

Abstract
Incorporation of the histone deacetylase (HDAC) inhibitor, suberoylanilide hydroxamic acid (SAHA), to a culture broth of the endophytic fungus Phoma sp. nov. LG0217 isolated from Parkinsonia microphylla changed its metabolite profile and resulted in the production of (10′S)-verruculide B (1), vermistatin (2) and dihydrovermistatin (3). When cultured in the absence of the epigenetic modifier, it produced a new metabolite, (S,Z)-5-(3′,4′-dihydroxybutyldiene)-3-propylfuran-2(5H)-one (4) together with nafuredin (5). The structure of 4 was elucidated by spectroscopic analyses and its absolute configuration was determined by application of the modified Mosher’s ester method. The absolute structure of (10′S)-verruculide B was determined as 5-[(10′S,2′E,6′E)-10′,11′-dihydroxy-3′,7′,11′-trimethyldodeca-2′,6′-dien-1′-yl]-(3R)-6,8-dihydroxy-3-methylisochroman-1-one (1) with the help of CD and NOE data. Compound 1 inhibited the activity of protein tyrosine phosphatases (PTPs) 1B (PTP1B), Src homology 2-containing PTP 1 (SHP1) and T-cell PTP (TCPTP) with IC50 values of 13.7±3.4, 8.8±0.6, and 16.6±3.8 μM, respectively. Significance of these activities and observed modest selectivity of 1 for SHP1 over PTP1B and TCPTP is discussed.
Keywords: Epigenetic, Phoma sp. nov. LG0217, Histone deacetylase inhibitor, (10′S)-verruculide B, Protein tyrosine phosphatases
Graphical Abstract
1. Introduction
Endophytic fungi are a group of endosymbiotic microorganisms that inhabit healthy plant tissues with no apparent pathogenic effects on their hosts.1 Recent studies have shown that endophytic fungi are prolific producers of bioactive and/or novel secondary metabolites.2 However, when cultured utilizing traditional fermentation techniques, fungi often express only a subset of biosynthetic genes which encode for their secondary metabolite production.3 Realization of this has recently led to the discovery of strategies to activate these silent biosynthetic gene clusters in fungi including the use of epigenetic modifiers to activate silent biosynthetic pathways,4 and simulation of natural gene cluster activating conditions by biotic (co-cultivation),5 and abiotic (elicitation by chemical and physical means)6 methods. Among these, incorporation of small-molecule modifiers into fungal culture media to manipulate epigenetic regulation of gene transcription has received considerable attention primarily focusing on inhibitors of histone deacetylase (HDAC),7 DNA methyltransferase (DNMT)8 and the proteasome.9
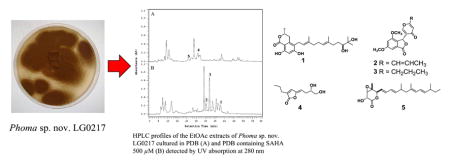
To investigate the effect of incorporation of the HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA), on production of secondary metabolites, we screened a small collection of endophytic fungi by culturing them in potato dextrose broth (PDB) medium with SAHA (100, 250, and 500 μM) and without this epigenetic modifier, and then by analyzing the resulting EtOAc extracts by HPLC. Among those investigated, Phoma sp. nov. LG0217, a fungal endophyte of Parkinsonia microphylla (foothills paloverde, family: Fabaceae), showed a significant difference in HPLC profiles of the extracts derived from a culture grown with SAHA (500 μM) and the control culture (Fig. 1) whereas no such effect was observed at SAHA concentrations of 100 and 200 μM. Chemical investigation of the culture broth treated with SAHA led to the isolation of (10′S)-verruculide B (1),10 vermistatin (2)12 and dihydrovermistatin (3)13 whereas the control culture broth afforded a new metabolite, (S,Z)-5-(3′,4′-dihydroxybutylidene)-3-propylfuran-2(5H)-one (4) together with nafuredin (5)14 (Fig. 2). Vermistatin (2) has been reported to act as an elastase inhibitor,15 phytotoxin on various banana cultivars,16 RNA synthesis inhibitor in Ehrlich ascites carcinoma,17 P-388 leukemia,17 weakly cytotoxic to KB tumor cell lines,18 and an inhibitor of α-glucosidase.19 Reported biological activities of nafuredin (5) include selective inhibition of helminth complex I and inhibition of NADH-fumarate reductase (NFRD) of Ascaris suum (pig roundworm) at nM concentrations.14a Verruculide B has been evaluated previously for its inhibitory activity against protein tyrosine phosphatase (PTP) 1B (PTP1B) and was found to exhibit 40% inhibition at 23.1 μM.11 Therefore, it was of interest to evaluate 1 for its activity against other PTPs. Herein we report the isolation of 1–5, structure elucidation and determination of absolute stereochemistry of 1 and 4 and evaluation of 1 for inhibition of PTPs including PTP1B, T-cell PTP (TCPTP), PTP non-receptor type 3 (PTPN3), PTP non-receptor type 5 (PTPN5), PTP non-receptor type 7 (PTPN7), Src homology 2-containing PTP 1 (SHP1), and Src homology 2-containing PTP 2 (SHP2).20
Figure 1.

(A) HPLC profiles of the EtOAc extracts of Phoma sp. nov. LG0217 cultured in PDB and (B) PDB containing 500 μM SAHA detected by UV absorption at 280 nm. Note: Those metabolites indicated above HPLC peaks have been isolated and characterized (see Fig. 2), but the remaining peaks in HPLC traces were found to be complex inseparable mixtures and/or undergo decomposition during isolation.
Figure 2.

Structures of metabolites 1–5 isolated from Phoma sp. nov. LG0217.
