

Abstract
Despite its remarkable importance in the arena of drug design, serotonin 1A receptor (5-HT1A) has been elusive to the x-ray crystallography community. This lack of direct structural information not only hampers our knowledge regarding the binding modes of many popular ligands (including endogenous neurotransmitter – serotonin), but also limits the search for more potent compounds. In this paper we shed new light on the 3D pharmacological properties of the 5-HT1A receptor by using a ligand-guided approach (ALiBERO) grounded in the Internal Coordinate Mechanics (ICM) docking platform. Starting from a homology template and set of known actives, the method introduces receptor flexibility via Normal Mode Analysis and Monte Carlo sampling, to generate a subset of pockets that display enriched discrimination of actives from inactives in retrospective docking. Here, we thoroughly investigated the repercussions of using different protein templates and the effect of compound selection on screening performance. Finally, the best resulting protein models were applied prospectively in a large virtual screening campaign, in which two new active compounds were identified that were chemically distinct from those described in the literature.
For Table of Contents use only
From homology models to a set of predictive binding pockets – a 5-HT1A receptor case study.
Dawid Warszycki, Manuel Rueda, Stefan Mordalski, Kurt Kristiansen, Grzegorz Satała, Krzysztof Rataj, Zdzisław Chilmonczyk, Ingebrigt Sylte, Ruben Abagyan, Andrzej J. Bojarski
1. INTRODUCTION
Ligand-guided receptor optimization has become a crucial tool in the search for new drugs for targets with an unknown experimental structure.1–3 The rationale underlying this approach is to reshape a similar (by homology) protein structure to accommodate known ligands. Within this context, the ICM-docking community has embraced the ALiBERO method (grounded in Ligand-guided Backbone Ensemble Receptor Optimization),4 a heuristic search method that maximizes the discrimination power of actives from inactives of a subset of pockets.5
In this paper, we used ALiBERO methodology to shed new light on the computational modeling of the 5-hydroxytryptamine 1a receptor (5-HT1AR),6–24 a therapeutic target for central nervous system drugs25,26 with no x-ray structure deposited to date. Sharing similar modeling difficulties with other class-A GPCRs,1–3 here we addressed several fundamental questions that affect the final Virtual Screening (VS) performance.
On the protein side, we examined the effect of using different initial templates on different aspects of screening performance. Until recently, homology models of 5-HT1AR were constructed based on bovine rhodopsin templates,9,27–32 which were replaced in recent years by the β2AR crystal structure (2RH1) – a first choice for homology modeling.20,33,34 Recently, Cappelli et al.35 also used β1-adrenergic and A2A adenosine receptor crystal structures as templates.36,37 In the course of this investigation, the crystal structure of the 5-HT1B receptor (4IAR) was determined,38 and since then it has been used as a template for 5-HT1AR models.39–45
Because ligand-guided approaches rely on a careful selection of “seed” actives, on the ligand side we investigated: (i) impact of the selection of active compounds, (ii) importance of truly inactive compounds used in training of the VS workflow and (iii) impact of the training set composition (active/inactive compounds ratio).
Although most of the questions raised were answered by carefully designed retrospective screening experiments, the best ensemble of models was successfully applied to a prospective screening campaign of 6.4 M compounds from seven commercial databases. As demonstrated by the results, the present analysis represents another successful application of ALiBERO in recent virtual screening campaigns.46–50
2. MATERIALS AND METHODS
2.1. Homology Modeling/Receptor preparation
All models were based on two templates, the β2 adrenergic receptor (Protein Data Bank ID 2RH1, antagonist bound state – inactive conformation of the receptor) and the closest homologue (50.34% identity between whole protein sequences; 42.80% for β2R) of 5-HT1AR, serotonin 1B receptor (PDB ID 4IAR, agonist bound state – semi-active conformation) crystal structures.34,38
Receptor Preparation
Receptors were prepared using the default ICM settings.51 Protein atom types were assigned, hydrogens and missing heavy atoms were added, and zero occupancy or added side chains and polar hydrogen atoms were optimized and assigned to the lowest energy conformation. Protein atom types and parameters were taken from a modified version of the ECEPP/3 force field. The binding pocket was described by five 0.5 Å spacing potential grid maps representing van der Waals potentials for hydrogens and heavy atoms, electrostatics, hydrophobicity, and hydrogen bonding. A truncated soft van der Waals potential was introduced, and the other potentials were rescaled accordingly to avoid atom overlap.
ALiBERO runs
Altogether, 22 different ALiBERO runs were performed, resulting in 20 final different receptor ensembles (See Tables 1 and 2). For all runs, the typical ALiBERO iterative procedure was applied.5 From each initial homology model, 100 random receptor pockets were generated. All of these 100 pockets were subjected to an unrestrained flexible-ligand static-receptor VS docking, that was repeated 3 times to account for ICM-VS intrinsic stochasticity. After the docking, up to 5 pockets (we set the maximum number of complementary pockets to 5) for which more than 75% of the active compounds formed charge-assisted H-bond with Asp116 (3.32 according to Ballesteros-Weinstein notation)52 that at the same time improved “uphill” NSQ_AUC values and the average docking score for the best half of the actives, (i.e., “nsaplus” fitness function in ALiBERO) were selected for additional side chain refinement. These pockets underwent to a round of Monte Carlo-based refinement together with the 3 top-scoring ligands, allowing for side chain flexibility. During the refinement step, a receptor-ligand distance restraint was imposed between the Asp116(3.32) oxygen(s) and a N+ in the ligand. After the refinement step, unrestrained VS were again performed, and the same selection criteria (see above) was applied. The best performing ensemble is then passed to the next generation. This iterative process was repeated 10 times (typically the best ensembles were generated in the first five repeats) to ensure that successive iterations no longer improved the results.
Table 1.
Evaluation of binding pocket ensembles developed for active compounds selected using different clustering approaches, templates, actives/inactives ratio and repeatability of ALiBERO runs.
| Run no. | Clustering method | Training set composition | Template | AUC | NSQ_AUC | Average NSQ_AUCa | ||
|---|---|---|---|---|---|---|---|---|
| Total | Actives | Inactives | ||||||
| 1 * | Manual | 54 | 27 | 27 | 4IAR | 0.851 | 0.675 | 0.657 (0.018) |
| 2 | M2D | 70 | 35 | 35 | 4IAR | 0.825 | 0.613 | 0.592 (0.026) |
| 3 | ICM | 56 | 28 | 28 | 4IAR | 0.796 | 0.554 | 0.526 (0.023) |
| 4 | Manual | 54 | 27 | 27 | 2RH1 | 0.826 | 0.625 | 0.600 (0.016) |
| 5 | M2D | 70 | 35 | 35 | 2RH1 | 0.861 | 0.666 | 0.629 (0.026) |
| 6 | ICM | 56 | 28 | 28 | 2RH1 | 0.843 | 0.646 | 0.628 (0.018) |
| 7 | Manual | 96 | 27 | 69 | 4IAR | 0.809 | 0.603 | 0.558 (0.026) |
| 8 | M2D | 104 | 35 | 69 | 4IAR | 0.824 | 0.610 | 0.572 (0.032) |
| 9 | ICM | 97 | 28 | 69 | 4IAR | 0.852 | 0.653 | 0.623 (0.017) |
| 10 | Manual | 96 | 27 | 69 | 2RH1 | 0.809 | 0.559 | 0.528 (0.027) |
| 11 | M2D | 104 | 35 | 69 | 2RH1 | 0.852 | 0.651 | 0.618 (0.033) |
| 12 | ICM | 97 | 28 | 69 | 2RH1 | 0.804 | 0.542 | 0.542 (0.027) |
| 13 | Manual | 54 | 27 | 27 | 4IAR | 0.828 | 0.633 | 0.610 (0.022) |
| 14 | Manual | 54 | 27 | 27 | 4IAR | 0.851 | 0.670 | 0.670 (0.038) |
The average NSQ_AUC value was calculated for all uphill generations of pockets within the run. The standard deviation is indicated in brackets.
Table 2.
Results for ensembles developed using raw and pre-optimized starting models.
| Run no. | Clustering method | Training set composition | Template | AUC | NSQ_AUC | Average NSQ_AUCa | ||
|---|---|---|---|---|---|---|---|---|
| Total | Actives | Inactives | ||||||
| 15 | – | – | – | – | 4IARb | 0.716 | 0.402 | – |
| 16 | – | – | – | – | 2HR1b | 0.682 | 0.308 | – |
| 17 | – | – | – | – | 4IARc | 0.775 | 0.541 | – |
| 18 | – | – | – | – | 2RH1c | 0.829 | 0.605 | – |
| 19 | Manual | 54 | 27 | 27 | 4IARb | 0.815 | 0.572 | 0.545 (0.016) |
| 20 | ICM | 97 | 28 | 69 | 4IARb | 0.823 | 0.596 | 0.559 (0.037) |
| 21 | Manual | 54 | 27 | 27 | 2RH1b | ALiBERO failedd | ||
| 22 | ICM | 97 | 28 | 69 | 2RH1b | ALiBERO failedd | ||
The average NSQ_AUC value was calculated for all uphill generation of pockets within the run. The standard deviation is indicated in brackets.
