetes and Firmicutes predominated in untreated mice. In contrast, a high relative abundance of Proteobacteria and Tenericutes were observed in antibiotic treated mice (Fig. 1). These data suggest a pronounced shift in community composition and structure following antibiotic treatment.10
Figure 1.
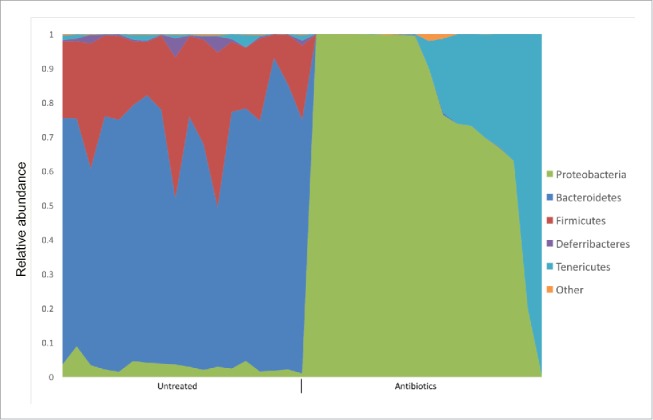
A cocktail of broad-spectrum antibiotics (AMNV) changes the composition of the microbiota in R161H mice. Distribution of the relative abundance of bacteria at the phylum level is graphically shown. Fecal pellets of R161H mice untreated (N = 18) or treated with antibiotics (N = 17, AMNV) were collected at various ages (3-4 wk, 6–7 wk, > 11 wk) and metagenome analyses was done by 16S rDNA sequencing.
In an attempt to narrow down the gut bacterial species that could be responsible for promoting disease in R161H mice, we treated them with each of four antibiotics (ampicillin, metronidazole, neomycin or vancomycin) individually to eliminate a more limited spectrum of microorganisms. Despite some subtle modifications in disease development, none of these four antibiotics individually replicated the results of drastic disease reduction that we saw with the treatment of all four antibiotics in combination (Fig. 2). Since the single antibiotics would result in different, partly non-overlapping, residual bacterial populations, this result may indicate that the bacterial source of the cross-reactive antigen is not confined to a single type of microorganism, and that multiple species of microbiota can contribute. In this context, it is also important to mention that R161H mice kept in other animal facilities, at NIH, elsewhere in the US and abroad, all develop spontaneous uveitis. This emphasized that the development of spontaneous uveitis in R161H mice is not restricted to a unique gut flora composition or a single animal facility.
Figure 2.
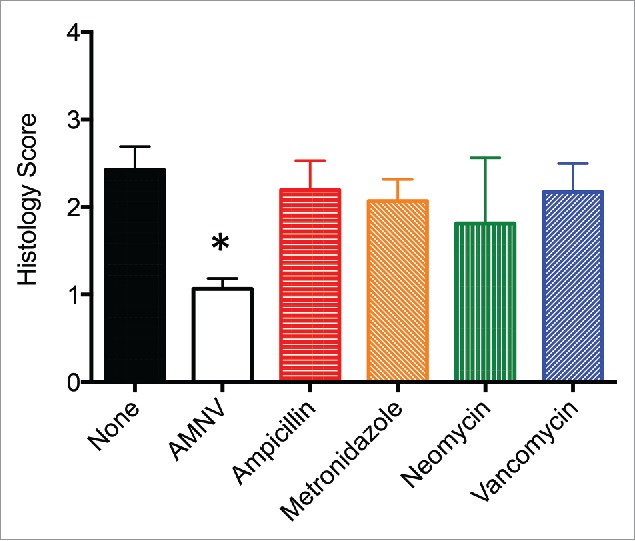
Single antibiotic treatment did not attenuate spontaneous uveitis in R161H mice. R161H mice were treated with Ampicillin (N = 13), Metronidazole (N = 15), Neomycin (N = 4), or Vancomycin (N = 14), and eyes were collected for histology between 11 and 14 weeks of age, and compared to age-matched R161H mice untreated (None, N = 13) or treated with a cocktail of all 4 antibiotics (AMNV, N = 40). Antibiotics were given in the drinking water at 1 g/L except for Vancomycin (0.5 g/L) and treatments started to the pregnant dam and continued indefinitely for the pups after weaning. There was statistical significance of *p < 0.0001 between the untreated group and the AMNV group by Mann-Whitney U test.
