

Abstract
Ubiquilin (UBQLN) proteins are adaptors thought to link ubiquitinated proteins to the proteasome. However, our lab has recently reported a previously unappreciated role for loss of UBQLN in lung cancer progression. In fact, UBQLN genes are lost in over 50% of lung cancer samples examined. However, a reason for the loss of UBQLN has not been proposed, nor has a selective pressure that could lead to deletion of UBQLN been reported. Diesel Exhaust Particles (DEP) are a major concern in the large cities of developing nations and DEP exposed populations are at an increased risk of developing a number of illnesses, including lung cancer. A connection between DEP and UBQLN has never been examined. In the present study, we determined the effect of DEP on lung cell lines and were interested to determine if UBQLN proteins could potentially play a protective role following treatment with DEP. Interestingly, we found that DEP treated cells have increased expression of UBQLN proteins. In fact, over-expression of UBQLN was capable of protecting cells from DEP toxicity. To investigate the mechanism by which DEP leads to increased UBQLN protein levels, we identified and interrogated microRNAs that were predicted to regulate UBQLN mRNA. We found that DEP decreases the oncogenic microRNA, MIR155. Further, we showed that MIR155 regulates the mRNA of UBQLN1 and UBQLN2 in cells, such that increased MIR155 expression increased cell invasion, migration, wound formation and clonogenicity in UBQLN-loss dependent manner. This is the first report of an environmental carcinogen regulating expression of UBQLN proteins. We show that exposure of cells to DEP causes an increase in UBQLN levels and that MIR155 regulates mRNA of UBQLN. Thus, we propose that DEP-induced repression of MIR155 leads to increased UBQLN levels, which in turn may be a selective pressure on lung cells to lose UBQLN1.
Abbreviations: DEP, diesel exhaust particle; MIR155m, MIR155 mimic; MIR155i, MIR155 inhibitor; NTC, non-targeting control; UBQLN, Ubiquilin; UTR, untranslated region; EMT, epithelial to mesenchymal transition
Introduction
Long-term exposure of diesel exhaust particles (DEP), generated by combustion engines, compromises lung function and increases susceptibility for lung cancer development. Large population based studies have clearly established the relationship between DEP exposure and lung cancer development [1]. DEP are a severe carcinogenic air pollutant that have large surface area due to their ultra-fine size and they have tendency to accumulate in lungs [2]. Earlier studies have shown that exposure to DEP deregulates expression of multiple genes related to inflammation, DNA repair, apoptosis and proteostasis in lung cells [3], [4], [5]. Ubiquilin proteins (UBQLN) are important regulators of proteostasis, working as adaptor proteins for the delivery of ubiquitinated proteins to the proteosome [6]. There are five closely related UBQLN that all contain a ubiquitin-like (UBL) domain at their N-termini and a ubiquitin-associated domain (UBA) at their C-termini [7]. UBQLN1 was first identified as a yeast homologue protein, which functionally links the proteasome and ubiquitination machinery [8]. Simultaneously, UBQLN1 was identified as a protein that interacted with presenilin, increasing its accumulation in neurofibral tangles and lewy bodies [9], [10]. After these discoveries, most studies have focused on the protective role of UBQLN1 expression in neurodegenerative disorders [10], [11]. A recent study utilizing UBQLN1 knockout and UBQLN1 over-expressing mice demonstrated a role for UBQLN1 in protecting cells from injury and oxidative stress [12]. Studies carried out in neuronal cells clearly indicated that increased expression of UBQLN helps to protect the cells from toxicity induced by protein mis-folding. However, our group has recently shown for the first time that down-regulation of UBQLN1 in lung cancer cells induces epithelial to mesenchymal transformation (EMT) [13]. Down-regulation of UBQLN1 in human lung cancer tissue samples and loss of UBQLN1 in a large percentage of human tumors also indicates a potential vital role of this gene in progression of lung cancer [13]. This has built on previous studies that identified differential regulation of UBQLN1 in primary human lung cancers and autoantibodies against UBQLN1 in the plasma of lung adenocarcinoma patients as a candidate biomarker for disease stage and progression [7], [14].
Mature microRNAs (miRNAs) are short (18-25 bp), endogenously transcribed, regulatory RNA molecules, which typically bind to 3′-UTR of protein coding mRNAs and negatively regulate their translation [15]. miRNAs are well established to regulate fundamental cellular processes like proliferation, differentiation, development, apoptosis and cell cycle [15], [16]. Cancer related genes and pathways have been identified as some of the major targets for miRNAs, and miRNAs, which induce cancer, are termed “oncomiRs” [17]. miRNA-induced reprogramming is emerging as a master regulator of EMT and cell proliferation [18]. TGF-β and its down-stream transcription factors like ZEB1, SNAIL, and TWIST are major miRNA-regulated genes during tumorogenesis [19]. MIR155 is a well-documented oncomiR, which is attracting interest due to its role in multiple cancers such as breast cancer, leukemia, lymphoma, lung, and liver [20]. In humans, MIR155 is located on chromosome 21q21.3, also known as B-cell integration cluster (BIC), which transcribes an exon of non-coding RNA containing MIR155 [21]. Several studies involving different types of cancers have correlated over-expression of MIR155 with cancer progression and pathogenesis [22], [23]. Moreover, studies of Liu [24] et al. have shown that MIR155 expression can be used as a biomarker for early diagnosis of esophageal cancer [24].
In the present study, we have demonstrated a role for UBQLN expression in modulating toxicity of lung cells to DEP. Moreover, we have also identified MIR155 as a regulator of cellular functions through down-regulation of UBQLN in lung cells. Also, we have demonstrated that up-regulation of UBQLN protects cells from DEP toxicity by inducing accumulation of DEP particles in vacuolar like sub-cellular structure. As in our earlier studies [13], we have found UBQLN1 proteins are frequently lost in lung cancer patients and decreased UBQLN levels induces EMT in lung cancer cells, in present study we have selected multiple cancer cell lines (A549 and H358) to study the effect of MIR155 mediated down-regulation of UBQLN in inducing EMT of lung cells. Moreover, using in vivo studies we demonstrate that MIR155 mediated down-regulation of UBQLN increases tumorigenic properties of cancer cells.
Materials and methods
Preparation and Characterization of DEP Particles
Diesel exhaust particles (DEP), a standard reference material, #2975 was prepared from a Forklift engine by U.S. National Institute of Standards and Technology, were procured from Sigma Aldrich, USA. DEP stock solutions were prepared by suspending it in Milli-Q water at concentration of 1 mg/ml and sonication at 20 kHz for 10 minutes with 45 seconds pulse and 15 sec resting interval.
Cell Culture, Cell Viability and siRNA/miRNA Transfections
A549, H358 and 293 T cell lines were procured from American Type Culture Collection (ATCC, Rockville, MD, USA). A549 and H358 were cultured in RPMI medium, while 293 T was cultured in DMEM medium. Both RPMI and DMEM media were supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 1% antibiotic/antimycotic (Sigma) and ciprofloxacin HCl (5 μg/ml). The cell lines were routinely sub-cultured every 3 to 4 days and checked once a month for mycoplasma contamination. MIR155 mimic (Assay ID:MC12601 cat. #4464066) and inhibitor (Assay ID:MH12601 Cat. #4464084) were purchased from Thermo Fisher. All transfections were performed using Dharmafect1 #T-2001-03 (Thermo Fisher Scientific Inc., Pittsburgh, PA, USA) as per the manufacturer's protocol. Cell viability assays were performed using Alamar Blue reagent as per manufacturer protocol. Briefly, 10% Alamar Blue was added in each well of 96 well plates, which are seeded with equal number (1000) of cells at the time points indicated before Alamar Blue was added. Fluorescence was measured using a plate reader.