Raw models.
Optimized models.
The criterion of 75% of actives forming charge-assisted H-bond with Asp3.32 was not achieved (in fact even less than 50% of actives formed this interaction).
Single models
In parallel to the ALiBERO approach, single models on both templates were also prepared in ICM (runs 15 and 16). Since VS docking of known binders was problematic and only for a few ligands interactions with Asp116(3.32) were observed, these models were further optimized. At first, Asn386(7.39) and Tyr390(7.43) side chains were substituted with alanine, and a set of arylpiperazine ligands were docked into this alanine-mutated model. Next, the Ala386(7.39)Asn and Ala390(7.43)Tyr were mutated back to the wild type sequence, and the 10 top-scored LR complexes of a highly active arylpiperazine derivative NBUMP (4-[4-(1-noradamantanecarboxamido)butyl]-1-(2-methoxyphenyl)piperazine, (Ki = 0.1 nM))53 were refined by applying the refinement option in ICM-Pro and used in runs 17 and 18. As the optimization protocol uses potent ligand as a guide for receptor model refinement, it resembles both an induced fit docking approach and a ligand-guided model selection first proposed by Evers et al.54
2.2. Ligand Selection and Preparation
2.2.1. Training sets
All compounds with measured activity towards 5-HT1AR were fetched from the ChEMBL database40 utilizing a previously described approach.23 The ligands were defined as active when their binding constant was lower than or equal to 100 nM; the threshold of inactivity was set at 1000 nM. Finally, the resulting sets consisted of 3616 active (due to sparse data on agonistic/antagonistic properties available in ChEMBL database, functional profile was not considered) and 438 inactive (decoy) compounds. The full chemical space of 5-HT1A ligands was investigated using three clustering methods: manual, MOLPRINT 2D fingerprint based (M2D) and ICM clustering. The manual and M2D approaches have been described previously.56,57 M2D clustering was performed using the Hierarchical Clustering tool in Canvas under default settings.58,59 For all clusters generated, only centroids were used in the experiments (27 from manual clustering, 35 from M2D and 28 from ICM clustering). All inactives were also clustered in Canvas (using the Kelly criterion),60 resulting in 69 non-singleton clusters. From this set, depending on the experiment, 27, 28, 35, or the full set of 69 cluster centroids were chosen to compose the training set of inactives. If the number of inactives needed to compose such a set was less than 69, the compounds were selected using the diversity-based selection tool in Canvas (similarity metric – Soergel distance; compound selection algorithm – sphere exclusion; sphere size – 0.5; initialization – random with random seed).
2.2.2. Test sets
Each scenario was evaluated in the same retrospective screening experiment with 100 diverse 5-HT1AR ligands (centroids of non-singleton clusters) that were not used in the training set and 900 DUD-like decoys selected from the ZINC database using an in-house script.61,62 All DUD-like decoys possessed protonable nitrogen – the most characteristic structural feature of aminergic GPCR ligands.
An additional set of 40 5-HT1A receptor ligands (arylpiperazines, indoles and tetralines; see Supplementary Information) with a putative binding mode proposed in the literature27–30,33,35,63–78 were docked to the top-scoring ensemble of pockets. The binding modes were analyzed using SIFt methodology.79,80
All compounds were ionized at pH=7.4, and all possible tautomers were generated prior to docking.
2.3. ICM Docking
Each docking experiment was performed as a standard ICM-VS docking procedure. ICM ligand docking uses biased probability Monte Carlo (BPMC) optimization of the ligand internal variables in the set of grid potential maps of the receptor.34 Flexible ligands are automatically placed into the binding pocket in several random orientations used as starting points for Monte Carlo optimization. The optimized energy function includes the ligand internal strain and a weighted sum of the grid map values in the ligand atom centers. The ligand binding poses were evaluated with an all-atom ICM ligand binding score that had been derived from a multi-receptor screening benchmark as a compromise between the approximated Gibbs free energy of binding and numerical errors.35 To improve the convergence of the docking predictions, three independent runs of the docking procedure were performed. In total, every ALiBERO run comprises 10 gen × 100 models × 3 repetitions = 3000 independent VS. All calculations were performed at our local cluster (mostly Intel Xeon X3370 3.00GHz) located at the San Diego Supercomputer Center. Screened compounds scores from individual pockets were merged, numerically sorted, and only the best score was kept to compute discrimination of active compounds from inactives/decoys.81,82 Such discrimination is quantified by AUC – the area under the receiver operating characteristics (ROC) curve. This parameter characterizes the cumulative ability of the docking protocol to correct the classification of instances. The AUC ranges from 1.0 (perfect performance) to 0.0 (inverse classification), whereas 0.5 indicates random activity class assignment. Recently, the Normalized Square root AUC (NSQ_AUC) metric was introduced,83 which is especially sensitive for early hit enrichment. This parameter utilizes the effective area under the curve (AUC*) which is defined for the ROC curve plotted with the abscissa coordinate calculated as the square root of the false positive rate (ratio between number of inactives classified as actives and all inactives). NSQ_AUC values range from 1.0 (perfect separation) to 0.0 (random selection). The NSQ_AUC is calculated as follows:
2.4. In vitro pharmacology
2.4.1. Cell culture and preparation of cell membranes
HEK293 cells stably expressing human 5-HT1AR, 5-HT2AR, 5-HT6R or 5-HT7bR (prepared using Lipofectamine 2000) were maintained at 37 °C in a humidified atmosphere with 5% CO2 and grown in Dulbecco’s Modified Eagle’s Medium containing 10% dialyzed fetal bovine serum and 500 μg/ml G418 sulfate. For membrane preparations, the cells were subcultured in 10-cm-diameter dishes, grown to 90% confluence, washed twice with phosphate-buffered saline (PBS), pre-warmed to 37 °C and pelleted by centrifugation (200 g) in PBS containing 0.1 mM EDTA and 1 mM dithiothreitol. Prior to the membrane preparations, the pellets were stored at −80 °C.
2.4.2. Radioligand binding assays
Cell pellets were thawed and homogenized in 20 volumes of assay buffer using an Ultra Turrax tissue homogenizer and centrifuged twice at 35000 g for 20 min at 4 °C, with incubation for 15 min at 37 °C in between rounds of centrifugation. The composition of the assay buffers was as follows: for 5-HT1AR: 50 mM Tris–HCl, 0.1 mM EDTA, 4 mM MgCl2, 10 μM pargyline and 0.1% ascorbate; for 5-HT2AR: 50 mM Tris–HCl, 0.1% ascorbate, 4 mMCaCl2; for 5-HT6R: 50 mM Tris–HCl, 0.5 mM EDTA and 4 mM MgCl2; for 5-HT7bR: 50 mM Tris–HCl, 4 mM MgCl2, 10 μM pargyline and 0.1% ascorbate.
All assays were incubated in a total volume of 200 μl in 96-well microliter plates for 1 h at 37 °C, except for 5-HT1AR, which was incubated at room temperature for 1 h. The process of equilibration was terminated by rapid filtration through Unifilter plates with a 96-well cell harvester, and the radioactivity retained on the filters was quantified on a Microbeta plate reader.
For the displacement studies, the assay samples contained the following as radioligands: 1.5 nM [3H]-8-OH-DPAT (187 Ci/mmol) for 5-HT1AR; 1.0 nM [3H]-Ketanserin (52 Ci/mmol) for 5-HT2AR; 2 nM [3H]-LSD (85.2 Ci/mmol for 5-HT6R; 0.6 nM [3H]-5-CT (39.2 Ci/mmol) for 5-HT7bR.
Non-specific binding was defined with 10 μM 5-HT in the 5-HT1AR and 5-HT7bR binding experiments, whereas 10 μM methiothepin and 1 μM (+)butaclamol were used for 5-HT6R, and 100 μM mianserin for5-HT2AR binding. The purchased compounds were initially screened using two compound concentrations: 10−6 and 10−7 M. The active compounds were then tested in triplicate at 7 different concentrations (10−11–10−4 M). The inhibition constants (Ki) were calculated from the Cheng-Prusoff equation.84 The results are expressed as the means of at least two separate experiments.