2. Results and Discussion
HPLC analyses of EtOAc soluble portions of MeOH/CHCl3 extracts derived from freeze-dried cultures of Phoma sp. nov. LG0217 cultured in PDB and PDB+SAHA (500 μM) differed markedly in their metabolite profiles (Fig. 1). Thus, each extract was fractionated separately resulting in the isolation of metabolites 1–3 from the PDB+SAHA culture broth extract and 4–5 from the PDB culture broth (control) extract. The spectroscopic (UV, IR, MS, 1H and 13C NMR) data for 1 were similar to those reported for verruculide B,11 except for the [α]D data (+2.0 for 1; −45.2 for verruculide B11). Although the absolute configuration of one of the two chiral centers (C-3) of verruculide B has been determined as R by the application of CD spectroscopy and comparison with the related 6,8-dihydroxy-3-methylisochroman-1-one [(R)-6- hydroxymellein], application of the modified Mosher’s ester method to define the absolute configuration of the remaining chiral center (C-10′) located in the side chain has been reported to be unsuccessful.11 Based on differences in [α]D data for 1 and verruculide B (see above), it was essential to determine the absolute configurations of both chiral centers (C-3 and C-10′) of 1. The absolute configuration of C-3 of 1 was established from the Cotton effect observed in its CD spectrum. The negative Cotton effect ascribed to the K-absorption band at 270 nm in the CD spectrum confirmed the R absolute configuration at C-3 of 1 (see Supplementary Fig. 12).21 The availability of a hydroxy group attached to the carbon atom (C-11′) adjacent to the side chain chiral center (C-10′) of 1 made it possible to apply in situ dimolybdenum CD method developed for vicinal diols by Snatzke and Frelek22 for the determination of the absolute configuration of this chiral center. The positive Cotton effect at 305 nm observed in the Mo2(OAc)4-induced CD spectrum of 1 (Fig. 3) suggested the absolute configuration of C-10′ to be S.22 Thus, absolute structure of 1 was determined as 5-[(10′S,2′E,6′E)-10′,11′-dihydroxy-3′,7′,11′-trimethyldodeca-2′,6′-dien-1′-yl]-(3R)-6,8-dihydroxy-3-methylisochroman-1-one [(10′S)-verruculide B].
Figure 3.

(A) CD spectrum of 1 with dimolybdinum tetraacetate in DMSO solution (after subtracting the inherent CD spectrum of 1), and (B) preferred conformation of the diol in the chiral Mo-complex.
Metabolite 4, obtained as a yellow oil, was determined to have the molecular formula C11H16O4 by a combination of HRFABMS and NMR data, indicating four degrees of unsaturation. Analysis of 1H, 13C and HSQC NMR data revealed that 4 contained a primary methyl [δH 0.94 (t, J = 7.0 Hz); δC 13.7], four methylenes of which one was oxygenated [δH 3.68 (dd, J = 11.0, 3.0 Hz), 3.49 (dd, J = 11.0, 7.0 Hz); δC 66.3], one oxymethine [δH 3.85 (m); δC 71.3], five sp2 carbons, two of which were protonated [δH 6.97 (s); δC 136.7 and δH 5.25 (t, J = 8.0 Hz); δC 109.2] and one was a lactone carbonyl (δC 170.5). Since two double bonds and carbonyl group accounted for three degrees of unsaturation, 4 should be monocyclic. The presence of two spin systems, CH3CH2CH2– and HOCH2CH(OH)–CH2CH=, in 4 were evident from its 1H-1H COSY spectrum (Fig. 4). The connectivities of these two spin systems and the remaining structural (two trisubstituted olefins and lactone) moieties in 4 were determined by the analysis of its HMBC correlations (Fig. 4). In the HMBC spectrum, the correlations between terminal CH2 protons [H2-1″ (δH 2.31)] of the CH3CH2CH2– spin system and lactone carbonyl carbon (δC 170.5), olefinic carbon [C-3 (δC 134.2)] and protonated olefinic carbon [C-4 (δC 136.7)] were observed, which established the connectivity of C-1″ with C-3, lactone carbonyl (C-2) with C-3, and C-3 with C-4. The HMBC correlations of the olefinic proton [H-1′ (δH 5.25)] of the HOCH2CH(OH)–CH2CH= spin system with C-5 (δC 149.8) and C-4 (δC 136.7) established the connectivity between C-5 and C-1′. Taken together these data suggested that the lactone carbonyl should be linked to C-5 via an oxygen atom to form a five membered ring. The proton H-4 showed long-range coupling with H-1′ (J = 1.5 Hz) suggesting that both these protons are on the same side of the molecule. The absolute configuration of the chiral carbon (C-3′) of 4 was determined by the application of the modified Mosher’s ester method.23 Reaction of 4 with (R)- and (S)-α-methoxy-α-trifluoromethylphenylacetyl chloride (MTPA-Cl) afforded (S)- and (R)- MTPA bisesters 4a and 4b, respectively. Analysis of Δδ (δS−δR) values of these Mosher’s esters confirmed the S absolute stereochemistry for C-3′ of 4 (see Fig. 5 and Supplementary Fig. 13). Thus, the absolute structure of 4 was determined as (S,Z)-5-(3′,4′-dihydroxybutylidene)-3-propylfuran-2(5H)-one [(S,Z)-7,8-dihydroxy-2-propyl-octa-2,4-dien-4-olide]. Metabolites 2, 3 and 5 were identified as vermistatin,12 dihydrovermistatin,13 and nafuredin,14 respectively by comparison of their spectroscopic (1H and 13C NMR) and [α]D data with those reported.
Figure 4.

1H–1H COSY and selected HMBC correlations for 4.
Figure 5.

ΔδH values [(Δδ in ppm) = δS−δR] obtained for (S)- and (R)-bis MTP esters (4a and 4b, respectively) of (S,Z)-5-(3′,4′-dihydroxybutylidene)-3-propylfuran-2(5H)-one (4) in d5-pyridine.