Fluorescence-Activated Cell Sorting
Fluorescence-activated cell sorting was performed by the flow cytometry core facility at the James Graham Brown Cancer Center or using BD Influx flow cytometer at CSIR-Indian Institute of Toxicology Research, Lucknow, India. A549 cells were infected with viruses containing MIG-RX (empty vector) or MIG-UBQLN1. The MIGRX vector, which is murine stem cell virus based retroviral vector derived from MIGR1 vector as described in our earlier studies was used for cloning UBQLN1 gene. Both MIGRX empty vector (MIG-EV) and MIGRX containing UBQLN1 (MIG-UBQLN1) express GFP. A549 cells infected with virus containing MIG-EV or MIG-UBQLN1 were sorted for GFP florescence and are referred to as MIG-EV or MIG-UBQLN1 respectively. For rescue experiments, above cells were transfected with NTC or MIR155 mimic.
TEM in DEP Exposed A549 Cells
Flow sorted A549 cells, which are infected with either empty vector (MIG-EV) or UBQLN1 over-expressing vector (MIG-UBQLN1) are exposed with either DEP or equal amount of autoclaved Milli-Q water. After completion of exposure, cells are trypsinized, washed with PBS and fixed for 2 h at 4 °C in 2.5% glutraldehyde solution prepared in sodium cacodylate buffer. After fixation, cells were washed three times with sodium cacodylate buffer and post-fixed in 1% Osmium tetroxide for 4 hours. Post-fixed cells were washed with sodium cacodylate buffer, dehydrated in acetone series (15–100%) and embedded in araldite-dodecenyl succinic anhydrite (DDSA; hardner) mixture. Cells are backed at 60° and blocks were cut by ultra-microtome (Leica EM UC7) into 60–80 nm thin sections, and mounted on TEM grids. Then sections were stained by Uranyl acetate and Lead citrate and analyzed by FEI Tecnai G2 spirit TWIN TEM equipped with Gatan digital CCD camera at 80Kv.
Apoptosis Assay by Flow Cytometer
A549 cells over-expressed with UBQLN1 (MIG-UBQLN1) or control vector (MIG-ER) and flow sorted for GFP using by flow cytometer are utilized to study the effect of DEP on apoptosis of lung cells. Cells are seeded equally and exposed with either 125, 250 or 500 μg/ml DEP for 72 hours. Apoptosis was measured using Annexin V-APC and propidium iodide (PI) labeling. After 72 hours of exposure, cells were washed twice with PBS and cell pelleted in 100 μl binding buffer. In each sample, 5 μl AnnexinV-APC was added and incubated for 15 minutes in dark at room temperature. After 15 minutes, cells are incubated for 10 minutes in PI. After PI incubation, cells were diluted with 400 μl binding buffer and apoptosis was measured using flow cytometer.
Luciferase Assay
The interaction of MIR155 with UBQLN1 or UBQLN2 was studied by co-transfection of 3′-UTR of UBQLN1 or UBQLN2 and mimic or inhibitors of MIR155. 3′-UTR of UBQLN1 or UBQLN2 was cloned in the pEZX-MTO1 vector of Genecopeia, USA, which contains firefly luciferase as the reporter gene (controlled by the SV40 promoter) and Renilla luciferase as the tracking gene (controlled by the CMV promoter). Both firefly and Renilla luciferase activities were measured by means of the Dual-Glo luciferase assay kit #E2920 (Promega, Inc. Madison, WI) as per manufacturer's protocol. Maximal luciferase activity was calculated by normalizing firefly luciferase activity to Renilla luciferase activity within each sample.
Clonogenic Assay, Scratch Assay, Cell Migration and Cell Invasion Assay
For clonogenic or colony formation assay, cells were transfected with MIR155 mimic or MIR155 inhibitor or NTC. After 24 hours of transfection, equal numbers (1000 cells) of cells were plated in triplicate in each well of six-well plates and grown for 10 days to allow the growth and colony formation. After colonies were formed, cells were fixed with 70% methanol and stained with 0.25% Coomassie blue stain. After destaining, six-well plates containing colonies were scanned and colonies were counted. Scratch assays, cell migration and cell invasion assays were performed exactly as previously described [26].
Immunoblotting and Immunofluorescence Staining
Western blots and immunofluorescence were performed exactly as described previously [26].
Real Time PCR
Total RNA was isolated using miRVana miRNA kit, of Thermo Scientific, USA as described by manufacturer. Reverse transcription was performed using High Capacity RNA-to-cDNA Kit from Thermo Scientific, USA. Briefly, each reaction consists of 10 μl of 2× RT buffer (includes dNTPs, random octamers, and oligo dT-16), 1 μl of 20× Enzyme mix (MuLV and RNase inhibitor protein) and 1 μg of total RNA in 20 μl final volume. Reaction mixture was incubated at 37 °C for 1 hour and reaction mixture was heat inactivated by incubating at 95 °C for 5 minutes. Real time PCR was carried out using Taqman chemistry and specific primers procured from ABI, USA for UBQLN1 (Assay ID: Hs00923840_m1) and β-actin (Assay ID: Hs99999903_m1) using conditions described by manufacturer. Sequence of real time PCR primers used for UBQLN2 are FP-5′-GGAGGCGACATCAATGCAG-3′; Probe – FAM – 5′-ACAGAAATGTGATTACGATGGCTGGGAG-3′-TAMRA; RP – 5′-AAATGAAAGTAAAGAACGATTTTGTGT-3′. Detection of mature MIR155 was performed by using taqman assay from ABI (cat. #4427975; Assay ID#002623) targeting sequence 5′-UUAAUGCUAAUCGUGAUAGGGGU-3′.
Animal Experiment
All animal experiments were done in accordance and under the approval of the University of Louisville IACUC protocol #15005. The NRG mice, which are immunodeficient were procured from ‘The Jackson Laboratory, USA’ and housed in animal facility of University of Louisville. Twenty mice were divided into four cohorts of five mice each according to the type of A549 cells received. Group 1: MIG-EV + NTC; Group 2: MIG-EV+ MIR155M; Group 3: MIG-UBQLN1 + NTC; Group 4: MIG-UBQLN1+ MIR155M. Each mouse was injected with 1 × 106 cells into the tail vein. 30 days post injection, mice were sacrificed and lungs were dissected out. Fresh lungs were weighed and imaged for fluorescence of GFP using AMI from Spectral Instrument Imaging (Tucson, AZ) and total photons were measured for fluorescence.