3. RESULTS AND DISCUSSION
3.1 Impact of the training set and crystal template
Three different approaches to active compounds selection based on different clustering methods (manual, M2D and ICM) and two different templates for pockets generation were applied with actives/inactives ratio set at 1:1. Inactives were selected from a set of 69 centroids of inactives as the most diverse. The outcomes of the ALiBERO models obtained with different sets of active compounds as well as different crystal templates for a homology modeling step were evaluated in retrospective screening experiments with DUD-like decoys. The overall performance of the runs confirmed the value of the ALiBERO approach, which reached ~0.8 AUC and ~0.6 NSQ_AUC, being significantly better than random classification (Table 1, runs 1–6). The spread of the obtained screening parameters varied from 6% for AUC to 18% when using NSQ_AUC. Among the three clustering methods, M2D and manual were the most useful, yet the difference varied between runs and for different templates. In addition, VS data do not demonstrate the superiority of either of the templates used – both top and bottom ranked complexes were derived from the 5-HT1BR template.
3.2 Impact of the training set composition
These experiments were aimed to evaluate the impact of the ratio between varying subsets of active compounds and full representation of inactives on the retrospective screening performance. The performed runs (Table 1, runs 1–12) differed in terms of the number of active compounds used, whereas the inactives encompassed the whole chemical space of known low affinity to non-binders. The obtained average NSQ_AUC values ranged from 0.526 to 0.657. Typically, models trained on actives/inactives ratio of 1 outperformed those developed using the complete set of inactives. The only exception was observed for the approach utilizing ICM clustering and the 5-HT1B template (4IAR), which reached one of the highest values of NSQ_AUC. Within runs 7–12, the β2AR template (2RH1) was more useful even when the manual clustering procedure was applied. Among the clustering approaches it was again hard to choose the best one – the best and the worst approach was based on the same method of grouping compounds – (ICM clustering).
Because ALiBERO is a heuristic algorithm, the best run achieved (Run 1) was repeated two more times to test the repeatability of the results and to evaluate the variability of the average NSQ values. The results varied from 0.610 to 0.670, with the initial value in between (Table 1 Runs 13–14).
3.4 Raw and pre-optimized input models vs ALiBERO ensembles
These experiments were designed to compare raw 5-HT1AR models (runs 15, 16, two templates applied) and those optimized with NBUMP (a compound with subnanomolar (Ki = 0.1 nM) affinity toward the target,53 runs 17, 18) with ALiBERO-generated pocket ensembles, both of which are described above (runs 1–14), and those obtained for raw templates (runs 19, 20 and 21, 22, respectively; Table 2). The results demonstrated the superiority, in most cases, of the ALiBERO ensembles. Because they are random samples of a conformational space in the receptor, the raw models (15, 16) did not provide optimal discriminating efficiency. The results showed, however, that pre-optimized models (run 17) could compete with the ALiBERO ensembles (e.g., runs 3, 7, 10 and 12, Table 1). It is also worth noting that, in case of pre-optimized models, the β2AR template resulted in a better model than the ALiBERO method, yet the model was not suitable input for pocket generation because it led to subsequent algorithm failure (less than 75% of the active compounds formed charge-assisted H-bond with Asp3.32).
3.5 Binding mode and conformational space of the pockets
A set of 40 5-HT1A receptor ligands (arylpiperazines, indoles and tetralines) with putative binding modes proposed in the literature was docked to the top-scoring ensemble of pockets (Run 1). The binding modes were analyzed using Structural Interaction Fingerprints (SIFt) profiles methodology.79,80 All interactions present in less than 50% of the complexes were discarded.
SIFt profiles were constructed for both training and literature sets, first to evaluate the consistency of ligand-receptor interactions, and second, to compare the obtained complexes with the data in the literature. The results revealed a high level of consistency for the obtained binding modes, however, not all interactions described in the literature were found (e.g., with T5.39, Table 3 and Figure 1).
Table 3.
A comparison between binding modes determined from ALiBERO runs and literature data (see Supporting Information for detailed data about reference ligands interacting with particular residue).
| Residue positiona | Frequency of contacts (training)b | Frequency of contacts (literature)c | Specific interactiond | Interaction from literature data |
|---|---|---|---|---|
| A932.60x60 | 0.70 | 0.70 | Hydrophobic | vdW27,64 |
| Y962.63x63 | 0.80 | 0.88 | Aromatic | vdW64 AFE64 |
| Q972.64x64 | 0.74 | 0.78 | Polar | vdW27 |
| N100 ecl1 | <0.50 | 0.53 | Polar | |
| F1123.28x28 | 0.89 | 0.85 | Aromatic | AFE28,74,77 AFF76 vdW27,72 |
| D1163.32x32 | 1.00 | 0.97 | H-bond | H-bond9,27,28,29,33,35,63, 64,66,67,70,72,73,74,76,77,78 |
| V1173.33x33 | 0.96 | 0.97 | Hydrophobic | vdW27,63,64,70,74,77 |
| C1203.36x36 | 0.93 | 0.95 | Hydrophobic | vdW9,63,64,70,72 H-bond63 |
| T1213.37x37 | 0.85 | 0.93 | Polar | H-bond9,64,67,78 |
| I1243.40x40 | 0.67 | 0.78 | Hydrophobic | |
| I1674.56x56 | 0.65 | 0.53 | Hydrophobic | |
| C187 ecl2 | 0.56 | <0.50 | Polar | vdW70 |
| T188 ecl2 | 0.65 | 0.53 | Polar | vdW63,64 |
| I189 ecl2 | 0.93 | 0.95 | Hydrophobic | vdW63,64 |
| S1995.43x43 | 0.80 | 0.93 | Polar | H_bond27,28,33,66,67,76,78 vdW9,27, 63,73,74 |
| T2005.44x44 | 0.78 | 0.90 | Polar | vdW27,63,74 H-bond28,66,67,78 |
| A2035.46x461 | 0.87 | 0.95 | Hydrophobic | vdW63,64,74 |
| W3586.48x48 | 0.98 | 0.90 | Aromatic | AFE28,64 H-bond28 vdW63,72 |
| F3616.51x51 | 0.98 | 0.97 | Aromatic | AFE28,29,63,64,70,74,77 vdW72 |
| F3626.52x52 | 0.93 | 0.97 | Aromatic | AFE9,27,33,35,63,64,73,74,76 |
| A3837.35x35 | 0.59 | 0.68 | Hydrophobic | |
| N3867.38x38 | 1.00 | 0.93 | Polar | vdW27,35,70,72,73,74,77 H-bond9,27,64,67 |
| W3877.39x39 | 0.56 | 0.53 | Aromatic | AFE28 H-bond9,66,76 |
| Y3907.41x41 | 1.00 | 0.97 | Aromatic | vdW27,63,70,72,74,77 H-bond9,64,76 AFE28,29 |
Residue positions use sequence numbers and GPCRdb generic numbers in superscript.85 Some positions from publications did not match the current GPCRdb positions; in such cases, the latter were used (see Discussion).
The most frequently interacting residues for the training set.
The most frequently interacting residues for the literature set.
Specific contacts from the literature.
Figure 1.
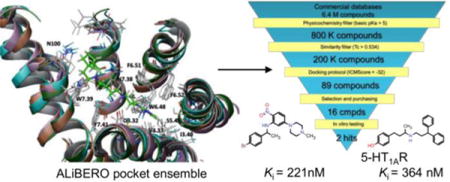
SIFt profile generated for the training set docking results. Residue colors correspond to dominating interactions (Table 3.): blue – polar, green – hydrophobic, orange – aromatic.
Serotonin, a representative of indole-like ligands was docked in accordance to the binding mode described by Seeber and interacted with TM3, TM5 and TM6.27 A strong, charge-assisted hydrogen bond was formed between the NH3+ group and D3.32. The OH group interacted with T5.39; however, contacts with S5.42 were also detected. The indole moiety formed face-to-edge stacking with F6.52. Buspirone, a member of a vast group of arylpiperazines, was docked in a similar way to the pose proposed by Bronowska and Sylte (Figure 2).74,77 The protonated nitrogen atom created a charge-assisted hydrogen bond with D3.32. The pyrimidine moiety was located between TM5 and TM6, but the interactions with TM6 were weak. The azaspirone part was more directed toward TM7 than described in previously published complexes, forming a hydrogen bond with N7.39. The 8-OH-DPAT, a tetraline-containing ligand, was docked in a pose rather similar to that proposed by Sylte than the binding mode described by Seeber.27,74 The tetralin moiety was perpendicular to the membrane surface and formed a face-to-edge stacking interaction with the aromatic cluster from TM6. The OH group interacted with TM5 (S5.42). The protonated nitrogen atom created a charge-assisted hydrogen bond with D3.32, whereas the n-propyl chains had contacts with Y7.43 and EL2.