PTPs are key regulatory enzymes that dephosphorylate phosphotyrosine residues and are critical regulators of signal transduction under normal and pathological conditions. In conjunction with protein tyrosine kinases (PTKs), they regulate the reversible phosphorylation of tyrosine residues in proteins. Thus, PTPs control fundamental physiological processes such as cell growth and differentiation, cell cycle, metabolism, immune responses and cytoskeletal function. In addition, interfering with the delicate balance between counteracting PTKs, PTPs are involved in the development of numerous inherited and acquired human diseases such as autoimmunity, diabetes and cancer.24 When evaluated for inhibition of a small panel of protein tyrosine phosphatases PTP1B, TCPTP, PTPN3, PTPN5, PTPN7, SHP1, and SHP2 in our screening assays, (10′S)-verruculide B (1) showed significant inhibitory activity for SHP1 and moderate activity for PTP1B and TCPTP (Fig. 6). It was found that 1 inhibited SHP1 with an IC50 = 8.8 ± 0.6 μM and showed modest selectivity for SHP1 versus PTP1B (1.6 fold) and TCPTP (1.9 fold). PTP1B is a negative regulator of insulin and leptin signaling pathway and as such is predicted to be an attractive target for metabolic diseases such as type 2 diabetes and obesity.25
Figure 6.

Inhibition of PTPs by (10′S)-verruculide B (1). (A) Percentage inhibition of a panel of protein tyrosine phosphatases by 1. (B) Dose-response curves of 1 for inhibition of PTP1B, TCPTP, and SHP1.
Compromised TCPTP function has been associated with autoimmune diseases like type 1 diabetes and Crohn’s disease,26 and obesity related metabolic disorders through antagonism of insulin and leptin signaling.27 Conversely, SHP1 has been linked to a number of cancer correlated phenotypes with a complex relationship. For instance, it is known to antagonize growth related tyrosine kinases,28 and to promote responsiveness and antitumor functions of natural killer (NK) cells.29 However, SHP1 activity has also been linked to metastasis via the epithelial-mesenchymal transition.30 Although 1 had moderate to weak inhibitory activity for PTPs tested with modest selectivity for SHP1, it offers an interesting lead molecule that could be optimized through strategies involving epigenetic culture manipulation and/or medicinal chemistry.
3. Experimental
3.1. General Experimental Procedures
Optical rotations were measured with a JASCO Dip-370 digital polarimeter using CHCl3 as solvent. UV spectra were recorded on a Shimadzu UV-1601 UV-vis spectrophotometer. IR spectra for KBr disks were recorded on a Shimadzu FTIR-8300 spectrometer. NMR spectra were recorded in CDCl3 with a Bruker Avance III 400 spectrometer at 400 MHz for 1H NMR and 100 MHz for 13C NMR, using residual CHCl3 as internal reference. High-resolution MS were recorded on Bruker LCQ-Fleet spectrometer. CD spectra were recorded in MeOH or DMSO on a Jasco 810 spectrometer at 25 °C, using a quartz cell with 1 mm optical path length. Whatman LRP-2 was used for reversed-phase column chromatography. Analytical and preparative thin-layer chromatography (TLC) was carried out on RP-18 F254S Aluminum-backed TLC plates (Merck) and compounds were visualized with short-wavelength UV and by spraying with anisaldehyde-sulfuric acid spray reagent and heating until the spots appeared. Analytical HPLC was performed on a Hitachi instrument equipped with L-6200A intelligent pump, L-4500 diode array detector, and P-6000 interface utilizing Hitachi Model D-7000 chromatography data station software using a RP column (Phenomenex 5μm, C-18, 4.6 × 250 mm). Injections (10 μL) were made with AS-4000 intelligent auto sampler. Preparative HPLC was performed on a Waters Delta Prep 400 preparative chromatography system equipped with Waters 996 photodiode array detector and a Water Prep LC controller utilizing Empower Pro software and using a RP column (Phenomenex Luna 5μm, C18, 10 × 250 mm). Dimolybdenum tetracetate was purchased from Sigma-Aldrich. DMSO was purchased from Sigma-Aldrich and dried with 4 Å molecular sieves. cDNA constructs and primers were purchased from DNAsu and Integrated DNA Technologies (IDT), respectively. A Techne Prime Thermocycler was used for all polymerase chain reactions (PCR). The Microfluidizer used was purchased from Microfluidics. Talon Cobalt Resin was purchased from Clontech. A BioTek Synergy 2 plate reader was used to measure absorbance of 96-well plates, data were recorded and processed using Gen 5 1.11 software. Graphs were generated with Kaleidagraph 4.5. Culture dishes were purchased from Greiner Bio-One. Gradient SDS PAGE (4–20%) gels were purchased from Invitrogen. Life Technologies gel boxes were used for protein staining.
3.2. Fungal isolation and identification
In October 2011, branches of Parkinsonia microphylla (foothills palo verde) were collected from a semi-urban area in Tucson, Arizona. Healthy leaves and photosynthetic stems of these branches were washed in tap water and cut into ca. 2.0 mm2 segments that were surface-sterilized by agitating sequentially in 95% EtOH for 30 sec, 0.5 % NaOCl for 2 min, and 70% EtOH for 2 min.31 Tissue segments were surface-dried under sterile conditions and then placed individually onto 2% malt extract agar in 1.5 mL microcentrifuge tubes. Tubes were sealed with Parafilm and incubated under ambient light/dark condition for one year. Emergent fungi were isolated into pure culture on 2% MEA, vouchered in sterile water, and deposited as living vouchers at the Robert L. Gilbertson Mycological Herbarium at the University of Arizona. One fungus of interest was used for the present study: isolate LG0217, which has been deposited at the Robert L. Gilbertson Mycological Herbarium with accession number LG0217. Total genomic DNA was extracted from fresh mycelium32 and the internal transcribed spacers and 5.8S gene (ITSrDNA) was amplified and sequenced by PCR.32 The ITSrDNA sequence was compared against the GenBank database using BLAST.33 The top BLAST matches were to unnamed fungi isolated from other desert plants,32 with several species of Phoma and allied taxa that were studied by Davey and Currah.34 We therefore downloaded the alignment published by Davey and Currah,34 trimmed it to include closely related taxa that were readily alignable with LG0217, and integrated two representative sequences.32 The resulting alignment consisted of species of Phoma, Ascochyta, Didymella, and Westerdykella (outgroup), for a total of 24 ingroup taxa and one outgroup taxon. The data set was aligned automatically using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) with default parameters and verified by eye prior to analysis. The data set was analyzed using maximum likelihood in PAUP 4.0a14735 followed by a bootstrap analysis with 100 replicates. The strain was placed with strong support in the Phoma/Ascochyta/Didymella complex identified by Davey and Currah.34 This strain was thus designated as Phoma sp. nov. LG0217 (Pleosporaceae, Pleosporales, Dothideomycetes, Ascomycota), pending morphological description.