Pan-Cancer DNA Copy Number and Expression Data Analysis
Information regarding DNA copy number alterations affecting UBQLN1 and UBQLN2, as well as hsa-miR-155, were obtained from colon adenocarcinoma (COAD), 191 acute myeloid leukemia (LAML), 239 bladder urothelial carcinoma (BLCA), 1033 breast invasive carcinoma (BRCA), 306 neck squamous cell carcinoma (HNSC), 436 kidney renal clear cell carcinoma (KIRC), 230 lung adenocarcinoma (LUAD), 179 lung squamous cell carcinoma (LUSC), 569 ovarian serous cystadenocarcinoma (OV), 237 prostate adenocarcinoma (PRAD), and 363 uterine corpus endometrial carcinoma (UCEC) tumors available through The Cancer Genome Atlas Project (TCGA).
Correlation of DNA-Level Alterations with Gene Expression in Pan-Cancer Tissues
In order to assess the effect of DNA-level alteration on gene expression of UBQLN1 and UBQLN2, pan-cancer tissues were divided into three groups based on copy number alteration status (copy number loss, copy number neutral, copy number gain) and expression was compared between groups using GraphPad Prism 6 (La Jolla, CA, USA). A Mann–Whitney U test compared the differences in the median expression values between copy number loss and diploid groups with 95% confidence. In all comparisons, a p value <.05 was considered significant.
Correlation between hsa-miR-155 gene expression and UQBLN1/2 expression was assessed using GraphPad Prism 6. Samples with available copy number and expression data were binned into two groups based on a percentile rank of hsa-miR-155 expression; then UBQLN1/2 expression in the 90th percentile of samples was compared to the 10th percentile using a Mann–Whitney test to identify significant differences between median expression values.
Survival Analysis
Publicly available clinical information, including follow-up times and vital status data for patients across 11 cancer types were obtained from the TCGA via the data portal (https://tcga-data.nci.nih.gov/) on January 21, 2015. Survival curves were generated with GraphPad Prism 6 software, using the Kaplan–Meier method and RNA Sequencing data. Survival curves for patients containing UBQLN1/2 expression in the 90th percentile were compared to those with expression in the 10th percentile. Any p values <.05 were considered significant.
Results
DEP Induced UBQLN Protects A549 Cells by Accumulating DEP Particles in Tightly Packed Vacuoles
We have recently demonstrated that loss of UBQLN1 increases metastatic properties and EMT of human lung adenocarcinoma cells. Although papers have shown that UBQLN1 can be induced by stress, physiological/pathological regulators of UBQLN1 expression are largely unknown. We therefore were interested to know if a suspected environmental carcinogen, DEP, could somehow alter the expression of UBQLN family members in A549 lung carcinoma cells. A549 and H358 cells are derived from lung adenocarcinoma patients and are epithelial in origin. We thus chose these cells to represent the response of lung epithelial cells to DEP and for characterization of downstream mechanistic studies. Exposure of cells to DEP (250 μg/ml) for 72 hours significantly induced the protein levels and mRNA expression of UBQLN1/2 in A549 cells (Figure 1, A and B). To determine if DEP-induced UBQLN1 expression alters the effect of DEP on apoptosis of A549 cells, we engineered cells that over-expressed UBQLN1. To demonstrate that A549 cells expressing UBQLN1 are protects from DEP-induced cell death, we carried out flow cytometry analysis of AnnexinV and propidium iodide following treatment of cells with DEP (Figure 1C and Supplemental Figure S1). With the increasing DEP concentrations, the difference in ratio of live cells to AnnexinV positive dead cells increased between UBQLN1 and control vector over-expressing cells. At the highest concentration of DEP (500 μg/ml), UBQLN1 over-expressing were approximately 40% more viable than control vector expressing cells (Figure 1C).
Figure 1.

DEP induced UBQLN protects cells by promoting accumulation of DEP in vacuole-like structures of A549 cells. A) Effect of DEP exposure on protein levels of UBQLN1/2 in A549 cells; B) Effect of DEP exposure on expression of UBQLN1/2 mRNAs in A549 cells; C) Effect of DEP exposure on apoptosis of UBQLN1 over-expressing cells using flow cytometric using anti-AnnexinV and PI staining; D) TEM images of DEP exposed A549 cells over-expressing UBQLN1 or empty vector (EV).
Supplemental Figure S1.

(supporting Figure 1C). Effect of DEP exposure on apoptosis of UBQLN1 over-expressing cells using flow cytometric using anti-AnnexinV and PI staining. Cells were treated with the indicated concentration of DEP for 72 hours. Cells were treated in triplicate and representative FACS profiles are shown on cells stained with APC-AnnexinV and PI.
In order to confirm that DEP is penetrating into the cells and identify the effect of UBQLN1 over-expression in differential response of A549 cells to DEP, we carried out transmission electron microscopy (TEM) (Figure 1D). Images taken on TEM showed accumulation of DEP particles in tightly closed cytoplasmic vesicle-like structure in A549 cells over-expressing UBQLN1 (Figure 1D). In contrast, A549 cells stably expressing control vector (MIG-ER) had DEP in more loosely associated form (Figure 1D).
Identification of UBQLN1 and UBQLN2 Regulating miRNAs
Since miRNAs are critical regulators of gene expression, we wondered if there were miRNAs that could regulate UBQLN1 expression and thus regulate role of UBQLNs in toxicity or metastatic properties of cancer cells. Analysis of the 3′-UTR of UBQLN1 using TargetScan web portal predicted more than 100 miRNA binding sites. Among the top predicted miRNAs, MIR200 and MIR425 are well-documented regulators of tumorigenesis [25], [26], [27]. However, transfection of A549 or H358 cells with either MIR200 or MIR425 mimics did not alter levels of UBQLN1 (Supplemental Figure S2). Further, cross-referencing the TargetScan algorithm with the miRDB database suggested MIR155 among the top predicted miRNA regulators of UBQLN1 (Figure 2A). Interestingly, MIR155 was predicted to target the 3′-UTR of both UBQLN1 and UBQLN2 in humans and mice (Figure 2B). Exposure of DEP down-regulated expression of MIR155, which correlates with DEP induced up-regulation of UBQLN1 and UBQLN2 in A549 cells (Figs. 1A and 2C). Our previous work demonstrated that UBQLN genes are frequently lost, mutated and under-expressed in human lung adenocarcinoma patient samples. Our new finding that MIR155 is capable of regulating the expression of UBQLN1 and UBQLN2 prompted us to examine whether or not the expression of MIR155 could predict survival in human cancer patients and whether or not the expression of MIR155 inversely correlates with mRNA expression of UBLQLN1 and UBQLN2. We examined 11 different cancer types available for analysis through The Cancer Genome Atlas (TCGA) and found that MIR155 expression does not predict poor survival in human lung adenocarcinoma (Figure 2D), nor did the expression of MIR155 inversely correlate with UBQLN1 or UBQLN2 in lung adenocarcinoma (Figure 2E). In fact, we find that high expression of MIR155 does not predict overall survival in nine of the eleven datasets examined (Supplemental Figure S3). In addition, when samples with high or low MIR155 were examined there was no consistent correlation with the expression of UBQLN1 or UBQLN2 (Supplemental Figure S3). Kidney clear cell carcinoma was the one striking exception (Figure 2, D and E). Expression of MIR155 clearly was a predictive biomarker for overall survival and samples with high MIR155 had significantly lower mRNA expression of both UBQLN1 and UBQLN2.
Supplemental Figure S2.