Figure 2.
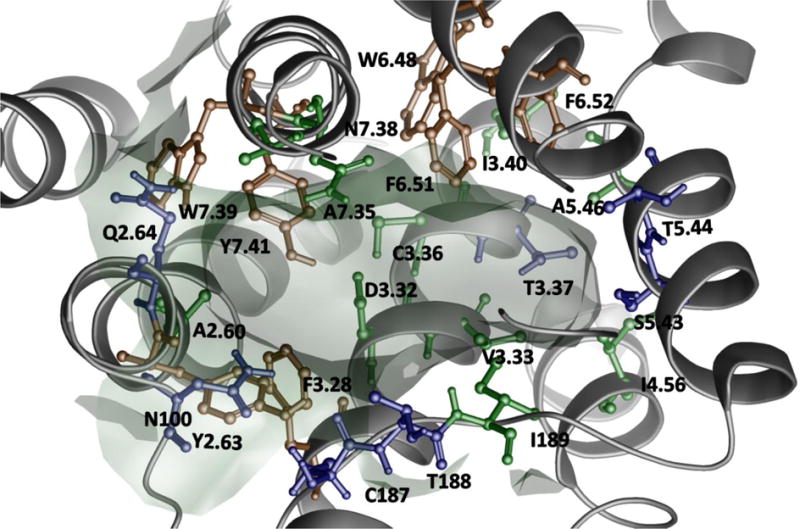
Binding pose of buspirone in the ensemble of the best models of 5-HT1AR (run 1). The compound is rendered as a ball and stick representation. A solid, transparent compound surface was generated. Only residues situated less than 4Å from the partial agonist are shown.
3.7 Virtual screening
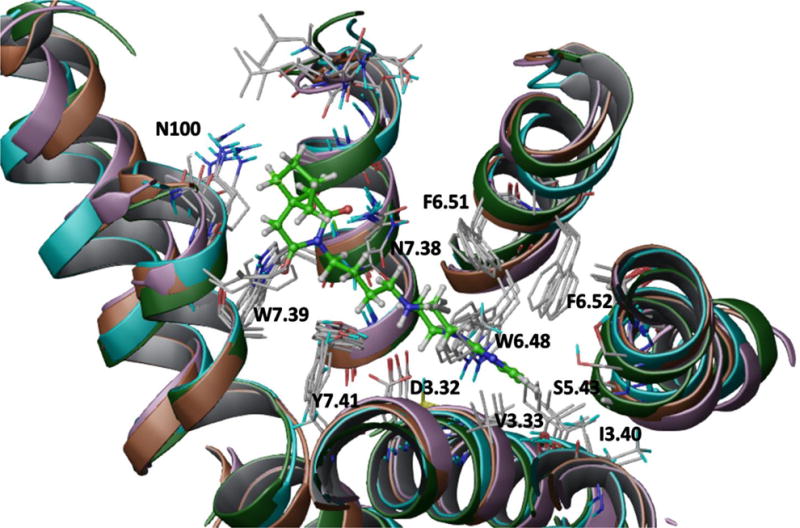
Seven commercial databases (ChemBridge, ChemDiv, Enamine, Maybridge, Specs, UORSY, VitasM) containing approximately 6.4 M compounds were utilized for the virtual screening campaign. All compounds were ionized at pH=7.4, and all possible tautomers were generated. The applied protocol (Figure 3) consisted of the following three filters: physicochemical, similarity to known 5-HT1AR ligands and docking protocol.
Figure 3.
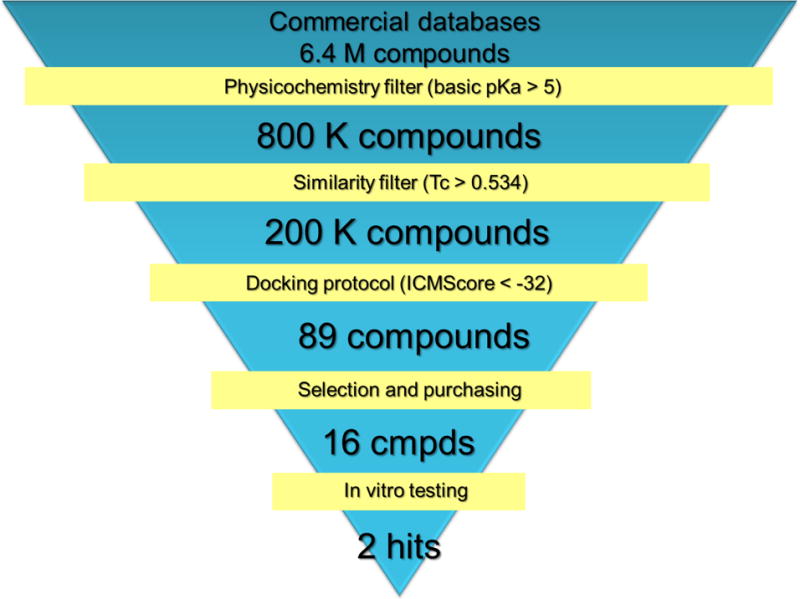
Workflow for the virtual screening protocol. The best ensemble derived from run 1 (Table 1) was used for docking.
First, the criterion of the strongest basic pKa >5 was applied (Calculator Plugins, JChem)86 which narrowed down the number of ligands to 800 K. Removal of 75% of the most dissimilar compounds to any known 5-HT1A receptor ligands (stored in ChEMBL) resulted in 200 K structures with a Tanimoto similarity metric greater than 0.534. After generation of the 3D structures (Ligprep, Schrödinger)87 for the remaining 200 K compounds, they were docked to the best models ensemble (Run 1). Eighty nine compounds with an ICMscore <−32 were clustered (Hierarchical Clustering Tool, Canvas)58 resulting in 15 groups (including a doubleton and two singletons). The clusters were evaluated by the team members to select the structures with the most diverse and novel chemotypes for biological investigation (for this reason singletons, doubleton and three clusters of common 5-HT1AR scaffolds were excluded). Finally, 16 compounds, covering 89% of the clustered structures, were purchased and biologically evaluated (Table 4.). Among them, two compounds (6216810 and 5464140) showed significant affinity for 5-HT1A receptor (Ki = 221 and 364 nM, respectively). Moreover, compound 6216810 is a dual ligand that acts also on the 5-HT6 receptor (Ki = 37 nM). Among the remaining 14 structures, one strong 5-HT2AR binder was identified (39866030, Ki = 21 nM).
Table 4.
Affinities of the hits from the virtual screening cascade.
| Compound ID | Chemical structure | Ki [nM] | ICMscore | |||
|---|---|---|---|---|---|---|
| 5-HT1A | 5-HT2A | 5-HT6 | 5-HT7 | |||
| 5464140a |
|
221 | 2850 | 1326 | 380 | −33.68 |
| 6216810a |
|
364 | 1853 | 37 | 878 | −33.74 |
| 26560725a |
|
1498 | 5518 | 4842 | 21650 | −34.08 |
| 18774467a |
|
2704 | 21070 | 8739 | 16520 | −37.81 |
| 18136999a |
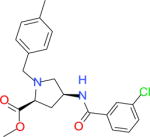
|
3243 | 7033 | 11220 | 10390 | −35.25 |
| 12438168a |
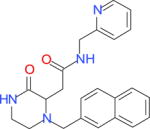
|
4291 | 21230 | 11270 | 19260 | −35.01 |
| G370-1604b |
|
6099 | 10560 | 10480 | 7567 | −33.55 |
| 39866030a |
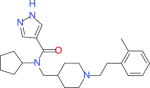
|
7732 | 21 | 6147 | 484 | −32.41 |
| 66929343a |
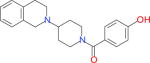
|
9964 | 2206 | 3793 | 2783 | −37.00 |
| G500-0869b |
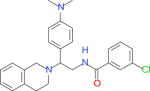
|
11760 | 23360 | 10170 | 2665 | −33.75 |
| 38222805a |
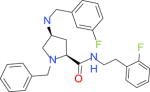
|
15000 | 2241 | 6696 | 3870 | −36.23 |
| 56468062a |
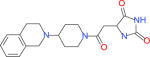
|
17440 | 16340 | 3131 | 7204 | −38.94 |
| D174-0581b |
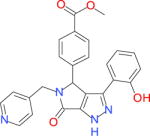
|
17750 | 3755 | 13520 | 6476 | −34.47 |
| E787-1093b |
|
19410 | 19270 | 4111 | 20630 | −33.74 |
| 17132328a |
|
22260 | 8544 | 17010 | 17700 | −35.28 |
| 9228210a |
|
23950 | 18610 | 14470 | 833 | −35.95 |
Chembridge database id,
Chemdiv database id
4. CONCLUSIONS
As mentioned above, ALiBERO is a robust and convenient workflow for structure-based drug design.5 The algorithm addresses the issue of insufficient conformational space of a single homology model or a crystal structure leading to VS that is either insufficiently or excessively strict in terms of active/inactive discrimination.40 The present findings show, that an ensemble of binding pockets provides conformational pseudoflexibility to accommodate multiple distinct classes of active compounds that is superior to single models.