3.3. Cultivation and isolation of metabolites
A seed culture of Phoma sp. nov. LG0217 grown on PDA for two weeks was used for inoculation. Mycelia were scraped out, mixed with sterile water, and filtered through a 100 μm filter to separate spores from the mycelia. Absorbance of the spore solution was measured (at 600 nm) and adjusted to between 0.8 and 1.0. This spore solution was used to inoculate 4 × 2.0 L Erlenmeyer flasks, each holding 1.0 L of the medium (PDB) containing 500 μM of SAHA and 5 × 2.0 L Erlenmeyer flasks, each holding 1.0 L of the medium (PDB) and incubated at 160 rpm and 28 °C. The glucose level in the medium was monitored using glucose strips (URISCAN glucose strips). On day 14, the strip gave a blue color for the glucose test, indicating the absence of glucose in the medium. After incubation for 14 days at 28 °C, the culture broth was freeze-dried and extracted with MeOH/CHCl3 (1:1, 1.0 L), and the resulting extract was filtered through Whatman No. 1 filter paper. The filtrate was evaporated under reduced pressure and the resulting residue was suspended in H2O (500 mL) and extracted with EtOAc (3 × 1.0 L). Combined EtOAc extract was evaporated under reduced pressure to afford an orange semisolid from the fungus cultured in PDB containing 500.0 μM of SAHA (1.54 g) and a dark brown semisolid from the control (3.82 g). A portion (1.33 g) of EtOAc extract from the fungus cultured in PDB containing SAHA was chromatographed over a column of C18 (66 g) made up in MeOH/H2O (1:9) and eluted with MeOH/H2O (1:9, 270 mL), MeOH/H2O (2:8, 270 mL), MeOH/H2O (3:7, 270 mL), MeOH/H2O (4:6, 270 mL), MeOH/H2O (1:1, 270 mL), MeOH/H2O (6:4, 270 mL), MeOH/H2O (7:3, 270 mL), MeOH/H2O (8:2, 270 mL), MeOH (270 mL) and finally with CHCl3 (270 mL) to afford 10 fractions [A (32.1 mg), B (14.8 mg), C (13.6 mg), D (47.8 mg), E (50.7 mg), F (28.7 mg), G (29.9 mg), H (17.7 mg), I (562.8 mg) and J (285.3 mg)]. Fraction F (28.7 mg) was purified by preparative HPLC (CH3CN/H2O, 35:65, 3.0 mL/min) to give 2 (1.6 mg, tR = 28.33 min) and 3 (0.8 mg, tR = 29.73 min). Fraction H (17.7 mg) was purified by preparative HPLC (CH3CN/H2O, 55:45, 2.0 mL/min) to give 1 (2.1 mg, tR = 33.07 min).
The EtOAc extract (3.82 g) from the Phoma sp. nov. LG0217 cultured in PDB was partitioned between hexanes and 80% aqueous MeOH. The 80% aqueous MeOH fraction was diluted to 50% aqueous MeOH by addition of water and then extracted with CHCl3. Evaporation of CHCl3 under reduced pressure yielded a dark brown semisolid (450 mg). A large portion (400 mg) of this was chromatographed over a column of silica gel (13.6 g) made up in hexanes/EtOAc (8:2) and eluted with hexanes/EtOAc (8:2, 70 mL), hexanes/EtOAc (7:3, 70 mL), hexanes/EtOAc (6:4, 70 mL), hexanes/EtOAc (1:1, 70 mL), hexanes/EtOAc (4:6, 70 mL), hexanes/EtOAc (3:7, 70 mL), hexanes/EtOAc (2:8, 70 mL), EtOAc (70 mL) and finally with MeOH (70 mL). A total of 95 fractions (5 mL each) were collected and combined on the basis of their TLC patterns to yield 13 fractions [A (58.3 mg), B (13.6 mg), C (5.0 mg), D (27.8 mg), E (2.1 mg), F (19.6 mg), G (12.6 mg), H (24.3 mg), I (9.7 mg), J (20.9 mg), K (13.1 mg), L (27.3 mg) and M (125.7 mg)]. Fraction F (6.7 mg) was purified by preparative HPLC (MeOH/H2O, 9:1, 3.0 mL/min) to give 5 (3.8 mg, tR=10.35 min). Fraction K (13.2 mg) was purified by preparative HPLC (CH3CN/H2O, 3:7, 2.5 mL/min) to give 4 (2.9 mg, tR=12.93 min).