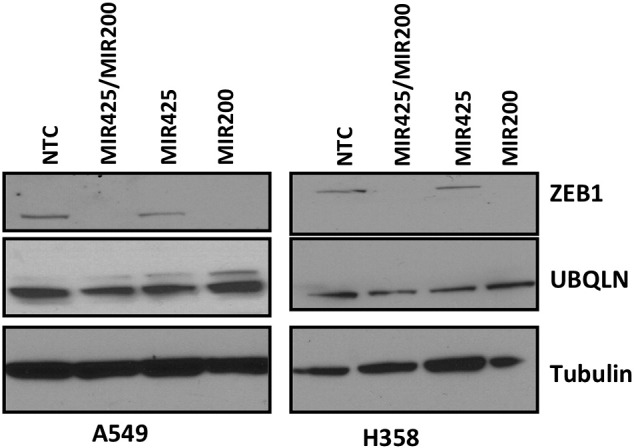
(supporting Figure 2A). Transfection of cells with MIR200 or MIR425 mimics does not regulate UBQLN1 or UBQLN2 levels. H358 and A549 cells were transfected with the indicated miRNA mimic and 72 hours later lysates were prepared and western blotted with the indicated antibodies.
Figure 2.

MIR155 is down-regulated by DEP and is predicted to bind and regulate UBQLN1 and UBQLN2 mRNA. A) Ranking of UBQLN1 targeting miRNAs using miRDB web portal according to their functional annotations (higher the score more the probability of binding); B) Targeting site of MIR155 in 3′-UTR of UBQLN1 and UBQLN2; C) Effect of DEP exposure on expression of MIR155 in A549 cells; D) Kaplan–Meier plot of overall survival of lung adenocarcinoma patients (top) and renal cell carcinoma patients (bottom) stratified by high (dashed) and low (solid) MIR155 expression. E) Box and whiskers plots comparing RSEM of UBQLN expression values in samples with high MIR155 expression (90th percentile) to samples with low MIR155 expression (10th percentile). The box extends from the 25th to the 75th percentile, with the median shown, whereas the whiskers range from 10th to 90th percentile. (Mann–Whitney test, one asterisk P < .05, two asterisks P < .01). Cancer types with a negative correlation between MIR155 and UBQLN expression also had MIR155 expression profiles significantly associated with overall patient survival.
Supplemental Figure S3.

(supporting Figure 2E). Correlation between MIR155 expression levels and overall survival in sets of cancer patients from TCGA. See methods for details on analysis.
MIR155 Regulates Protein Levels and mRNA Expression of UBQLN1 and UBQLN2
We next wanted to determine if the regulation of UBQLN1 and UBQLN2 by MIR155 was at the level of mRNA and if it was direct. First, we transfected A549 and H358 with a MIR155 mimic, a MIR155 inhibitor or a non-targeting miRNA control (NTC) and measured UBQLN1 and UBQLN2 levels by immunoblotting (Figure 3A). Transfection of MIR155 mimic substantially down-regulated levels of UBQLN1 and UBQLN2 in both A549 and H358 cells, whereas the MIR155 inhibitor did not significantly alter the expression of UBQLN1 or UBQLN2 in these cells (Figure 3A). Similar to immunoblots, transfection of MIR155 mimics significantly down-regulated mRNA expression of UBQLN1 and UBQLN2 as measured by real time PCR (Figure 3B). Immunofluorescence staining of MIR155 mimic or inhibitor transfected cells with a pan-UBQLN antibody also showed decreased UBQLN1/2 in cells transfected with MIR155 mimic (Figure 3C).
Figure 3.

Levels of UBQLN1/2 protein and mRNA expression are regulated by MIR155. A) Effect of transfection of MIR155 mimic (MIR155m) and inhibitor (MIR155i) on protein levels of UBQLN1/2 in A549 and H358 cells; B) Real time PCR of UBQLN1/2 in A549 and H358 cells transfected with MIR155m and MIR155i; C) Confocal images of A549 and H358 cells transfected with MIR155 mimic and inhibitor and probed with anti—UBQLN1/2. Green florescence is represents UBQLN1/2, red florescence represents actin. Cells are counterstained with DAPI for nuclear staining.
Luciferase assays were carried out in A549 and 293 T cells to establish the regulation of UBQLN1/2 mRNA by MIR155 (Figure 4). Cells co-transfected with MIR155 and UBQLN1 or UBQLN2 3″-UTR-luciferase showed significant decrease in relative luciferase activity in comparison to cells co-transfected with UBQLN1 or UBQLN2 3″-UTR plasmid and NTC or MIR155 inhibitor (Figure 4). Interestingly, transfection of equimolar amounts of MIR155 inhibitor prevented MIR155 mediated decrease in relative luciferase activity of UBQLN1 or UBQLN2 3″-UTR plasmid, further demonstrating the specificity of MIR155 for UBQLN1 mRNA (Figure 4). Decreased relative luciferase activity and mRNA expression of UBQLN1 or UBQLN2 in MIR155 transfected cells confirms regulation of UBQLN1/2 mRNAs by MIR155.
Figure 4.

3′-untranslated regions (UTR) of UBQLN1 and UBQLN2 are regulated by MIR155. Dual luciferase assay in A549 and 293 T cells transfected with luciferase reporter plasmids pEZX-MT01 (empty vector; EV), pEZX-UBQLN1 (contains 3′-UTR of UBQLN1; 3′-UTR) or pEZX-UBQLN2 (contains 3′-UTR of UBQLN2; 3′-UTR) and MIR155 mimic (155 m) or MIR155 inhibitor (155i) or NTC. Relative luciferase activity was calculated by normalizing firefly luciferase activity with Renilla luciferase activity (transfection control). Relative luciferase activities of cells transfected with 3′-UTR plasmid and NTC is considered as 1 (equals to 100%) and other activities are presented in comparison to them.
MIR155 Induces Invasion and Migration of Cancer Cells
To study the role of MIR155 in cellular processes, we introduced a MIR155 mimic into A549 and H358 cells and examined migration and invasion of the cells (Figure 5). Transfection of MIR155 mimic increased the number of cells capable of both migrating and invading through Boyden chamber transwell inserts, compared to NTC transfected A549 and H358 cells (Figure 5A). The effect of MIR155 on cell migration was further studied using a “wound healing” scratch assay in A549 and H358 cells (Figure 5B). Twenty-four hours after transfection of the miRNA, a wound was made of approximate equal size in all the samples and the size of wound was measured over the next 2 days (Figure 5B). Both, A549 and H358 cells showed increased healing when transfected with MIR155 mimic in comparison to NTC transfected cells (Figure 5B). After 2 days of wound formation, the wound was almost completely healed in cells transfected with MIR155 mimic, while it was approximately 50% healed in cells transfected with MIR155 inhibitor and NTC (Figure 5B).
Figure 5.

Expression of MIR155 induces cell invasion and migration. A) Cell migration and invasion assay in cells transfected with MIR155m, or MIR155i or NTC. After 24 hours of transfection, equal number of cells was seeded into Boyden chambers with (invasion) or without (migration) matrigel. The lower chamber contained the media with serum, whereas upper chamber contained media without chamber. After 48 hours, lower side of membrane were fixed and stained. Cells migrated or invaded are counted manually and presented as bar diagram. B) Scratch assay in A549 cells transfected with MIR155m, or MIR155i or NTC. After 24 hours of transfections, scratch was made using a pipette tip and cells were examined for scratch area, just after making scratches and on following two day. Microscopic images of scratches were analyzed using Image J software and changes in area were calculated considering 0 day as 100%. Values in % represent average scratch area remaining.