In addition, a number of different factors affecting the screening performance of ALiBERO were evaluated, including the composition of the training and test sets and the template selection. The results demonstrate that the composition of the training set is of great importance. The optimal actives to inactives ratio value of 1 indicates the significance of the difference between docking and method like machine learning, in which unbalanced sets generated the best models.88
The choice of the template does not have as much of an impact as in other methods,40 the differences in VS performances do not allow to point one of significant preference. The optimization of the structure and adaptation to the training compounds, underlying ALiBERO algorithm, allows neglecting the spatial orientation of the different crystal structures.
The ensembles of pockets obtained using this method are quite diverse (RMSD between conformations varied from 1.36 to 1.6 Å2, with maximum displacement of atoms of 5 Å), yet the binding modes were very consistent between the training and literature sets. The poses selected for each “classical” compound corresponded to the literature data and previously published binding modes.
The best ensemble of receptors was used as a final filter in a prospective virtual screening campaign picking up two active compounds. These novel structures (the Tanimoto similarity coefficient to any compound with defined 5-HT1AR affinity is 0.73 for 5464140 and 0.56 for 6216810 are a good foundation for further optimization. It is also worth noting that similarity of the found hits to ligands of other serotonin receptors was relatively low (Tc ≤ 0.69), except high similarity of 6216810 (Tc ≤ 0.91) to 1-methyl-4-(4-nitrophenyl)piperazines reported by Tasler et al. as 5-HT6R ligands.89 Indeed, compound 6216810 displayed high 5-HT6R affinity (Ki = 37 nM), so it could be classified as a dual 5-HT1A/5-HT6R ligand. Moreover, one of the tested compounds (39866030) is a potent 5-HT2AR ligand (Ki = 21 nM) with the new scaffold (Tanimoto similarity coefficient to any known 5-HT2AR binder is 0.5). It has to be stressed, that none of those three 5-HTR ligands were classified as PAINS, and only one of the tested VS hits (G500-0869) was recognized as potential PAINS.90,91
Given the above findings, ALiBERO bears great potential as a universal structure-based design tool. The repeatability of runs and immunity to template selection for homology models renders this all-in-one workflow capable of returning viable VS hits.
Supplementary Material
Acknowledgments
This study was partially supported by the Polish-Norwegian Research Programme operated by the National Centre for Research and Development under the Norwegian Financial Mechanism 2009–2014 in the frame of Project PLATFORMex (Pol-Nor/198887/73/2013) and by the NIH grant 1-R01-GM074832.
Footnotes
Supporting Information. The 2D structures of 5-HT1AR ligands for which binding poses have been determined in the literature. Extended version of Table 3 enhanced by reference ligands interacting with particular residues. Extended version of Table 4 supplemented with the most similar known 5-HT1AR ligand along with Tanimoto similarity coefficient to the respective hits.
References
- 1.Michino M, Abola E, Brooks CL, Dixon JS, Moult J, Stevens RC. Community-Wide Assessment of GPCR Structure Modelling and Ligand Docking: GPCR Dock 2008. Nat Rev Drug Discov. 2009;8:455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R, Yoshikawa Y, Furuya T, Lee H, Roy A, Grime J, Rebehmed J, Zhang Y, Roumen L, de Esch IJP, Leurs R, de Graaf C, Li Y, Hou T, Mysinger MM, Weiss DR, Irwin JJ, Shoichet BK, McRobb FM, Capuano B, Crosby IT, Chalmers DK, Yuriev E, Wang Q, Mach RH, Reichert DE, Chuang GY, Rognan D, Simms J, Sexton P, Wootten D, Latek D, Ghoshdastider U, Filipek S, LenServer, Kirkpatrick A, Trzaskowski B, Griffith A, Kim SK, Abrol R, Goddard WA, Vaidehi N, Lam A, Bhattacharya S, Li H, Balaraman G, Niesen M, Pal S, Solovyev V, Vorobjev Y, Bakulina N, Beuming T, Costanzi S, Shi L, Higgs C, Salam N, Lupyan D, Sherman W, Ding F, Kota P, Ramachandran S, Dokholyan NV, Carlsson J, Coleman RG, Fan H, Schlessinger A, Irwin JJ, Sali A, Shoichet BK, Tikhonova I, Pogozheva I, Lomize A, Hall NE, Muddassa M, Zhang Y, Nim Pae A, Lee J, Lopez L, Obiol-Pardo C, Selent J, Mahboob S, Werner T, Bret Church W, Brylinski M, Ando T, Guerler A, Zhou H, Skolnick J, Xhaard H, Jurkowski W, Elofsson A, Murad AK, Drwal M, Dupree TB, Griffith R, Ostopovici-Halip L, Bologa C, Chen KM, Sun J, Barth P, Yarov-Yarovoy V, Baker D, Vroling B, Sanders MPA, Nabuurs SB, Nikiforovich G. Status of GPCR Modeling and Docking as Reflected by Community-Wide GPCR Dock 2010 Assessment. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kufareva I, Katritch V, Stevens RC, Abagyan R. Advances in GPCR Modeling Evaluated by the GPCR Dock 2013 Assessment: Meeting New Challenges. Structure. 2014;22:1120–1139. doi: 10.1016/j.str.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katritch V, Rueda M, Abagyan R. Ligand-Guided Receptor Optimization. Methods Mol Biol. 2012;857:189–205. doi: 10.1007/978-1-61779-588-6_8. [DOI] [PubMed] [Google Scholar]
- 5.Rueda M, Totrov M, Abagyan R. ALiBERO: Evolving a Team of Complementary Pocket Conformations rather than a Single Leader. J Chem Inf Model. 2012;52:2705–2714. doi: 10.1021/ci3001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paluchowska MH, Mokrosz MJ, Bojarski A, Wesołowska A, Borycz J, Charakchieva-Minol S, Chojnacka-Wójcik E. On the Bioactive Conformation of NAN-190 (1) and MP3022 (2), 5-HT(1A) Receptor Antagonists. J Med Chem. 1999;42:4952–4960. doi: 10.1021/jm991045h. [DOI] [PubMed] [Google Scholar]
- 7.Bojarski AJ, Paluchowska MH, Duszyńska B, Kłodzińska A, Tatarczyńska E, Chojnacka-Wójcik E. 1-Aryl-4-(4-Succinimidobutyl)piperazines and Their Conformationally Constrained Analogues: Synthesis, Binding to Serotonin (5-HT1A, 5-HT2A, 5-HT7), alpha1-Adrenergic, and Dopaminergic D2 Receptors, and in Vivo 5-HT1A Functional Characteristics. Bioorg Med Chem. 2005;13:2293–2303. doi: 10.1016/j.bmc.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 8.Paluchowska MH, Bojarski AJ, Charakchieva-Minol S, Wesołowska A. Active Conformation of Some Arylpiperazine Postsynaptic 5-HT(1A) Receptor Antagonists. Eur J Med Chem. 2002;37:273–283. doi: 10.1016/s0223-5234(01)01312-5. [DOI] [PubMed] [Google Scholar]
- 9.Nowak M, Kolaczkowski M, Pawlowski M, Bojarski AJ. Homology Modeling of the Serotonin 5-HT(1A) Receptor Using Automated Docking of Bioactive Compounds with Defined Geometry. J Med Chem. 2006;49:205–214. doi: 10.1021/jm050826h. [DOI] [PubMed] [Google Scholar]
- 10.Hibert MF, Gittos MW, Middlemiss DN, Mir aK, Fozard JR. Graphics Computer-Aided Receptor Mapping as a Predictive Tool for Drug Design: Development of Potent, Selective, and Stereospecific Ligands for the 5-HT1A Receptor. J Med Chem. 1988;31:1087–1093. doi: 10.1021/jm00401a007. [DOI] [PubMed] [Google Scholar]
