

ABSTRACT
Nephrotoxicity is a major side effect during chemotherapy with cisplatin and related platinum compounds. Previous work unveiled a role of PRKCD/PKCδ (protein kinase C delta) in cisplatin-induced nephrotoxicity; however, the underlying mechanism was largely unknown. Our recent work showed that PRKCD may suppress macroautophagy/autophagy, a cytoprotective mechanism, to promote kidney tubule cell death during cisplatin treatment. Interestingly, PRKCD may do so by phosphorylating AKT, which further phosphorylates MTOR to repress ULK1.
KEYWORDS: AKT, autophagy, chemotherapy, cisplatin, kidney, MTOR, nephrotoxicity, PKCδ, side effect
Since its discovery in 1960s, cisplatin has been the mainstay of chemotherapy for a variety of solid tumors. However, cisplatin is also notorious for its side effects or toxicity in normal tissues, inducing nausea and vomiting, hearing loss, and kidney failure. A few derivatives of cisplatin with lower side effects are available, but they are only effective for very limited tumor types at low chemotherapy efficacy. As such, cisplatin continues to be widely used and it is desirable to identify approaches for protection of normal tissues or organs during cisplatin chemotherapy, which obviously depends on the understanding of the mechanism of cisplatin toxicity in normal cells and tissues. Kidney toxicity occurs in ∼30% of cancer patients receiving cisplatin treatment, leading to acute as well as chronic renal disorders. The pathogenesis of cisplatin nephrotoxicity involves vascular dysfunction, inflammation, and damage to kidney tubules. Interestingly, in response to cisplatin toxicity, renal tubular cells may mount a vigorous defense. In this regard, in 2008 we documented the activation of autophagy as a protective mechanism during cisplatin nephrotoxicity. While the initial observation has been verified and extended by both pharmacological and autophagy gene knockout studies, it remains largely elusive as to how autophagy is induced and regulated in cisplatin nephrotoxicity. Our latest work has demonstrated a critical role of PRKCD in the regulation of tubular cell autophagy during cisplatin treatment.
The focus on PRKCD was based on our study published in 2011 that unveiled PRKCD in a novel signaling pathway leading to renal tubular cell death in cisplatin nephrotoxicity. Remarkably, that study showed that inhibition of PRKCD can protect kidneys during cisplatin treatment and enhance the chemotherapy effect in tumors, suggesting a strategy of “killing two birds with one stone.” With that observation, we were intrigued to figure out how PRKCD participates in tubular cell death. In our most recent work, we show that PRKCD may do so by suppressing autophagy. Moreover, while PRKCD inhibitors are protective during cisplatin treatment of wild-type mice, the effect is lost in renal tubule autophagy-deficient mice, supporting a pivotal role of autophagy in the beneficial effect of PRKCD inhibitors. Indeed, blockade of PRKCD pharmacologically or genetically induces autophagy in renal tubular cells, whereas activation of PRKCD has opposite effects.
How does PRKCD repress autophagy? We showed that PRKCD may directly bind and phosphorylate AKT at Ser473, and upon activation, AKT phosphorylates and activates MTOR, which in turn phosphorylates ULK1 to prevent the formation of the autophagy initiating complex (Fig. 1). As a result, our study unveils a signaling pathway for autophagy suppression, consisting of PRKCD-AKT-MTOR-ULK1. The possibility of direct phosphorylation of AKT by PRKCD is particularly interesting, because it provides new insights into the mechanism of AKT phosphorylation. AKT activation depends on its phosphorylation at 2 specific sites: Thr308 and Ser473. PDPK1/PDK1 (3-phosphoinositide dependent protein kinase 1) is responsible for Thr308 phosphorylation, but it remains a mystery how AKT is phosphorylated at Ser473. It has been proposed that, following Thr308 phosphorylation and initial AKT activation, AKT may autophosphorylate Ser473; however, this hypothesis has been seriously challenged by several studies. Alternatively, there may be a PDPK2 that is responsible for AKT phosphorylation at Ser473. Our study demonstrated a good correlation between PRKCD activity and AKT phosphorylation at Ser473. Importantly, incubation of AKT with recombinant active PRKCD induces AKT phosphorylation specifically at Ser473. In addition, co-immunoprecipitation analysis further indicates the molecular interaction between PRKCD and AKT, which, notably, is enhanced during cisplatin treatment. Together, these data suggest that PRKCD may be a candidate of PDPK2 for AKT phosphorylation at Ser473. However, we emphasize that the observation was made during cisplatin treatment of kidney cells and tissues, and it remains unclear if PRKCD phosphorylates AKT Ser473 under other experimental conditions.