3.3.1. (10′S)-Verruculide B (1)
A yellow amorphous solid, +2.0 (c 0.2, CHCl3); UV (MeOH) λmax/nm (log ε) 311 (3.45), 272 (4.21), 207 (2.32); IR (KBr) νmax/cm−1 3385, 1660, 1615, and 1573, 1485, 1263, 1095; 1H NMR (400 MHz, CDCl3) δ 6.29 (1H, s, H-7), 5.09 (1H, t, J = 6.2 Hz, H-6′), 4.95 (1H, t, J = 6.0 Hz, H-2′), 4.58 (1H, m, H-3), 3.37 (1H, d, J = 10.0 Hz, H-10′), 3.20 (2H, brt, J = 4.7 Hz, H2-1′), 2.95 (1H, dd, J = 16.0, 3.0 Hz, Ha-4), 2.68 (1H, dd, J = 16.0, 11.5Hz, Hb-4), 2.14 (1H, m, Ha-8′), 2.11 (2H, dd, J =14.3, 6.9 Hz, H2-5′), 2.04 (2H, dd, J = 14.3, 6.9 Hz, H2-4′), 2.03 (1H, m, Hb-8′), 1.69 (3H, s, H3-14′), 1.61 (1H, m, Ha-9′), 1.55 (3H, s, H3-15′), 1.50 (3H, d, J = 6.0 Hz, H3-9), 1.39 (1H, m, Hb-9′), 1.21 and 1.18 (3H each, s, H3-12′/13′); 13C NMR (100 MHz, CDCl3) δ 170.4 (C, C-1), 162.8 (C, C-8), 161.9 (C, C-6), 138.6 (C, C-4a), 135.9 (C, C-3′), 134.7 (C, C-7′), 124.4 (CH, C-6′), 122.8 (CH, C-2′), 117.3 (C, C-5), 101.9 (CH, C-7), 101.3 (C, C-8a), 78.1 (C, C-10′), 74.8 (C, C-3), 73.1 (C, C-11′), 38.9 (CH2, C-4′), 36.4 (CH2, C-8′), 32.0 (CH2, C-4), 29.3 (CH2, C-9′), 26.5 (CH3, C-12′), 25.2 (CH2, C-5′), 24.4 (CH2, C-1′), 23.5 (CH3, C-13′), 20.9 (CH3, C-9), 16.2 (CH3, C-15′), 15.9 (CH3, C-14′); HRESIMS m/z 455.2397 [M+Na]+ (calcd for C25H36NaO4, 455.2410).
3.3.2. Vermistatin (2)
A white amorphous solid; −96 (c 0.3, CHCl3); 1H and 13C NMR data were consistent with those reported.12
3.3.3. Dihydrovermistatin (3)
A yellow gum; −110 (c 0.2, CHCl3) [lit13 −108 (c 0.1, CHCl3)]; 1H NMR data were consistent with those reported.13
3.3.4. (S,Z)-5-(3′,4′-Dihydroxybutylidene)-3-propylfuran-2(5H)-one (4)
A yellow gum, +10.9 (c 0.28, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.97 (1H, t, J = 1.5 Hz, H-4), 5.25 (1H, t, J = 8.0 Hz, H-1′), 3.85 (1H, m, H-3′), 3.68 (1H, dd, J =11.0, 3.0 Hz, Ha-4′), 3.49 (1H, dd, J = 11.0, 7.0 Hz, Hb-4′), 2.53 (2H, m, H2-2′), 2.31 (2H, t, J = 7.0 Hz, H2-1″), 1.58 (2H, qt, J = 7.5 Hz, H2-2″), 0.94 (3H, t, J = 7.5 Hz, H3-3″); 13C NMR (400 MHz, CDCl3) δ 170.5 (C, C-2), 149.8 (C, C-5), 136.7 (CH, C-4), 134.2 (C, C-3), 109.2 (CH, C-1′), 71.3 (CH, C-3′), 66.3 (CH2, C-4′), 30.1 (CH2, C-2′), 27.1 (CH2, C-1″), 20.8 (CH2, C-2″), 13.7 (CH3, C-3″); HRESIMS m/z 213.1087 [M+H]+ (calcd for C11H17O4, 213.1126).
3.3.5. Nafuredin (5)
A yellow amorphous solid; +90 (c 0.1, CHCl3) [lit14 +89.9 (c 0.1, CHCl3)]; 1H and 13C NMR data were consistent with those reported.14
3.4. Determination of absolute configuration at C-10′ of 1
According to the published procedure,12 diol/Mo2(OAc)4 mixture (ca. 1:1) was subjected to CD measurement of 1 at the concentration of 0.3 mg/mL. The first CD spectrum was recorded immediately after mixing, and its time evolution was monitored with a rate of about one spectrum every 10 min until stationary CD was reached (ca. 30 min after mixing). The inherent CD was subtracted. The observed sign of the diagnostic band at 305 nm in the induced CD spectrum was correlated to the absolute configuration of the vic-diol moiety.
3.5. Preparation of (S)- and (R)-MTPA bisesters of 4
Compound 4 (0.5 mg) was transferred into a clean NMR tube and was dried completely under the vacuum. Deuterated pyridine (0.4 mL) and (R)-MTPA-Cl (10.0 μL) was added into the NMR tube immediately under a stream of N2. The reaction in the NMR tube was monitored by 1H NMR and left at 25 °C for 2 h, to give the (S)-MTPA bisester derivative (4a) of 4. 1H NMR data of 4a: (400 MHz, pyridine-d5). δ 7.166 (1H, t, J = 1.5 Hz, H-4), 5.897 (1H, m, H-3′), 5.336 (1H, t, J = 8.0 Hz, H-1′), 5.018 (1H, dd, J = 12.3, 3.0 Hz, Ha-4′), 4.659 (1H, dd, J = 12.3, 5.5 Hz, Hb-4′), 2.979 (2H, m, H2-2′), 2.260 (2H, t, J = 7.3 Hz, H2-1″), 1.505 (2H, sextet, J = 7.3 Hz, H2-2″), 0.818 (3H, t, J = 7.3 Hz, H3-3″). In the same manner described for 4a another portion of 4 (0.5 mg) was reacted in a second NMR tube with (S)-MTPA-Cl (10.0 μL) at 25 ºC for 2 h using d5-pyridine (0.4 mL) as solvent, to give the (R)-MTPA bisester derivative 4b. 1H NMR data of 4b: (400 MHz, pyridine-d5). δ 7.082 (1H, t, J = 1.5 Hz, H-4), 5.843 (1H, m, H-3′), 5.146 (1H, t, J = 7.7 Hz, H-1′), 5.057 (1H, dd, J = 12.0, 2.5 Hz, Ha-4′), 4.708 (1H, dd, J = 12.0, 6.5 Hz, Hb-4′), 2.894 (2H, m, H2-2′), 2.261 (2H, t, J = 7.3 Hz, H2-1″), 1.503 (2H, sextet, J = 7.3 Hz, H2-2″), 0.834 (3H, t, J = 7.3 Hz, H3-3″).