- 11.Mellin C, Vallgårda J, Nelson DL, Björk L, Yu H, Andén NE, Csöregh I, Arvidsson LE, Hacksell U. A 3-D Model for 5-HT1A-Receptor Agonists Based on Stereoselective Methyl-Substituted and Conformationally Restricted Analogues of 8-Hydroxy-2-(Dipropylamino)tetralin. J Med Chem. 1991;34:497–510. doi: 10.1021/jm00106a004. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal A, Pearson PP, Taylor EW, Li HB, Dahlgren T, Herslof M, Yang Y, Lambert G, Nelson DL. Three-Dimensional Quantitative Structure-Activity Relationships of 5-HT Receptor Binding Data for Tetrahydropyridinylindole Derivatives: A Comparison of the Hansch and CoMFA Methods. J Med Chem. 1993;36:4006–4014. doi: 10.1021/jm00077a003. [DOI] [PubMed] [Google Scholar]
- 13.Orús L, Pérez-Silanes S, Oficialdegui A-M, Martínez-Esparza J, Del Castillo J-C, Mourelle M, Langer T, Guccione S, Donzella G, Krovat EM, Poptodorov K, Lasheras B, Ballaz S, Hervías I, Tordera R, Del Río J, Monge A. Synthesis and Molecular Modeling of New 1-Aryl-3-[4-Arylpiperazin-1-Yl]-1-Propane Derivatives with High Affinity at the Serotonin Transporter and at 5-HT(1A) Receptors. J Med Chem. 2002;45:4128–4139. doi: 10.1021/jm0111200. [DOI] [PubMed] [Google Scholar]
- 14.Sleight AJ, Peroutka SJ. Identification of 5-hydroxytryptamine1A Receptor Agents Using a Composite Pharmacophore Analysis and Chemical Database Screening. Naunyn Schmiedebergs Arch Pharmacol. 1991;343:109–116. doi: 10.1007/BF00168596. [DOI] [PubMed] [Google Scholar]
- 15.Chidester CG, Lin CH, Lahti RA, Haadsma-Svensson SR, Smith MW. Comparison of 5-HT1A and Dopamine D2 Pharmacophores. X-Ray Structures and Affinities of Conformationally Constrained Ligands. J Med Chem. 1993;36:1301–1315. doi: 10.1021/jm00062a001. [DOI] [PubMed] [Google Scholar]
- 16.Mokrosz MJ, Duszynska B, Bojarski AJ, Mokrosz JL. Structure-Activity Relationship Studies of CNS Agents–XVII. Spiro[piperidine-4′,1-(1,2,3,4-Tetrahydro-Beta-Carboline)] as a Probe Defining the Extended Topographic Model of 5-HT1A Receptors. Bioorg Med Chem. 1995;3:533–538. doi: 10.1016/0968-0896(95)00039-j. [DOI] [PubMed] [Google Scholar]
- 17.Langlois M, Brémont B, Rousselle D, Gaudy F. Structural Analysis by the Comparative Molecular Field Analysis Method of the Affinity of Beta-Adrenoreceptor Blocking Agents for 5-HT1A and 5-HT1B Receptors. Eur J Pharmacol. 1993;244:77–87. doi: 10.1016/0922-4106(93)90061-d. [DOI] [PubMed] [Google Scholar]
- 18.van Steen BJ, van Wijngaarden I, Tulp MT, Soudijn W. Structure-Affinity Relationship Studies on 5-HT1A Receptor Ligands. 2. Heterobicyclic Phenylpiperazines with N4-Aralkyl Substituents. J Med Chem. 1994;37:2761–2773. doi: 10.1021/jm00043a015. [DOI] [PubMed] [Google Scholar]
- 19.Bojarski AJ. Pharmacophore Models for Metabotropic 5-HT Receptor Ligands. Curr Top Med Chem. 2006;6:2005–2026. doi: 10.2174/156802606778522186. [DOI] [PubMed] [Google Scholar]
- 20.Franchini S, Prandi A, Sorbi C, Tait A, Baraldi A, Angeli P, Buccioni M, Cilia A, Poggesi E, Fossa P, Brasili L. Discovery of a New Series of 5-HT1A Receptor Agonists. Bioorg Med Chem Lett. 2010;20:2017–2020. doi: 10.1016/j.bmcl.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 21.Lepailleur A, Bureau R, Paillet-Loilier M, Fabis F, Saettel N, Lemaître S, Dauphin F, Lesnard A, Lancelot J-C, Rault S. Molecular Modeling Studies Focused on 5-HT7 versus 5-HT1A Selectivity. Discovery of Novel Phenylpyrrole Derivatives with High Affinity for 5-HT7 Receptors. J Chem Inf Model. 2005;45:1075–1081. doi: 10.1021/ci050045p. [DOI] [PubMed] [Google Scholar]
- 22.Chilmonczyk Z, Szelejewska-Wozniakowska A, Cybulski J, Cybulski M, Koziol AE, Gdaniec M. Conformational Flexibility of serotonin1A Receptor Ligands from Crystallographic Data. Updated Model of the Receptor Pharmacophore. Arch Pharm (Weinheim) 1997;330:146–160. doi: 10.1002/ardp.19973300507. [DOI] [PubMed] [Google Scholar]
- 23.Weber KC, Salum LB, Honório KM, Andricopulo AD, da Silva ABF. Pharmacophore-Based 3D QSAR Studies on a Series of High Affinity 5-HT1A Receptor Ligands. Eur J Med Chem. 2010;45:1508–1514. doi: 10.1016/j.ejmech.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 24.Sanders MPa, Barbosa AJM, Zarzycka B, Nicolaes GaF, Klomp JPG, de Vlieg J, Del Rio A. Comparative Analysis of Pharmacophore Screening Tools. J Chem Inf Model. 2012;52:1607–1620. doi: 10.1021/ci2005274. [DOI] [PubMed] [Google Scholar]
- 25.Lanfumey L, Hamon M. 5-HT1 Receptors. Curr Drug Targets CNS Neurol Disord. 2004;3:1–10. doi: 10.2174/1568007043482570. [DOI] [PubMed] [Google Scholar]
- 26.Hoyer D, Hannon JP, Martin GR. Molecular, Pharmacological and Functional Diversity of 5-HT Receptors. Pharmacol Biochem Behav. 2002;71:533–554. doi: 10.1016/s0091-3057(01)00746-8. [DOI] [PubMed] [Google Scholar]
- 27.Seeber M, De Benedetti PG, Fanelli F. Molecular Dynamics Simulations of the Ligand-Induced Chemical Information Transfer in the 5-HT(1A) Receptor. J Chem Inf Comput Sci. 2003;43:1520–1531. doi: 10.1021/ci034069c. [DOI] [PubMed] [Google Scholar]
- 28.López-Rodríguez ML, Morcillo MJ, Fernández E, Benhamú B, Tejada I, Ayala D, Viso A, Campillo M, Pardo L, Delgado M, Manzanares J, Fuentes Ja. Synthesis and Structure-Activity Relationships of a New Model of Arylpiperazines. 8. Computational Simulation of Ligand-Receptor Interaction of 5-HT(1A)R Agonists with Selectivity over alpha1-Adrenoceptors. J Med Chem. 2005;48:2548–2558. doi: 10.1021/jm048999e. [DOI] [PubMed] [Google Scholar]
- 29.Zlatović MV, Sukalović VV, Schneider C, Roglić GM. Interaction of Arylpiperazine Ligands with the Hydrophobic Part of the 5-HT1A Receptor Binding Site. Bioorg Med Chem. 2006;14:2994–3001. doi: 10.1016/j.bmc.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 30.Dabrowska J, Brylinski M. Stereoselectivity of 8-OH-DPAT toward the Serotonin 5-HT1A Receptor: Biochemical and Molecular Modeling Study. Biochem Pharmacol. 2006;72:498–511. doi: 10.1016/j.bcp.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Jacoby E, Fauchère J-L, Raimbaud E, Ollivier S, Michel A, Spedding M. A Three Binding Site Hypothesis for the Interaction of Ligands with Monoamine G Protein-Coupled Receptors: Implications for Combinatorial Ligand Design. Quant Struct Relationships. 1999;18:561–572. [Google Scholar]
- 32.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The Retinal Conformation and Its Environment in Rhodopsin in Light of a New 2.2 Å Crystal Structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 33.Pessoa-Mahana H, Recabarren-Gajardo G, Temer JF, Zapata-Torres G, Pessoa-Mahana CD, Barría CS, Araya-Maturana R. Synthesis, Docking Studies and Biological Evaluation of Benzo[b]thiophen-2-Yl-3-(4-Arylpiperazin-1-Yl)-Propan-1-One Derivatives on 5-HT1A Serotonin Receptors. Molecules. 2012;17:1388–1407. doi: 10.3390/molecules17021388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cherezov V, Rosenbaum DM, Hanson Ma, Rasmussen SGF, Thian FS, Kobilka TS, Choi H-J, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-Resolution Crystal Structure of an Engineered Human beta2-Adrenergic G Protein-Coupled Receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cappelli A, Manini M, Valenti S, Castriconi F, Giuliani G, Anzini M, Brogi S, Butini S, Gemma S, Campiani G, Giorgi G, Mennuni L, Lanza M, Giordani A, Caselli G, Letari O, Makovec F. Synthesis and Structure-Activity Relationship Studies in Serotonin 5-HT1A Receptor Agonists Based on Fused Pyrrolidone Scaffolds. Eur J Med Chem. 2013;63:85–94. doi: 10.1016/j.ejmech.2013.01.044. [DOI] [PubMed] [Google Scholar]