Figure 1.
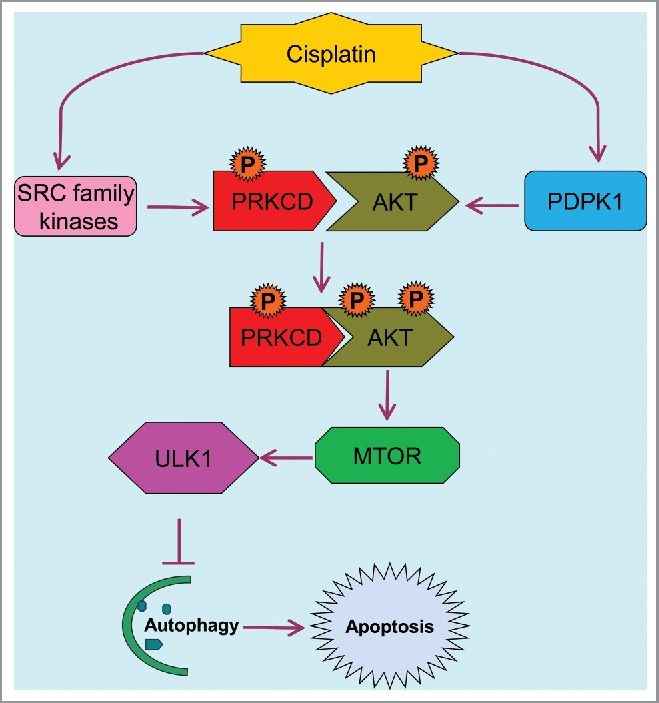
PRKCD-AKT-MTOR-ULK1 signaling pathway for autophagy suppression in cisplatin nephrotoxicity. Upon cisplatin treatment, PRKCD is phosphorylated and activated by SRC family kinases and AKT is phosphorylated at Thr308 by PDPK1, promoting PRKCD binding and phosphorylation of AKT at Ser473 and full activation of AKT. AKT then activate MTOR to repress ULK1 and associated autophagy, leading to apoptosis.
One such condition that is particularly relevant is cancer cells during cisplatin chemotherapy. Cancer cells respond to cisplatin treatment by activating autophagy and various mechanisms of cell death. Despite some conflicting data, it is generally accepted that autophagy protects against cell death in cancers during chemotherapy. Is PRKCD activated to suppress autophagy in cancer as it does in kidneys? If so, PRKCD inhibitors are expected to increase autophagy in cancer cells and protect them from death resulting in decreased effects from chemotherapy. Nonetheless, our previous work showed that PRKCD inhibitors enhance the cancer therapy effect of cisplatin. Thus, PRKCD may have pleiotropic functions in cancer cells. Although not an oncogene, PRKCD belongs to the class of “onco-addictive gene” products, which facilitate cancer cell growth and proliferation. Thus, the role of PRKCD in cancer cells is complex. Certainly, it would be interesting to investigate PRKCD in the regulation of autophagy, cell death, and proliferation in cancer cells and tumors during chemotherapy.
Obviously, delineation of the PRKCD-AKT-MTOR-ULK1 signaling pathway has shed new light on autophagy and kidney cell death regulation during cisplatin chemotherapy. But, the study also leaves out the answers to many important questions. For example, this pathway negatively regulates autophagy during cisplatin nephrotoxicity: What is responsible for autophagy induction under these conditions?