An anecdotal story worth recounting involves R161H mice monocolonized with what turned out to be a new Turicibacter sp. H121 strain.22 During a failed germ-free derivation attempt at Taconic Farms (Germantown, NY), the colony was contaminated with this Gram-positive bacterium. One of two contaminated R161H mice showed disease. We isolated this organism and performed whole genome sequencing22 in an attempt to identify common sequences that might serve as mimic antigens. Several candidate sequences with partial homology were identified, and peptides were synthesized and tested, but none gave consistent results in stimulating R161H T cells in vitro or eliciting EAU by immunization in vivo. We also monocolonized germ-free R161H mice with “our” Turicibacter H121 to test if it can promote spontaneous uveitis, but there was no significant enhancement of disease or of Th17 induction in the gut in the monocolonized R161H mice (unpublished data).
Finally, we performed extensive bioinformatics analyses searching for sequences related to the IRBP160-180 peptide in the microbial protein database, obtaining a number of sequences with partial homology with IRBP161-180. The search included the peptides derived from the Turicibacter sp H121, described above.22 We synthesized 16 peptides that were reasonable hits in terms of similarity (Table 1) and used them to stimulate R161H T cells. However, none of them induced T cell proliferation even at 300 μg/ml (unpublished data) and even the best candidate with the highest similarity (Turicibacter pep #7) did not induce disease upon immunization of WT mice. These results underscore the difficulties inherent in connecting particular candidate organisms and the putative antigens they encode to disease pathogenesis.
Table 1.
Commensal bacteria-derived peptides that were used for analysis based on homology search to human IRBP161-180 peptide.
| Pept # | Sequence | Protein | Organism |
|---|---|---|---|
| 161-180 | SGIPYIISYLHPGNTILHVD | IRBP | Homo sapiens |
| 1 | GMNIDYLSPGEIPNSTILK | Aryl sulfatase | Turicibacter sp |
| 2 | VVQPYIYIGKGNTISAEG | Dead box helicase | Turicibacter sp |
| 3 | KEHLYLLAHPGSVILDGE | Conserved domain | Turicibacter sp |
| 4 | SGEPYIIHPIEVAYILA | GTP diphosphokinase | Turicibacter sp |
| 5 | YGIPYIYHHVYGLQNTIDFI | Oxidoreductase | Turicibacter sp |
| 6 | TSVVEMVDYILPPDKV | Peptidase S41 | Turicibacter sp |
| 7 | YYIPFIISLHTGARRGEIL | Site-specific recombinase | Turicibacter sp |
| 8 | QSVAQLMSHFLPFGTDLLIN | IRBP | Citromicrobium sp |
| 9 | DQAERMASMFLKNGETIVQFE | Peptidase S41 | Enterococcus faecalis |
| 10 | INSYIINYKLLNI | Hypothetical Protein | Francisella tularensis |
| 11 | SGIPSAVIGVPARYIHSSNSILHVD | Glut-Aminopeptidase | Listeria monocytogenes |
| 12 | SMVAFITSYLFDSEPFHLN | Peptidase S41 | Paenibacillus vortex |
| 13 | SMVALLTSYLLPAYPPVHLT | Peptidase S41 | Synechococcus |
| 14 | YYIPFIIAIHTGARRGEIL | Site-specific recombinase | Turicibacter sp |
| 15 | SGIPYVSYNNLGNLIQNHF | Glycerophosphoryl diester phosphodiesterase | Lactococcus lactis |
| 16 | DSTYLHLRYPGNTILMDSI | ABC transporter permease | Lactococcus lactis |
Antigen or superantigen?
Nur77-GFP reporter mice, which turn on GFP in T cells when their antigen receptor is engaged23 demonstrated that the retina-specific R161H cells were receiving a TCR signal in the gut.10 However, before concluding that the TCR signal must come from cross-reactive antigen mimic, it was necessary to examine the possibility that the TCR could be engaged by a bacterial superantigen. Superantigens are protein toxins that activate T cells in an MHC class II dependent fashion by binding to particular classes of Vβ TCR chains outside of the antigen-binding groove.24 In this, they behave much like an antigen, only without antigenic specificity. To examine if R161H T cells can be activated by bacterial superantigens, we tested a number of commercially available superantigens (Staphylococcal enterotoxins B, D and I) that were known to bind the TCRVβ1,24 as IRBP specific-T cells express a Vα17/Vβ1 TCR.12 However, none of these superantigens activated R161H T cells. While we cannot completely discount the possibility of a yet to be identified superantigen, we believe that these findings make that possibility unlikely and a microbiota dependent non-cognate antigen mimic remains our preferred interpretation.