- 36.Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Structure of a beta1-Adrenergic G-Protein-Coupled Receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang X-P, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE. Structural Basis for Molecular Recognition at Serotonin Receptors. Science. 2013;340:610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salerno L, Pittalà V, Modica MN, Siracusa MA, Intagliata S, Cagnotto A, Salmona M, Kurczab R, Bojarski AJ, Romeo G. Structure-Activity Relationships and Molecular Modeling Studies of Novel Arylpiperazinylalkyl 2-Benzoxazolones and 2-Benzothiazolones as 5-HT(7) and 5-HT(1A) Receptor Ligands. Eur J Med Chem. 2014;85:716–726. doi: 10.1016/j.ejmech.2014.08.023. [DOI] [PubMed] [Google Scholar]
- 40.Rataj K, Witek J, Mordalski S, Kosciolek T, Bojarski AJ. Impact of Template Choice on Homology Model Efficiency in Virtual Screening. J Chem Inf Model. 2014;54:1661–1668. doi: 10.1021/ci500001f. [DOI] [PubMed] [Google Scholar]
- 41.Selent J, Marti-Solano M, Rodríguez J, Atanes P, Brea J, Castro M, Sanz F, Loza MI, Pastor M. Novel Insights on the Structural Determinants of Clozapine and Olanzapine Multi-Target Binding Profiles. Eur J Med Chem. 2014;77:91–95. doi: 10.1016/j.ejmech.2014.02.058. [DOI] [PubMed] [Google Scholar]
- 42.Canale V, Guzik P, Kurczab R, Verdie P, Satała G, Kubica B, Pawłowski M, Martinez J, Subra G, Bojarski AJ, Zajdel P. Solid-Supported Synthesis, Molecular Modeling, and Biological Activity of Long-Chain Arylpiperazine Derivatives with Cyclic Amino Acid Amide Fragments as 5-HT(7) and 5-HT(1A) Receptor Ligands. Eur J Med Chem. 2014;78:10–22. doi: 10.1016/j.ejmech.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 43.Canale V, Kurczab R, Partyka A, Satała G, Witek J, Jastrzębska-Więsek M, Pawłowski M, Bojarski AJ, Wesołowska A, Zajdel P. Towards Novel 5-HT7versus 5-HT1A Receptor Ligands among LCAPs with Cyclic Amino Acid Amide Fragments: Design, Synthesis, and Antidepressant Properties. Part II. Eur J Med Chem. 2015;92:202–211. doi: 10.1016/j.ejmech.2014.12.041. [DOI] [PubMed] [Google Scholar]
- 44.Tena-Campos M, Ramon E, Lupala CS, Pérez JJ, Koch K-W, Garriga P. Zinc Is Involved in Depression by Modulating G Protein-Coupled Receptor Heterodimerization. Mol Neurobiol. 2016;53:2003–2015. doi: 10.1007/s12035-015-9153-y. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura Y, Ishii J, Kondo A. Applications of Yeast-Based Signaling Sensor for Characterization of Antagonist and Analysis of Site-Directed Mutants of the Human Serotonin 1A Receptor. Biotechnol Bioeng. 2015;112:1906–1915. doi: 10.1002/bit.25597. [DOI] [PubMed] [Google Scholar]
- 46.Gabrielsen M, Kurczab R, Siwek A, Ravna AW, Kristiansen K, Kufareva I, Abagyan R, Nowak G, Sylte I, Bojarski AJ. Identi Fi Cation of Novel Serotonin Transporter Compounds by Virtual Screening. J Chem Inf Model. 2014:933–943. doi: 10.1021/ci400742s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan FY, Sun N, Neves MAC, Lam PCH, Chung WH, Wong LK, Chow HY, Ma DL, Chan PH, Leung YC, Chan TH, Abagyan R, Wong KY. Identification of a New Class of FtsZ Inhibitors by Structure-Based Design and in Vitro Screening. J Chem Inf Model. 2013;53:2131–2140. doi: 10.1021/ci400203f. [DOI] [PubMed] [Google Scholar]
- 48.McRobb FM, Kufareva I, Abagyan R. In Silico Identification and Pharmacological Evaluation of Novel Endocrine Disrupting Chemicals That Act via the Ligand-Binding Domain of the Estrogen Receptor Α. Toxicol Sci. 2014;141:188–197. doi: 10.1093/toxsci/kfu114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG, Beibel M, Renaud Na, Smith TM, Salcius M, Shi X, Hild M, Servais R, Jain M, Deng L, Bullock C, McLellan M, Schuierer S, Murphy L, Blommers MJJ, Blaustein C, Berenshteyn F, Lacoste A, Thomas JR, Roma G, Michaud Ga, Tseng BS, Porter Ja, Myer VE, Tallarico Ja, Hamann LG, Curtis D, Fishman MC, Dietrich WF, Dales Na, Sivasankaran R. SMN2 Splice Modulators Enhance U1-Pre-mRNA Association and Rescue SMA Mice. Nat Chem Biol. 2015;11:511–517. doi: 10.1038/nchembio.1837. [DOI] [PubMed] [Google Scholar]
- 50.Lückmann M, Holst B, Schwartz TW, Frimurer TM. In Silico Investigation of the Neurotensin Receptor 1 Binding Site: Overlapping Binding Modes for Small Molecule Antagonists and the Endogenous Peptide Agonist. Mol Inform. 2015;35:19–24. doi: 10.1002/minf.201500080. [DOI] [PubMed] [Google Scholar]
- 51.Abagyan R, Totrov M. Biased Probability Monte Carlo Conformational Searches and Electrostatic Calculations for Peptides and Proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 52.Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 53.El-Bermawy MA, Raghupathi RK, Ingher S, Teitler M, Maayani S, Glennon RA. 4-[4-(1-Noradamantanecarboxamido)butyl]-1-(2-Methoxyphenyl)piperazine: A High-Affinity 5-HT1A Selective Agent. Med Chem Res. 1992;2:88–95. [Google Scholar]
- 54.Evers A, Klebe G. Ligand-Supported Homology Modeling of G-Protein-Coupled Receptor Sites: Models Sufficient for Successful Virtual Screening. Angew Chem Int Ed Engl. 2004;43:248–251. doi: 10.1002/anie.200352776. [DOI] [PubMed] [Google Scholar]
- 55.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012;40:D1100–7. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warszycki D, Mordalski S, Kristiansen K, Kafel R, Sylte I, Chilmonczyk Z, Bojarski AJ. A Linear Combination of Pharmacophore Hypotheses as a New Tool in Search of New Active Compounds–an Application for 5-HT1A Receptor Ligands. PLoS One. 2013;8:e84510. doi: 10.1371/journal.pone.0084510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sastry M, Lowrie JF, Dixon SL, Sherman W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J Chem Inf Model. 2010;50:771–784. doi: 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- 58.Canvas, Version 2.2. Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 59.Duan J, Dixon SL, Lowrie JF, Sherman W. Analysis and Comparison of 2D Fingerprints: Insights into Database Screening Performance Using Eight Fingerprint Methods. J Mol Graph Model. 2010;29:157–170. doi: 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Kelly J. A New Interpretation of Information Rate. IEEE Trans Inf Theory. 1956;2:185–189. [Google Scholar]
- 61.Huang N, Shoichet BK, Irwin JJ. Benchmarking Sets for Molecular Docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paila YD, Kombrabail M, Krishnamoorthy G, Chattopadhyay A. Oligomerization of the serotonin(1A) Receptor in Live Cells: A Time-Resolved Fluorescence Anisotropy Approach. J Phys Chem B. 2011;115:11439–11447. doi: 10.1021/jp201458h. [DOI] [PubMed] [Google Scholar]
- 64.Prandi A, Franchini S, Manasieva LI, Fossa P, Cichero E, Marucci G, Buccioni M, Cilia A, Pirona L, Brasili L. Synthesis, Biological Evaluation, and Docking Studies of Tetrahydrofuran- Cyclopentanone- and Cyclopentanol-Based Ligands Acting at Adrenergic Α₁- and Serotonine 5-HT1A Receptors. J Med Chem. 2012;55:23–36. doi: 10.1021/jm200421e. [DOI] [PubMed] [Google Scholar]