MIR155 Increases Clonogenic Potential and Cell Proliferation
Clonogenic assays were carried out with A549 and H358 cells to study the effect of MIR155 on survival and growth capabilities of cancer cells seeded at low density (Figure 6A). A549 and H358 cells were transfected with MIR155 mimic, MIR155 inhibitor, KIF11 siRNA (positive control that is lethal in cells) and NTC, and 24 hours after transfection, 1000 cells/well were seeded in triplicate in 6 well plates. Counting of colonies after 10 days, showed that cells transfected with MIR155 mimics produced significantly more (2 fold) colonies then cells transfected with NTC (Figure 6A). Interestingly, transfection of MIR155 inhibitor significantly decreased colonies in A549 cells, however no significant differences were observed in H358 cells (Figure 6A). As expected knocking down KIF11 dramatically reduced colony formation capabilities of both A549 and H358 cells (Figure 6A). Cell proliferation assays were carried out using Alamar Blue conversion as a read-out to determine relative numbers of viable cells. Cells transfected with MIR155 showed a dramatic increase in cell growth, as compared with cells transfected with NTC transfected cells in both A549 and H358 cells (Figure 6B). These data indicate that MIR155 increases both clonogenic potential and proliferation of adenocarcinoma cells.
Figure 6.

Expression of MIR155 induced clonogenic capabilities and proliferation of lung carcinoma cells. A) Cells are transfected with MIR155m or MIR155i or NTC or KIff11 (positive control). After 24 hours of transfection, each well of 6-well dish was plated with 1000 cells and grown for 10 day. After 10 days, cells were fixed, stained and destained as described in methodology. Bar diagram represents average number of colonies counted after staining. B) Alamar blue assay in A549 and H358 cells transfected with MIR155m or MIR155i or Kiff11 or NTC. After 24 hours of transfection, 96 well plate were seeded with 1000 cells/well and Alamar blue assay was carried out on 0 day (day of cell seeding), and next 4 consecutive days. Day 0 was used to normalize the counts of next 4 days for any possible difference in cell seeding. Relative activities were then calculated each day against NTC (considered as 1).
Regulation of Epithelial and Mesenchymal Markers by MIR155 in A549 and H358 Cells
Increased invasion, migration and colony formation of MIR155 over-expressing cells prompted us to study whether MIR155 could induce EMT in cancer cells. Immunoblotting of total cell lysates prepared from MIR155 mimic, MIR155 inhibitor and NTC transfected A549 and H358 cells, showed dramatic decrease of epithelial markers with a concomitant increase in mesenchymal markers, indicating an EMT-like phenotypic transition (Figure 7). Immunoblotting for mesenchymal markers Vimentin and Snail, showed substantial increase in cells transfected with MIR155 mimic, compared with NTC transfected cells, whereas epithelial markers E-cadherin, and claudin decreased significantly in cells transfected with MIR155 mimic, compared with NTC transfected cells (Figure 7A). MIR155 mediated regulation of E-cadherin and Vimentin, two classical markers of EMT, were also studied by immunofluorescence imaging of cells to validate the western blot results. Similar to immunoblotting, immunofluorescence imaging demonstrated up-regulation of Vimentin and down-regulation of E-cadherin in cells transfected with MIR155 mimic (Figure 7B).
Figure 7.

Expression of MIR155 regulates levels of epithelial and mesenchymal markers. A) Immunoblotting of epithelial (E-cadherin and Claudin1) and mesenchymal (Vimentin and Snail) markers in lysates of A549 or H358 cells transfected with MIR155m, or MIR155i or NTC. B) Immunocytochemical detection of vimentin and E-cadherin in A549 and H358 cells transfected with MIR155m, or MIR155i or NTC. (Florescence: green represents Vimentin or E-Cadherin, red represents actin and blue is from nucleus).
Over-Expression of Exogenous UBQLN1 Lacking 3′ UTR Rescues MIR155 Phenotypes in A549 Cells
To demonstrate that the phenotypes observed following introduction of MIR155 are, at least in part, through its ability to decrease UBQLN1, we engineered cells that stably over-express a FLAG-tagged-UBQLN1 cDNA lacking the 3′-UTR (Figure 8A). Briefly, A549 cells were infected with a virus expressing UBQLN1 and GFP (MIG-UBQLN1) or GFP only (MIG-EV) and cells were sorted by flow cytometry followed by FLAG western blot to detect the exogenous UBQLN1 (Figure 8A). Transfection of MIR155 mimic into MIG-UBQLN1 cells decreased endogenous UBQLN1, however exogenous FLAG-tagged UBQLN1 was still abundantly expressed (Figure 8B). Transfection of MIR155 mimic, up-regulated Vimentin and down-regulated E-cadherin levels in MIGRX-EV cells, while in MIG-UBQLN1 cells their levels remain unchanged (Figure 8B). Further, MIR155-induced wound healing and clonogenic potential was significantly suppressed in MIG-UBQLN1 cells (Figure 8, C and D). In addition, increased proliferation rates of A549 cells transfected with MIR155 was also blocked in cells stably expressing UBQLN1 (Figure 8E). These data strongly suggest that MIR155 is capable of regulating tumorigenic properties of cancer cells, in large part through its ability to target UBQLN1.
Figure 8.

Over-expression UBQLN1 lacking a 3′-UTR inhibits MIR155 mediated phenotypes. A549 cells are infected with UBQLN1 (U1) or empty vector (EV) and sorted by flow cytometer as described in methodology section. Flow cytometer sorted cells are transfected with MIR155m or NTC and used for different assays: (A) Western blots were performed on Flow sorted cells with anti-FLAG antibody to detect over-expressed exogenous UBQLN1. (B) Western blots were performed after 72 hours following miRNA transfection for EMT markers. (C) Wound healing/scratch assay (D) Colony formation assay, (E) Alamar Blue assay (F) Transfection of MIR155m increased growth and colonization of A549 cells in mice lungs. A549 cells were infected with viruses containing MIG-RX (empty vector; EV) or MIG-UBQLN1 (U1) and sorted with flow cytometer using GFP as marker as described in methods. GFP sorted cells are transfected with NTC or MIR155m. After 24 hours of transfection, five mice in each group are injected with 1×106/mice. After 30 days of injection, mice were scarified and total lung weight was measured and GFP florescence was measured using animal imager.