3.6. Ligation independent cloning of PTP expression constructs
Amplifications via PCR were carried out (5.0 μL 10X Pfx buffer, 1.0 μL forward primer (50.0 μM), 1.0 μL reverse primer (50.0 μM), 1.0 μL template (~20.0 nM) 1 μL dNTPs (10.0 mM), 1.0 μL MgSO4 (50 mM), 39.6 μL of H2O, and 0.4 μL of Pfx polymerase). All reaction materials were pipetted into a PCR tube on ice with Pfx polymerase added last. Reaction mixtures were then placed in a thermal cycler and heated to 95 ºC for 5 min, 55 ºC for 45 sec, and 68 ºC for 6 min for 30 cycles. PCR products were analyzed by 1% agarose gel electrophoresis. Gels were stained with ethidium bromide and then washed with buffer. The gels were then examined by UV trans-illuminator. The PCR products (10.0 μL, 100 ng/μL) were then mixed with pSpeedET vector (10.0 μL, 100.0 ng/μL) for 10 min at 25 ºC and used to transform E. coli DH5α cells. A single colony was picked and used to inoculate 3.0 mL of Terrific Broth (TB) media. Plasmids were purified via a Qiagen miniprep kit. The correct sequences were confirmed by Sanger sequencing.
3.7. PTP expression and purification
BL21 DE3 E. coli cells were transformed with the desired plasmid. A single colony was picked and used to inoculate 10.0 mL of LB media supplemented with kanamycin (50.0 μg/mL). When the culture reached OD600 = 0.5, 10.0 mL were transferred to one liter of LB media supplemented with kanamycin (50.0 μg/mL) and grown at 37 ºC to an OD600 = 0.3. Cultures were transferred to a 16 ºC incubator and grown for 60 min before induction with 500.0 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) at OD600 = 0.8 and incubated for 20 h. Cells were centrifuged (3000 × g, 15 min) and liquid was decanted. Cells were re-suspended in lysis buffer (50.0 mM HEPES, pH 7.4, 150.0 mM KCl, 5.0 mM MgCl2, 5% glycerol, 1.0 mM phenyl methanesulfonyl fluoride (PMSF), 2.0 mM 2-mercaptoethanol). Cells were lysed in a microfluidizer at 12,000 psi. Cobalt resin was washed with water and equilibrated with lysis buffer. Lysate of cells were spun down (11,180 × g, 1 h) and the supernatant was incubated with Talon resin for 1 h at 4 ºC. The resin was then transferred to a column, the supernatant allowed to flow through and then washed with 1.0 M KCl in lysis buffer. The protein was eluted with imidazole in a step-wise fashion (5.0, 20.0, 100.0, 250.0 mM imidazole in lysis buffer). The protein containing fractions, as confirmed by 12% SDS PAGE, were sealed in 12–14 KDa MWCO dialysis tubing and dialyzed against 4 L dialysis buffer ( 20.0 mM HEPES, 150.0 mM KCl, 1.0 mM MgCl2) at 4 ºC. Dialysates were concentrated in a 30,000 kDa MWCO protein concentrator. Aqueous solution of 50% glycerol was added at an equal volume as dialysate and proteins frozen using liquid nitrogen. All proteins were stored at −80 ºC until needed.
3.8. Phosphatase pNPP assay
The para-nitro phenol phosphate (pNPP) assay was carried out in 96-well transparent plates. Each well contained a 50.0 μL reaction in phosphatase buffer (50.0 mM Tris pH 7.4, 50.0 mM NaCl, 1.0 mM MgCl2, 2.0 mM β-mercaptoethanol) with 1.0 mM pNPP and 250.0–500.0 nM protein. DMSO and 100.0 μM sodium orthovanadate were used as negative and positive controls, respectively.36 Protein was added to the 96-well plates and incubated with 1.0 μL compound 1 (20.0 μM) or control at 37 ºC for 15 min. pNPP (1.0 mM) in phosphatase buffer was then added to the plate and incubated for 10 min at 37 ºC. Absorbance was measured at 405 nm at 25 ºC. Data were exported to Microsoft Excel for analysis. The inhibition percentage was calculated according to the following equation: Inhibition [%] = 100−(OD405 compound 1−OD405 DMSO)/(OD405 Na3VO4−OD405 DMSO)*100.
3.9. Dose-response studies
Dose responses were carried out in phosphatase buffer (as above) using ten-point serial dilution of compound 1 (100.00, 50.00, 25.00, 12.50, 6.25, 3.13, 1.56, 0.78, 0.39 and 0.20 μM). To 96-well transparent plates containing 24.0 μL protein solution (250.0 nM for PTP1B, TCPTP, PTPN3, PTPN5 and PTPN7, 500.0 nM for SHP1 and SHP2), 1.0 μL compound 1 for each of 10 concentrations, DMSO control, and sodium orthovanadate were added. After incubation of the resulting mixture at 37 ºC for 15 min, 24.0 μL phosphatase buffer with 2.0 mM pNPP was added. The assay plate was incubated for 10 min at 37 ºC. Absorbance was measured with a plate reader at 405 nm at 25 ºC. Data was exported to Microsoft excel for analysis. Logarithmic regression in KaleidaGraph was used to calculate the IC50 values.