- 65.Bronowska A, Leś A, Chilmonczyk Z, Filipek S, Edvardsen Ø, Østensen R, Sylte I. Molecular Dynamics of Buspirone Analogues Interacting with the 5-HT1A and 5-HT2A Serotonin Receptors. Bioorganic Med Chem. 2001;9:881–895. doi: 10.1016/s0968-0896(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 66.López-Rodríguez ML, Morcillo MJ, Fernández E, Rosado ML, Pardo L, Schaper K. Synthesis and Structure-Activity Relationships of a New Model of Arylpiperazines. Study of the 5-HT(1a)/alpha(1)-Adrenergic Receptor Affinity by Classical Hansch Analysis, Artificial Neural Networks, and Computational Simulation of Ligand Recognition. J Med Chem. 2001;44:198–207. doi: 10.1021/jm000930t. [DOI] [PubMed] [Google Scholar]
- 67.López-Rodríguez ML, Vicente B, Deupi X, Barrondo S, Olivella M, Morcillo MJ, Behamú B, Ballesteros Ja, Sallés J, Pardo L. Design, Synthesis and Pharmacological Evaluation of 5-hydroxytryptamine(1a) Receptor Ligands to Explore the Three-Dimensional Structure of the Receptor. Mol Pharmacol. 2002;62:15–21. doi: 10.1124/mol.62.1.15. [DOI] [PubMed] [Google Scholar]
- 68.López-Rodríguez ML, Morcillo MJ, Fernández E, Benhamú B, Tejada I, Ayala D, Viso A, Olivella M, Pardo L, Delgado M, Manzanares J, Fuentes JA. Design and Synthesis of S-(-)-2-[[4-(Napht-1-Yl)piperazin-1-Yl]methyl]-1,4-dioxoperhydropyrrolo[1,2-A]pyrazine (CSP-2503) Using Computational Simulation. A 5-HT1A Receptor Agonist. Bioorg Med Chem Lett. 2003;13:1429–1432. doi: 10.1016/s0960-894x(03)00160-4. [DOI] [PubMed] [Google Scholar]
- 69.Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, Bar-Haim S, Warshaviak D, Fichman M, Noiman S. G Protein-Coupled Receptors: In Silico Drug Discovery in 3D. Proc Natl Acad Sci U S A. 2004;101:11304–11309. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chilmonczyk Z, Cybulski M, Iskra-Jopa J, Chojnacka-Wójcik E, Tatarczyńska E, Kłodzińska A, Leś A, Bronowska A, Sylte I. Interaction of 1,2,4-Substituted Piperazines, New Serotonin Receptor Ligands, with 5-HT1A and 5-HT2A Receptors. Farmaco. 2002;57:285–301. doi: 10.1016/s0014-827x(02)01205-3. [DOI] [PubMed] [Google Scholar]
- 71.Nowak M, Kołaczkowski M, Pawłowski M, Bojarski AJ. Homology Modeling of the Serotonin 5-HT1A Receptor Using Automated Docking of Bioactive Compounds with Defined Geometry. J Med Chem. 2006;49:205–214. doi: 10.1021/jm050826h. [DOI] [PubMed] [Google Scholar]
- 72.Strzelczyk AA, Jarończyk M, Chilmonczyk Z, Mazurek AP, Chojnacka-Wójcik E, Sylte I. Intrinsic Activity and Comparative Molecular Dynamics of Buspirone Analogues at the 5-HT(1A) Receptors. Biochem Pharmacol. 2004;67:2219–2230. doi: 10.1016/j.bcp.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 73.Becker OM, Dhanoa DS, Marantz Y, Chen D, Shacham S, Cheruku S, Heifetz A, Mohanty P, Fichman M, Sharadendu A, Nudelman R, Kauffman M, Noiman S. An Integrated in Silico 3D Model-Driven Discovery of a Novel, Potent, and Selective Amidosulfonamide 5-HT1A Agonist (PRX-00023) for the Treatment of Anxiety and Depression. J Med Chem. 2006;49:3116–3135. doi: 10.1021/jm0508641. [DOI] [PubMed] [Google Scholar]
- 74.Sylte I, Bronowska a, Dahl SG. Ligand Induced Conformational States of the 5-HT(1A) Receptor. Eur J Pharmacol. 2001;416:33–41. doi: 10.1016/s0014-2999(01)00860-3. [DOI] [PubMed] [Google Scholar]
- 75.Jacoby E. A Novel Chemogenomics Knowledge-Based Ligand Design Strategy—Application to G Protein-Coupled Receptors. Quant Struct Relationships. 2001;20:115–123. [Google Scholar]
- 76.Siracusa MA, Salerno L, Modica MN, Pittalà V, Romeo G, Amato ME, Nowak M, Bojarski AJ, Mereghetti I, Cagnotto A, Mennini T. Synthesis of New Arylpiperazinylalkylthiobenzimidazole, Benzothiazole, or Benzoxazole Derivatives as Potent and Selective 5-HT1A Serotonin Receptor Ligands. J Med Chem. 2008;51:4529–4538. doi: 10.1021/jm800176x. [DOI] [PubMed] [Google Scholar]
- 77.Bronowska A, Leś A, Chilmonczyk Z, Filipek S, Edvardsen O, Ostensen R, Sylte I. Molecular Dynamics of Buspirone Analogues Interacting with the 5-HT1A and 5-HT2A Serotonin Receptors. Bioorg Med Chem. 2001;9:881–895. doi: 10.1016/s0968-0896(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 78.López-Rodríguez ML, Morcillo MJ, Fernández E, Benhamú B, Tejada I, Ayala D, Viso A, Olivella M, Pardo L, Delgado M, Manzanares J, Fuentes Ja. Design and Synthesis of S-(−)-2-[[4-(Napht-1-Yl)piperazin-1-Yl]methyl]-1,4-dioxoperhydropyrrolo[1,2-A]pyrazine (CSP-2503) Using Computational Simulation. A 5-HT1A Receptor Agonist. Bioorg Med Chem Lett. 2003;13:1429–1432. doi: 10.1016/s0960-894x(03)00160-4. [DOI] [PubMed] [Google Scholar]
- 79.Deng Z, Chuaqui C, Singh J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein-Ligand Binding Interactions. J Med Chem. 2004;47:337–344. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 80.Mordalski S, Kosciolek T, Kristiansen K, Sylte I, Bojarski AJ. Protein Binding Site Analysis by Means of Structural Interaction Fingerprint Patterns. Bioorg Med Chem Lett. 2011;21:6816–6819. doi: 10.1016/j.bmcl.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 81.Totrov M, Abagyan R. Flexible Protein-Ligand Docking by Global Energy Optimization in Internal Coordinates. Proteins Struct Funct Genet. 1997;29:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 82.Gaillard P, Carrupt Pa, Testa B, Schambel P. Binding of Arylpiperazines, (Aryloxy)propanolamines, and Tetrahydropyridylindoles to the 5-HT1A Receptor: Contribution of the Molecular Lipophilicity Potential to Three-Dimensional Quantitative Structure-Affinity Relationship Models. J Med Chem. 1996;39:126–134. doi: 10.1021/jm950410b. [DOI] [PubMed] [Google Scholar]
- 83.Katritch V, Kufareva I, Abagyan R. Structure Based Prediction of Subtype-Selectivity for Adenosine Receptor Antagonists. Neuropharmacology. 2011;60:108–115. doi: 10.1016/j.neuropharm.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yung-Chi C, Prusoff WH. Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 85.Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin J-P, Stevens RC, Vriend G, Gloriam DE. Generic GPCR Residue Numbers - Aligning Topology Maps While Minding the Gaps. Trends Pharmacol Sci. 2015;36:22–31. doi: 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marvin 6.2.2. ChemAxon; 2014. Calculator Plugins Were Used for Structure Property Prediction and Calculation. Http://www.chemaxon.com. [Google Scholar]
- 87.Schrödinger Release 2016-1: LigPrep, Version 3.7. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 88.Kurczab R, Smusz S, Bojarski AJ. The Influence of Negative Training Set Size on Machine Learning-Based Virtual Screening. J Cheminform. 2014;6:32–40. doi: 10.1186/1758-2946-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tasler S, Kraus J, Wuzik A, Müller O, Aschenbrenner A, Cubero E, Pascual R, Quintana-Ruiz J-R, Dordal A, Mercè R, Codony X. Discovery of 5-HT6 Receptor Ligands Based on Virtual HTS. Bioorg Med Chem Lett. 2007;17:6224–6229. doi: 10.1016/j.bmcl.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 90.Baell JB, Holloway GA. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 91.Baell J, Walters MA. Chemistry: Chemical Con Artists Foil Drug Discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.