MIR155 Transfected A549 Cells Colonize and Grow in Lungs of Mice
Our earlier studies have shown that siRNA mediated down-regulation of UBQLN1 induces EMT and carcinogenesis in cellular models. In the present study, we explored the effect of MIR155 mediated UBQLN1 down-regulation on tumorigenic properties of lung adenocarcinoma cells, in vivo (Figure 8F). A549 cells stably expressing either GFP and UBQLN1 (U1) or GFP only (MIG) were transfected with MIR155 mimic or NTC and injected into the tail veins of immunodeficient NRG mice. 30 days after injection mice were euthanized and analyzed. Lung weights and GFP fluorescence were measured. The cohort of mice injected with MIR155 mimic transfected MIGRX-EV cells showed increased lung weights (average lung wt. 1.18 g) compared to mice injected with NTC transfected MIGRX-EV (average lung wt. 0.86 g) cells (Supplemental Figure S4). However, transfection of MIR155 mimic did not produce substantial alterations in lung size in cells over-expressing exogenous UBQLN1 lacking a 3′ UTR (Supplemental Figure S4). Further, support for these findings were obtained by measuring the GFP fluorescence in isolated lungs. There was a significant increase in GFP signal in lungs isolated from mice injected with MIR155 mimic transfected MIGRX-EV cells (Figure 8F). Similar to lung weight, no significant alterations were observed in GFP fluorescence of lungs isolated from mice injected with MIR155 mimic transfected MIGRX+-UBQLN1 cells (Figure 8F). These data demonstrate the potential in vivo importance of the ability of MIR155 to increase tumorigenic properties of human cancer cells through targeting UBQLN1 expression.
Supplemental Figure S4.
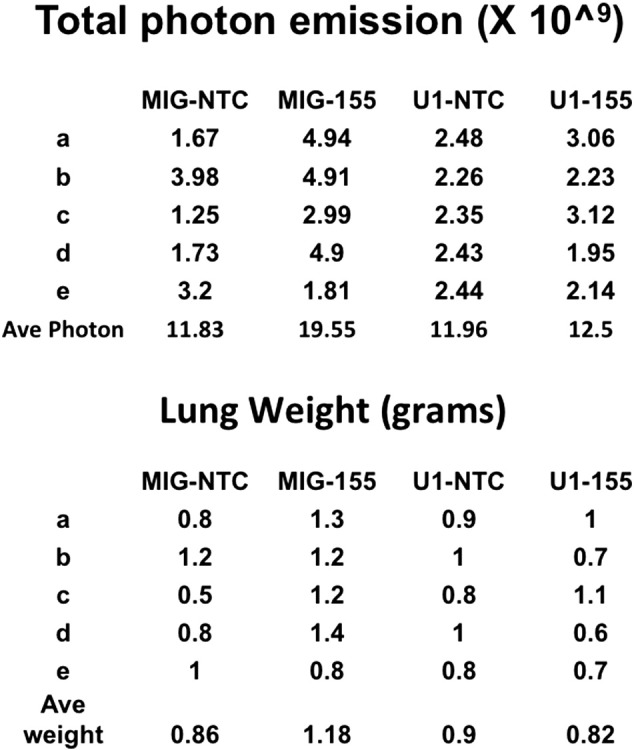
(supporting Figure 8F). Raw data for all mice graphed in Figure 8F showing photon emission and actual wet lung weights.
Discussion
International Agency for Research on Cancer (IARC) and the US National Toxicology Program (NTP) have categorized DEPs as known human carcinogen and lung as is identified the most severely affected organ [28]. UBQLNs are primarily studied for their role in degradation of ubiquitinated proteins, management of ER stress, or autophagy dysfunction of neuronal cells [6]. However, recent studies from our group have shown novel role of UBQLN in lung cancer cells [13]. Our studies have shown that in lung cancer cells, loss of UBQLN1 does not induce ER stress or alter autophagy, but instead, induces EMT. In the present study, we have demonstrated that increased expression of UBQLN1 protects A549 cells from DEP toxicity, while MIR155 mediated down-regulation of UBQLN1 and UBQLN2 induces EMT in lung cells. A recently published study showed that different particulate matters like DEP, urban dust, and carbon black utilize different mechanisms to induce cell death in lung cells and they identified a role for a ubiquitin-independent autophagy pathway in DEP induced cell death [29]. Our studies have shown that DEP exposure to A549 cells induces UBQLN1 and UBQLN2, which is likely a protective mechanism against DEP-induced toxicity. Exposure of DEP is well known to induce oxidative stress in different types of cells and tissues [30]. Our present study has shown that increased expression of UBQLN1 accumulates DEP particles in vesicle like cytoplasmic structure, which protects cells from oxidative stress imbalance induced by DEP. A recent study has shown that UBQLN2 clears tightly aggregated cellular aggregates in autophagy independent manner via HSP70 [31].
As our earlier studies [13] have shown that down-regulation of UBQLN1/2 induces EMT in lung cancer cells, in present study, we identified MIR155 as a regulator of UBQLN1 and UBQLN2. Using in silico approaches, we identified widely conserved miRNA sites for MIR200 and MIR425 in the 3′ UTR of UBQLN1. However, transfection of MIR200b/c and/or MIR425 did not modulated levels of UBQLN1, whereas, over-expression of MIR200 dramatically down-regulated levels of ZEB1 (Supplemental Figure S2), a transcription factor already demonstrated as target of MIR200 [32]. In contrary to our findings it was recently reported that, in breast cancer cells, over-expression of MIR200c could down-regulate UBQLN1 levels [33]. Studies of Sun [33] et al., have also shown that MIR200c mediated down-regulation of UBQLN1 enhances radio-sensitivity of breast cancer cells by suppressing autophagy. A recent study also demonstrated that down-regulation of UBQLN1 modulates the radio-sensitivity of the nasopharyngeal cells. Interestingly, they have also identified a role for MIR155 in regulating UBQLN1 expression [34]. However, our earlier studies have demonstrated that in lung cancer cells, knock down of UBQLN1 does not cause ER stress or alter autophagy [13]. It seems that both the role and regulation of UBQLN1 may be cell type specific. Even more intriguing, it may be possible that different cell types express different UBQLN mRNA with alternative 3′-UTRs. This possibility has never been explored, but warrants investigation.
Our previous studies have shown that knock-down of UBQLN1 in lung adenocarcinoma cells induces EMT transition [13], so we extended our studies by measuring the effect of MIR155 on migratory and invasive behavior of lung cancer cells. Similar to our previous report, migration and invasion assays have shown that over-expression of MIR155 induces EMT in both A549 and H358 cells. Analysis of A549 and H358 cells after transfection of MIR155 mimic, further confirmed the inverse relationship between expression of UBQLN1 and cell migration. MIR155 transfections also increased colony formation capabilities of A549 and H358 cells, which is an indicator of cell survival capacity when seeded at low density. Similar to our findings, recent studies have also shown a role for MIR155 up-regulation in inducing cell invasion and migration of breast, liver and hepatic cancers [35], [36], [37]. Zhang et al., found higher expression of MIR155 in colorectal cancer tissue in comparison to normal adjacent mucosa, which also correlates with advanced clinical stage and metastases [36]. Although, our analysis did not find a statistical difference in overall survival of colon cancer patients with high MIR155 expression, there was a significant decrease in UBQLN2 mRNA in some samples with high MIR155 (Supplemental Figure S3).