Unresolved questions and future perspectives
Our data provide strong support for the notion that T cells first primed in the gut reach the eye and elicit disease. That said, we have not actually shown that a T cell which has been in the gut actually ends up in the eye. There is a method to endoscopically photolabel T cells within the intestine25 and they remain fluorescent for several days. Nevertheless, given that fewer than 10 activated T cells entering the eye can precipitate uveitis,26 the dynamics of the system and the level of detectability would make this an extremely challenging task. Future technological advances will hopefully make this more achievable.
As is clear from what was discussed above, our attempts to identify the antigenic mimic for uveitogenic T cells in the model of spontaneous uveitis have not been successful thus far. The identification of such a mimic and its source could uncover a trigger of uveitis and provide a potential approach for the development of potential probiotic and prebiotic therapies or vaccines. To this end, we will continue bioinformatic analyses using new algorithms and evolving databases, to include fungi and viruses. A potential source for new microbial candidates could be the small intestine. In our published article,10 we observed that more T cells were being activated in the LP of the small intestine (ileum) than in the large intestine. However, our 16S rDNA sequencing analysis of the microbiota was done using fecal pellets (Fig. 1). Given that the population of microbiota in the ileum is less complex than in the colon,27,28 its analysis could yield useful new information regarding the putative mimic.
While it is important to identify the putative microbial mimic(s) in mouse commensal flora, finding one in human microbiota would be even more so. Our clinical colleagues are already cataloguing the flora of uveitis patients compared to healthy controls, however, such studies can only show associations. In the next step, we will reconstitute germ-free R161H mice with human commensals, from healthy donors and from patients, to examine whether they can support development of disease. We will then proceed to identify the involved microorganisms and analyze them using the methodologies described above.
As a final puzzle, one would think that under circumstances where there are activated T cells in the gut, antigen is being presented, and there is no evidence for expansion of T regulatory cells, there could (should?) be development of colitis. We performed careful histological evaluation of small and large intestines of R161H mice from 3 weeks to 6 months of age and did not uncover detectable evidence of intestinal inflammation (Fig. 3 and data not shown). The reasons for this are unclear, and can be several. Since most of the bacterial antigen is in the lumen of the gut, perhaps there is insufficient antigen in the tissue itself, or the T cells may emigrate from the gut very soon after activation, or (as has been shown for commensal-specific Th17 cells in the gut29) they may adopt a “nonpathogenic Th17” phenotype, and only become tissue destructive subsequent to migrating through certain tissue sites (e.g., spleen, lung)30,31 and/or upon the cognate Ag being re-presented within the retinal tissue. This is currently under study in our lab.
Figure 3.
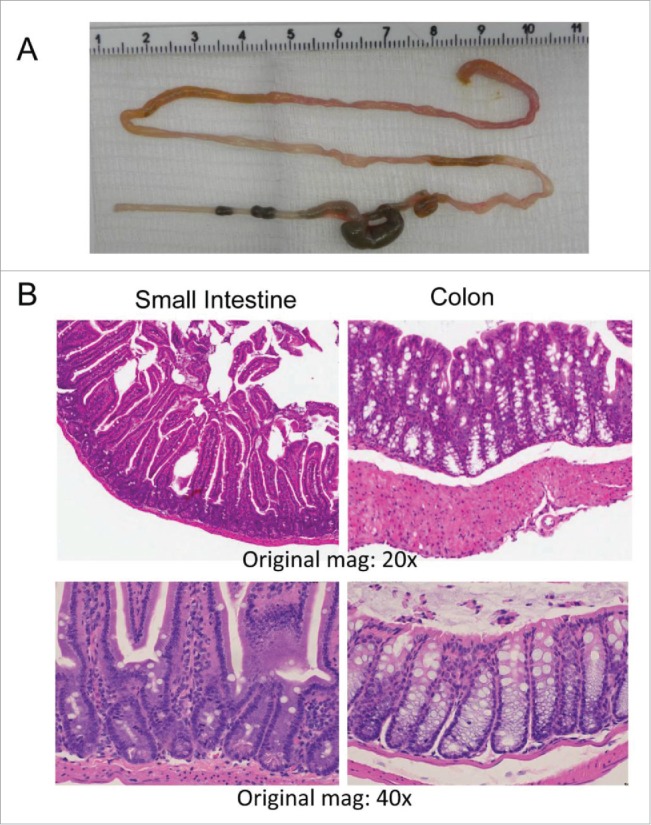