Supplementary Material
Acknowledgments
This work was supported by grants from National Cancer Institute (Grant No. R01 CA090265) and National Institute of General Medical Sciences (Grant No. P41 GM094060). CAPES, Brazil, and China Scholarship Council are thanked for the award of graduate fellowships to J.R.G. and T.S., respectively. We also thank Nicholas C. Massimo for assistance with isolation of the fungus and Ms. Patricia Espinosa-Artiles for her help with culturing of the fungus.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Arnold AE. Fungal Biol Rev. 2007;21:51–56. [Google Scholar]; (b) Rodriguez RJ, White JF, Arnold AE, Redman RS. New Phytologist. 2009;182:314–330. doi: 10.1111/j.1469-8137.2009.02773.x. [DOI] [PubMed] [Google Scholar]
- 2.(a) Gunatilaka AAL. J Nat Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kusari S, Spiteller M. Nat Prod Rep. 2011;28:1203–1207. doi: 10.1039/c1np00030f. [DOI] [PubMed] [Google Scholar]
- 3.Hertweck C. Nat Chem Biol. 2009;5:450–452. doi: 10.1038/nchembio0709-450. [DOI] [PubMed] [Google Scholar]
- 4.Cichewicz RH. Nat Prod Rep. 2010;27:11–22. doi: 10.1039/b920860g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Pettit RK. Appl Microbiol Biotechnol. 2009;83:19–25. doi: 10.1007/s00253-009-1916-9. [DOI] [PubMed] [Google Scholar]; (b) Rateb ME, Hallyburton I, Houssen WE, Bull AT, Goodfellow M, Santhanam R, Jaspars M, Ebel R. RSC Advances. 2013;3:14444–14450. [Google Scholar]
- 6.Pimentel-Elardo SM, Sorensen D, Ho L, Ziko M, Bueler SA, Lu S, Tao J, Moser A, Lee R, Agard D, Fairn G, Rubinstein JL, Shoichet BK, Nodwell JR. ACS Chem Biol. 2015;10:2616–2623. doi: 10.1021/acschembio.5b00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Vervoort HC, Drašković M, Crews P. Org Lett. 2011;13:410–413. doi: 10.1021/ol1027199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shwab EK, Bok JW, Tribus M, Galehr J, Graessle S, Keller NP. Eukaryotic Cell. 2007;6:1656–1664. doi: 10.1128/EC.00186-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Henrikson JC, Hoover AR, Joyner PM, Cichewicz RH. Org Biomol Chem. 2009;7:435–438. doi: 10.1039/b819208a. [DOI] [PubMed] [Google Scholar]; (d) Albright JC, Henke MT, Soukup AA, McClure RA, Thomson RJ, Keller NP, Kelleher NL. ACS Chem Biol. 2015;10:1535–1541. doi: 10.1021/acschembio.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Fisch KM, Gillaspy AF, Gipson M, Henrikson JC, Hoover AR, Jackson L, Najar FZ, Wägele H, Cichewicz RH. J Ind Microbiol Biotechnol. 2009;36:1199–1213. doi: 10.1007/s10295-009-0601-4. [DOI] [PubMed] [Google Scholar]; (b) Williams RB, Henrikson JC, Hoover AR, Lee AE, Cichewicz RH. Org Biomol Chem. 2008;6:1895–1897. doi: 10.1039/b804701d. [DOI] [PubMed] [Google Scholar]; (c) Yakasai AA, Davison J, Wasil Z, Halo LM, Butts CP, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. J Am Chem Soc. 2011;133:10990–10998. doi: 10.1021/ja204200x. [DOI] [PubMed] [Google Scholar]; (d) Beau J, Mahid N, Burda WN, Harrington L, Shaw LN, Mutka T, Kyle DE, Barisic B, van Olphen A, Baker BJ. Mar Drugs. 2012;10:762–774. doi: 10.3390/md10040762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang X, Filho JGS, Hoover AR, King JB, Ellis TK, Powell DR, Cichewicz RH. J Nat Prod. 2010;73:942–948. doi: 10.1021/np100142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.VanderMolen KM, Darveaux BA, Chen W-L, Swanson SM, Pearce CJ, Oberlies NH. RSC Adv. 2014;4:18329–18335. doi: 10.1039/C4RA00274A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.After this work was completed, a paper describing the isolation and structure elucidation of verruculide B with the same gross structure as 1 appeared in the literature (see ref. 11); however, these authors reported that attempted determination of the absolute stereochemistry of verruculide B proved to be unsuccessful.