Transfection of MIR155 mimic regulated the levels of EMT markers in favor of an EMT-like state. Significantly increased level of mesenchymal markers (vimentin and snail) and decreased level of epithelial markers (E-cadherin and claudin-1) in MIR155 mimic transfected cells confirmed a role for MIR155 in EMT transition of A549 and H358 cells. Increased EMT by MIR155 could be inhibited by the constitutive expression of a UBQLN1 cDNA lacking the 3′-UTR, indicating that the ability of MIR155 to induce EMT is largely due to its ability to target UBQLN1 mRNA. Scanning of 3′-UTR of claudin shows the presence of a potential target site for MIR155, however, this MIR155 site scores very low (53% and 59th position), which indicates that levels of claudin-1 are likely not directly regulated by MIR155. Studies of Zhang et al., have identified claudin-1 as potential target of MIR155, but they did not perform reporter assays to validate direct interaction between claudin-1 3″-UTR and MIR155. However, we have also found down-regulation of claudin-1 by MIR155, but our earlier studies have shown that siRNA mediated down-regulation of UBQLN1 down-regulates claudin-1 in lung cancer cells [13]. Overall it seems that, at least in lung cancer cells, levels of claudin-1 are down-regulated majorly due to induction of EMT by UBQLN1 knockdown and not directly by MIR155. This finding will be explored in more detail in future studies.
How could an initial increase in UBQLN1 and decrease of MIR155 contribute to tumorigenesis? We hypothesize that an acute stress response is responsible for the up-regulation of UBQLN1 following DEP treatment. This is caused by an as yet unknown mechanism that induces down-regulation of MIR155. This initial stress response must be overcome, thus deletion of UBQLN1 locus may result. Initially we were surprised that high MIR155 expression did not correlate with low UBQLN1 and UBQLN2 mRNA in lung adenocarcinoma samples. However, this finding is in agreement with other observations that we had previously made. For example, UBQLN1 is heterozygously lost at the genomic level in ~50% of human lung adenocarcinoma patients and UBQLN2 is lost in another 15% of samples. We do not see this level of allelic loss in many other cancers, such as renal clear cell carcinoma. Furthermore, our data suggest that MIR155 may not be highly expressed in human lung adenocarcinoma cells because introduction of the MIR155 inhibitor does not alter UBQLN1 or UBQLN2 levels, nor does it alter cellular phenotypes. We propose that loss of UBQLN family members will be a common phenomenon contributing to neoplastic transformation. We suggest at least two mechanisms for UBQLN expression loss. First, allelic loss, such is the case in lung cancer, and second increased expression of miRNA, such as MIR155, which may be the case in kidney cancer.
Conclusion
In summary, we have identified different roles for UBQLN in lung cancer cells. Our studies have shown that ultrafine DEP particles up-regulate UBQLN1/2, which protects A549 cells from DEP toxicity by accumulating DEP particles in cytoplasmic vesicles. Our studies have also identified a role for MIR155 mediated down-regulation of UBQLN1/2 in inducing EMT in A549 cells. Down-regulation of UBQLN1/2 by MIR155 induces invasion and migration of cancer cells and regulates the expression of epithelial and mesenchymal markers to induce an EMT-like phenotype. Over-expression of UBQLN1, which lacks MIR155 binding site, rescued the effect of MIR155 on EMT, which confirmed the role of MIR155 mediated down-regulation of UBQLN1 in EMT. Increased growth rate of MIR155 mimic transfected MIG-EV cells in mice lungs have validated our in vitro findings, that knocking down UBQLN1 induces tumorigenesis in lung cells. In conclusion, up-regulation of UBQLN1/2 protects lung cells from toxicity, while down-regulation of UBQLN1 induces EMT transformation and increases tumorigenic properties of cancer cells.
The following are the supplementary data related to this article.
Acknowledgements
We would like to sincerely thank the members of the Beverly Lab and the members of the Siskind Lab for thoughtful input and assistance.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
The work was supported by an NCI RO1 R01CA193220 to LJB, funds from James Graham Brown Cancer Center, University of Louisville and Kosair Pediatric Cancer Program and Molecular Targets COBRE 8P20GM103482-10 from NIH, an award from the Lung Cancer Research Foundation to LJB, an award from the Rounsavall Foundation to LJB and an Indo-U.S. fellowship from the Indian Government to S.Y. Funding from the Canadian Institutes of Health Research (CIHR) (Operating Grant to W. Lam and W. Lockwood), BC Cancer Foundation (W. Lockwood), Canadian Cancer Society Research Institute (W. Lam), a CIHR Banting an Best Graduate Scholarship (D. Rowbotham) and a Michael Smith Foundation for Health Research Scholar Award (W. Lockwood)
Contributor Information
Sanjay Yadav, Email: sanjay@iitr.res.in.
Nishant Singh, Email: Nishant.Singh@iitr.res.in.
Parag P. Shah, Email: Parag.Shah@Louisville.edu.
David A. Rowbotham, Email: Drowbotham@bccrc.ca.
Danial Malik, Email: Danial.Malik@Louisville.edu.
Ankita Srivastav, Email: AnkitaBiochem@gmail.com.
Jai Shankar, Email: Jai.Shankar@iitr.res.in.
Wan L. Lam, Email: WanLam@bccrc.ca.
William W. Lockwood, Email: Wlockwood@bccrc.ca.
Levi J. Beverly, Email: Levi.Beverly@Louisville.edu.
References
- 1.Attfield MD, Schleiff PL, Lubin JH, Blair A, Stewart PA, Vermeulen R, Coble JB, Silverman DT. The Diesel Exhaust in Miners study: a cohort mortality study with emphasis on lung cancer. J Natl Cancer Inst. 2012;104:869–883. doi: 10.1093/jnci/djs035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wichmann HE. Diesel exhaust particles. Inhal Toxicol. 2007;19(Suppl. 1):241–244. doi: 10.1080/08958370701498075. [DOI] [PubMed] [Google Scholar]
- 3.Srivastava A, Sharma A, Yadav S, Flora SJ, Dwivedi UN, Parmar D. Gene expression profiling of candidate genes in peripheral blood mononuclear cells for predicting toxicity of diesel exhaust particles. Free Radic Biol Med. 2014;67:188–194. doi: 10.1016/j.freeradbiomed.2013.10.820. [DOI] [PubMed] [Google Scholar]
- 4.Srivastava A, Yadav S, Sharma A, Dwivedi UN, Flora SJ, Parmar D. Similarities in diesel exhaust particles induced alterations in expression of cytochrome P-450 and glutathione S-transferases in rat lymphocytes and lungs. Xenobiotica. 2012;42:624–632. doi: 10.3109/00498254.2011.650732. [DOI] [PubMed] [Google Scholar]
- 5.Kipen HM, Gandhi S, Rich DQ, Ohman-Strickland P, Laumbach R, Fan ZH, Chen L, Laskin DL, Zhang J, Madura K. Acute decreases in proteasome pathway activity after inhalation of fresh diesel exhaust or secondary organic aerosol. Environ Health Perspect. 2011;119:658–663. doi: 10.1289/ehp.1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DY, Brown EJ. Ubiquilins in the crosstalk among proteolytic pathways. Biol Chem. 2012;393:441–447. doi: 10.1515/hsz-2012-0120. [DOI] [PubMed] [Google Scholar]