R161H mice with uveitis have no evidence of inflammation in the intestine. (A) A freshly extracted intestine of an 11 week-old R161H mouse. (B) Hematoxylin and Eosin-stained sections of small intestine and colon (original magnification x20 and x40). No evidence of histological abnormality is apparent.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Caspi RR, Silver PB, Luger D, Tang J, Cortes LM, Pennesi G, Mattapallil MJ, Chan CC. Mouse models of experimental autoimmune uveitis. Ophthalmic Res 2008; 40(3-4):169-74; PMID:18421234; http://dx.doi.org/ 10.1159/000119871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gery I, Wiggert B, Redmond TM, Kuwabara T, Crawford MA, Vistica BP, Chader GJ. Uveoretinitis and pinealitis induced by immunization with interphotoreceptor retinoid-binding protein. Invest Ophthalmol Vis Sci 1986; 27(8):1296-300; PMID:3488297 [PubMed] [Google Scholar]
- [3].Caspi RR, Roberge FG, Chan CC, Wiggert B, Chader GJ, Rozenszajn LA, Lando Z, Nussenblatt RB. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol 1988; 140(5):1490-5; PMID:3346541 [PubMed] [Google Scholar]
- [4].Pennesi G, Mattapallil MJ, Sun SH, Avichezer D, Silver PB, Karabekian Z, David CS, Hargrave PA, McDowell JH, Smith WC, et al.. A humanized model of experimental autoimmune uveitis in HLA class II transgenic mice. J Clin Invest 2003; 111(8):1171-80; PMID:12697736; http://dx.doi.org/ 10.1172/JCI15155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med 2007; 13(6):711-8; PMID:17496900; http://dx.doi.org/ 10.1038/nm1585 [DOI] [PubMed] [Google Scholar]
- [6].Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med 2008; 205(4):799-810; PMID:18391061; http://dx.doi.org/ 10.1084/jem.20071258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Peng Y, Han G, Shao H, Wang Y, Kaplan HJ, Sun D. Characterization of IL-17+ interphotoreceptor retinoid-binding protein-specific T cells in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci 2007; 48(9):4153-61; PMID:17724201; http://dx.doi.org/ 10.1167/iovs.07-0251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gery I, Mochizuki M, Nussenblatt RB. Retinal specific antigens and immunopathogenic processes they provoke. Prog Retinal Res 1986; 5:75-109. [Google Scholar]
- [9].Zhou R, Horai R, Silver PB, Mattapallil MJ, Zárate-Bladés CR, Chong WP, Chen J, Rigden RC, Villasmil R, Caspi RR. The living eye "Disarms" uncommitted autoreactive T cells by converting them to Foxp3(+) regulatory cells following local antigen recognition. J Immunol 2012; 188(4):1742-1750; PMID:22238462; http://dx.doi.org/ 10.4049/jimmunol.1102415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Horai R, Zárate-Bladés CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, Jittayasothorn Y, Chan CC, Yamane H, Honda K, et al.. Microbiota-dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site. Immunity 2015; 43(2):343-53; PMID:26287682; http://dx.doi.org/ 10.1016/j.immuni.2015.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bordon Y. Autoimmunity: The bug eye bandits. Nat Rev Immunol 2015; 15(10):595; PMID:26338264; http://dx.doi.org/ 10.1038/nri3911 [DOI] [PubMed] [Google Scholar]
- [12].Horai R, Silver PB, Chen J, Agarwal RK, Chong WP, Jittayasothorn Y, Mattapallil MJ, Nguyen S, Natarajan K, Villasmil R, et al.. Breakdown of immune privilege and spontaneous autoimmunity in mice expressing a transgenic T cell receptor specific for a retinal autoantigen. J Autoimmun 2013; 44:21-33; PMID:23810578; http://dx.doi.org/ 10.1016/j.jaut.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004; 118(2):229-41; PMID:15260992; http://dx.doi.org/ 10.1016/j.cell.2004.07.002 [DOI] [PubMed] [Google Scholar]
- [14].Dillenburg-Pilla P, Z.-B.C. SP, Horai R, Caspi R. Preparation of protein-containing extracts from microbiota-rich intestinal contents. Bio-protocol 2016; 6(18):e1936; bio-protocol.org/e1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2011; 108 Suppl 1:4615-22; PMID:20660719; http://dx.doi.org/ 10.1073/pnas.1000082107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, Kasper LH. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol 2009; 183(10):6041-50; PMID:19841183; http://dx.doi.org/ 10.4049/jimmunol.0900747 [DOI] [PubMed] [Google Scholar]