- 11.Yamazaki H, Nakayama W, Takahashi O, Kirikoshi R, Izumikawa Y, Iwasaki K, Toraiwa K, Ukai K, Rotinsulu H, Wewengkang DS, Sumilat DA, Mangindaan REP, Namikoshi M. Bioorg Med Chem. 2015;25:3087–3090. doi: 10.1016/j.bmcl.2015.06.026. [DOI] [PubMed] [Google Scholar]
- 12.Fuska J, Uhrín D, Proksa B, Voticky Z, Ruppeldt J. J Antibiot. 1986;39:1605–1608. doi: 10.7164/antibiotics.39.1605. [DOI] [PubMed] [Google Scholar]
- 13.Komai S, Hosoe T, Itabashi T, Nozawa K, Yaguchi T, Fukushima K, Kawai K. Heterocycles. 2005;65:2771–2776. [Google Scholar]
- 14.(a) Ui H, Shiomi K, Yamaguchi Y, Masuma R, Nagamitsu T, Takano D, Sunazuka T, Namikoshi M, Omura S. J Antibiot. 2001;54:234–238. doi: 10.7164/antibiotics.54.234. [DOI] [PubMed] [Google Scholar]; (b) Takano D, Nagamitsu T, Ui H, Shiomi K, Yamaguchi Y, Masuma R, Kuwajima I, Omura S. Tetrahedron Lett. 2001;42:3017–3020. doi: 10.1021/ol010089t. [DOI] [PubMed] [Google Scholar]
- 15.Sturdikova M, Proksa B, Fuska J, Stancikova M. Biologia (Bratislava) 1995;50:233–236. [Google Scholar]
- 16.Upadhyay RK, Strobel GA, Coval SJ, Clardy J. Experientia. 1990;46:982–984. [Google Scholar]
- 17.Fuska J, Fuskova A, Nemec P. Biologia (Bratislava, Slovakia) 1979;34:735–739. [Google Scholar]
- 18.Xia X-K, Huang H-R, She Z-G, Cai J-W, Lan L, Zhang J-Y, Fu LW, Vrijmoed LLP, Lin Y-C. Helv Chim Acta. 2007;90:1925–1931. [Google Scholar]
- 19.Liu Y, Xia G, Li H, Ma L, Ding B, Lu Y, He L, Xia X, She Z. Planta Med. 2014;80:912–917. doi: 10.1055/s-0034-1382859. [DOI] [PubMed] [Google Scholar]
- 20.Tautz L, Critton DA, Grotegut S. Methods Mol Biol. 2013;1053:179–221. doi: 10.1007/978-1-62703-562-0_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Shibano M, Naito H, Taniguchi M, Wang N-H, Baba K. Chem Pharm Bull. 2006;54:717–718. doi: 10.1248/cpb.54.717. [DOI] [PubMed] [Google Scholar]; (b) Krohn K, Bahramsari R, Flörke U, Ludewig K, Kliche-Spory C, Michel A, Aust H-J, Draeger S, Schulz B, Antus S. Phytochemistry. 1997;45:313–320. doi: 10.1016/s0031-9422(96)00854-0. [DOI] [PubMed] [Google Scholar]
- 22.(a) Di Bari L, Pescitelli G, Pratelli C, Pini D, Salvadori P. J Org Chem. 2001;66:4819–4825. doi: 10.1021/jo010136v. [DOI] [PubMed] [Google Scholar]; (b) Górecki M, Jablońska E, Kruszewska A, Suszczyńska A, Urbańczyk-Lipkowska Z, Gerards M, Morzycki JW, Szczepek WJ, Frelek J. J Org Chem. 2007;72:2906–2916. doi: 10.1021/jo062445x. [DOI] [PubMed] [Google Scholar]; (c) Politi M, De Tommasi N, Pescitelli G, Di Bari L, Morelli I, Braca A. J Nat Prod. 2002;65:1742–1745. doi: 10.1021/np020260p. [DOI] [PubMed] [Google Scholar]; (d) Tang W-Z, Ma S-G, Yu S-S, Qu J, Liu Y-B, Liu J. J Nat Prod. 2009;72:1017–1021. doi: 10.1021/np9001702. [DOI] [PubMed] [Google Scholar]
- 23.(a) Su BN, Park EJ, Mbwambo ZH, Santarsiero BD, Mesecar AD, Fong HHS, Pezzuto JM, Kinghorn AD. J Nat Prod. 2002;65:1278–1282. doi: 10.1021/np0202475. [DOI] [PubMed] [Google Scholar]; (b) Seco JM, Quiñoá E, Riguera R. Chem Rev. 2004;104:17–118. doi: 10.1021/cr2003344. [DOI] [PubMed] [Google Scholar]
- 24.(a) Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]; (b) Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, Tonks NK, Møller NPH. FASEB J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]; (c) Bialy L, Waldmann H. Angew Chem Int Ed Engl. 2005;44:3814. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein BJ. Curr Drug Targets Immune Endocr Metabol Disord. 2001;1:265–275. doi: 10.2174/1568008013341163. [DOI] [PubMed] [Google Scholar]
- 26.Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, Tremblay ML, Russell SM, Godfrey DI, Tiganis T. J Clin Invest. 2011;121:4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiganis T. FEBS J. 2013;280:445–458. doi: 10.1111/j.1742-4658.2012.08563.x. [DOI] [PubMed] [Google Scholar]
- 28.Wu C, Sun M, Liu L, Zhou GW. Gene. 2003;306:1–12. doi: 10.1016/s0378-1119(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 29.Viant C, Fenis A, Chicanne G, Payrastre B, Ugolini S, Vivier E. Nat Comm. 2014;5 doi: 10.1038/ncomms6108. [DOI] [PubMed] [Google Scholar]
- 30.Fan L-C, Shiau C-W, Tai W-T, Hung M-H, Chu P-Y, Hsieh F-S, Lin H, Yu H-C, Chen K-F. Oncogene. 2015;34:5252–5263. doi: 10.1038/onc.2014.445. [DOI] [PubMed] [Google Scholar]
- 31.U’Ren JM, Lutzoni F, Miadlikowska J, Arnold AE. Microb Ecol. 2010;60:340–353. doi: 10.1007/s00248-010-9698-2. [DOI] [PubMed] [Google Scholar]
- 32.Massimo NC, Devan MMN, Arendt KR, Wilch MH, Riddle JM, Furr SH, Steen C, U’Ren JM, Sandberg DC, Arnold AE. Microb Ecol. 2015;70:61–76. doi: 10.1007/s00248-014-0563-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 34.Davey ML, Currah RS. Am J Bot. 2009;96:1281–1288. doi: 10.3732/ajb.0900010. [DOI] [PubMed] [Google Scholar]
- 35.Swofford DL. Phylogenetic analysis using parsimony (*and other methods), Version 4. Sinauer Associates; Sunderland, MA: 2002. [Google Scholar]
- 36.(a) Martin KR, Narang P, Xu Y, Kauffman AL, Petit J, Xu HE, Meurice N, MacKeigan JP. PLoS One. 2012;7:e50217. doi: 10.1371/journal.pone.0050217. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen C, Cao M, Zhu S, Wang C, Liang F, Yan L, Luo D. Sci Rep. 2015;5 doi: 10.1038/srep17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.