- 7.Beverly LJ, Lockwood WW, Shah PP, Erdjument-Bromage H, Varmus H. Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proc Natl Acad Sci U S A. 2012;109:E119–E126. doi: 10.1073/pnas.1119167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell. 2000;6:409–419. doi: 10.1016/s1097-2765(00)00040-x. [DOI] [PubMed] [Google Scholar]
- 9.Mah AL, Perry G, Smith MA, Monteiro MJ. Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol. 2000;151:847–862. doi: 10.1083/jcb.151.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiltunen M, Lu A, Thomas AV, Romano DM, Kim M, Jones PB, Xie Z, Kounnas MZ, Wagner SL, Berezovska O. Ubiquilin 1 modulates amyloid precursor protein trafficking and Abeta secretion. J Biol Chem. 2006;281:32240–32253. doi: 10.1074/jbc.M603106200. [DOI] [PubMed] [Google Scholar]
- 11.Lu A, Hiltunen M, Romano DM, Soininen H, Hyman BT, Bertram L, Tanzi RE. Effects of ubiquilin 1 on the unfolded protein response. J Mol Neurosci. 2009;38:19–30. doi: 10.1007/s12031-008-9155-6. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Lu L, Hettinger CL, Dong G, Zhang D, Rezvani K, Wang X, Wang H. Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J Neurosci. 2014;34:2813–2821. doi: 10.1523/JNEUROSCI.3541-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah PP, Lockwood WW, Saurabh K, Kurlawala Z, Shannon SP, Waigel S, Zacharias W, Beverly LJ. Ubiquilin1 represses migration and epithelial-to-mesenchymal transition of human non-small cell lung cancer cells. Oncogene. 2014 doi: 10.1038/onc.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen G, Wang X, Yu J, Varambally S, Yu J, Thomas DG, Lin MY, Vishnu P, Wang Z, Wang R. Autoantibody profiles reveal Ubiquilin 1 as a humoral immune response target in lung adenocarcinoma. Cancer Res. 2007;67:3461–3467. doi: 10.1158/0008-5472.CAN-06-4475. [DOI] [PubMed] [Google Scholar]
- 15.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 16.Singh T, Jauhari A, Pandey A, Singh P, Pant AB, Parmar D, Yadav S. Regulatory triangle of neurodegeneration, adult neurogenesis and microRNAs. CNS Neurol Disord Drug Targets. 2014;13:96–103. doi: 10.2174/18715273113126660190. [DOI] [PubMed] [Google Scholar]
- 17.Adams BD, Kasinski AL, Slack FJ. Aberrant Regulation and Function of MicroRNAs in Cancer. Curr Biol. 2014;24:R762–R776. doi: 10.1016/j.cub.2014.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouyssou JM, Manier S, Huynh D, Issa S, Roccaro AM, Ghobrial IM. Regulation of microRNAs in cancer metastasis. Biochim Biophys Acta. 2014;1845:255–265. doi: 10.1016/j.bbcan.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sivadas VP, Kannan S. The microRNA networks of TGFbeta signaling in cancer. Tumour Biol. 2014;35:2857–2869. doi: 10.1007/s13277-013-1481-9. [DOI] [PubMed] [Google Scholar]
- 20.Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 21.Tam W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene. 2001;274:157–167. doi: 10.1016/s0378-1119(01)00612-6. [DOI] [PubMed] [Google Scholar]
- 22.Liu L, Chen Q, Lai R, Wu X, Wu X, Liu F, Xu G, Ji Y. Elevated expression of mature miR-21 and miR-155 in cancerous gastric tissues from Chinese patients with gastric cancer. J Biomed Res. 2010;24:187–197. doi: 10.1016/S1674-8301(10)60028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang M, Shen H, Qiu C, Ni Y, Wang L, Dong W, Liao Y, Du J. High expression of miR-21 and miR-155 predicts recurrence and unfavourable survival in non-small cell lung cancer. Eur J Cancer. 2013;49:604–615. doi: 10.1016/j.ejca.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 24.Liu R, Liao J, Yang M, Shi Y, Peng Y, Wang Y, Pan E, Guo W, Pu Y, Yin L. Circulating miR-155 expression in plasma: a potential biomarker for early diagnosis of esophageal cancer in humans. J Toxicol Environ Health A. 2012;75:1154–1162. doi: 10.1080/15287394.2012.699856. [DOI] [PubMed] [Google Scholar]
- 25.Humphries B, Yang C. The microRNA-200 family: small molecules with novel roles in cancer development, progression and therapy. Oncotarget. 2015;6:6472–6498. doi: 10.18632/oncotarget.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hummel R, Wang T, Watson DI, Michael MZ, Van der Hoek M, Haier J, Hussey DJ. Chemotherapy-induced modification of microRNA expression in esophageal cancer. Oncol Rep. 2011;26:1011–1017. doi: 10.3892/or.2011.1381. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Hu X, Miao X, Zhu K, Cui S, Meng Q, Sun J, Wang T. MicroRNA-425-5p regulates chemoresistance in colorectal cancer cells via regulation of Programmed Cell Death 10. J Cell Mol Med. 2016;20:360–369. doi: 10.1111/jcmm.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vermeulen R, Silverman DT, Garshick E, Vlaanderen J, Portengen L, Steenland K. Exposure-response estimates for diesel engine exhaust and lung cancer mortality based on data from three occupational cohorts. Environ Health Perspect. 2014;122:172–177. doi: 10.1289/ehp.1306880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai CH, Lee CN, Bai KJ, Yang YL, Chuang KJ, Wu SM, Chuang HC. Protein oxidation and degradation caused by particulate matter. Sci Rep. 2016;6:33727. doi: 10.1038/srep33727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wan J, Diaz-Sanchez D. Antioxidant enzyme induction: a new protective approach against the adverse effects of diesel exhaust particles. Inhal Toxicol. 2007;19(Suppl. 1):177–182. doi: 10.1080/08958370701496145. [DOI] [PubMed] [Google Scholar]
- 31.Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, Sahu I, Varghese J, Wood N, Wightman M. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell. 2016;166:935–949. doi: 10.1016/j.cell.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Q, Liu T, Yuan Y, Guo Z, Xie G, Du S, Lin X, Xu Z, Liu M, Wang W. MiR-200c inhibits autophagy and enhances radiosensitivity in breast cancer cells by targeting UBQLN1. Int J Cancer. 2014 doi: 10.1002/ijc.29065. [DOI] [PubMed] [Google Scholar]
- 34.Yang F, Liu Q, Hu CM. Epstein–Barr virus-encoded LMP1 increases miR-155 expression, which promotes radioresistance of nasopharyngeal carcinoma via suppressing UBQLN1. Eur Rev Med Pharmacol Sci. 2015;19:4507–4515. [PubMed] [Google Scholar]
- 35.Han ZB, Chen HY, Fan JW, Wu JY, Tang HM, Peng ZH. Up-regulation of microRNA-155 promotes cancer cell invasion and predicts poor survival of hepatocellular carcinoma following liver transplantation. J Cancer Res Clin Oncol. 2012;138:153–161. doi: 10.1007/s00432-011-1076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang GJ, Xiao HX, Tian HP, Liu ZL, Xia SS, Zhou T. Upregulation of microRNA-155 promotes the migration and invasion of colorectal cancer cells through the regulation of claudin-1 expression. Int J Mol Med. 2013;31:1375–1380. doi: 10.3892/ijmm.2013.1348. [DOI] [PubMed] [Google Scholar]
- 37.Kong W, He L, Richards EJ, Challa S, Xu CX, Permuth-Wey J, Lancaster JM, Coppola D, Sellers TA, Djeu JY. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene. 2014;33:679–689. doi: 10.1038/onc.2012.636. [DOI] [PMC free article] [PubMed] [Google Scholar]