- [17].Dorozynska I, Majewska-Szczepanik M, Marcińska K, Szczepanik M. Partial depletion of natural gut flora by antibiotic aggravates collagen induced arthritis (CIA) in mice. Pharmacol Rep 2014; 66(2):250-5; PMID:24911078; http://dx.doi.org/ 10.1016/j.pharep.2013.09.007 [DOI] [PubMed] [Google Scholar]
- [18].Nakamura YK, Metea C, Karstens L, Asquith M, Gruner H, Moscibrocki C, Lee I, Brislawn CJ, Jansson JK, Rosenbaum JT, et al.. Gut microbial alterations associated with protection from autoimmune Uveitis. Invest Ophthalmol Vis Sci 2016; 57(8):3747-58; PMID:27415793; http://dx.doi.org/ 10.1167/iovs.16-19733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al.. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011; 331(6015):337-41; PMID:21205640; http://dx.doi.org/ 10.1126/science.1198469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al.. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139(3):485-98; PMID:19836068; http://dx.doi.org/ 10.1016/j.cell.2009.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al.. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013; 500(7461):232-6; PMID:23842501; http://dx.doi.org/ 10.1038/nature12331 [DOI] [PubMed] [Google Scholar]
- [22].Auchtung TA, Holder ME, Gesell JR, Ajami NJ, Duarte RT, Itoh K, Caspi RR, Petrosino JF, Horai R, Zárate-Bladés CR. Complete genome sequence of turicibacter sp. Strain H121, isolated from the feces of a contaminated germ-free mouse. Genome Announc 2016; 4(2):e00114-16; PMID:27013036; http://dx.doi.org/ 10.1128/genomeA.00114-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med 2011; 208(6):1279-89; PMID:21606508; http://dx.doi.org/ 10.1084/jem.20110308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fraser JD, Proft T. The bacterial superantigen and superantigen-like proteins. Immunol Rev 2008; 225:226-43; PMID:18837785; http://dx.doi.org/ 10.1111/j.1600-065X.2008.00681.x [DOI] [PubMed] [Google Scholar]
- [25].Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc Natl Acad Sci U S A 2014; 111(18):6696-701; PMID:24753589; http://dx.doi.org/ 10.1073/pnas.1405634111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Caspi RR. Ocular autoimmunity: the price of privilege? Immunol Rev 2006; 213:23-35; PMID:16972894; http://dx.doi.org/ 10.1111/j.1600-065X.2006.00439.x [DOI] [PubMed] [Google Scholar]
- [27].Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014; 14(10):667-85; PMID:25234148; http://dx.doi.org/ 10.1038/nri3738 [DOI] [PubMed] [Google Scholar]
- [28].Geuking MB, Köller Y, Rupp S, McCoy KD. The interplay between the gut microbiota and the immune system. Gut Microbes 2014; 5(3):411-8; PMID:24922519; http://dx.doi.org/ 10.4161/gmic.29330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Burkett PR, Meyer zu Horste G, Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J Clin Invest 2015; 125(6):2211-9; PMID:25961452; http://dx.doi.org/ 10.1172/JCI78085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen J, Vistica BP, Takase H, Ham DI, Fariss RN, Wawrousek EF, Chan CC, DeMartino JA, Farber JM, Gery I. A unique pattern of up- and down-regulation of chemokine receptor CXCR3 on inflammation-inducing Th1 cells. EurJ Immunol 2004; 34(10):2885-94. [DOI] [PubMed] [Google Scholar]
- [31].Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schläger C, Lodygin D, Heckelsmiller K, Nietfeld W, Ellwart J, Klinkert WE, et al.. T cells become licensed in the lung to enter the central nervous system. Nature 2012; 488(7413):675-9; PMID:22914092 [DOI] [PubMed] [Google Scholar]