

Abstract
Tuberculosis drug discovery has shifted in recent years from a primarily target-based approach to one that uses phenotypic high-throughput screens. As examples of this, through our EU-funded FP7 collaborations, New Medicines for Tuberculosis was target-based and our more-recent More Medicines for Tuberculosis project predominantly used phenotypic screening. From these projects we have examples of success (DprE1) and failure (PimA) going from drug to target and from target to drug, respectively. It is clear that we still have much to learn about the drug targets and the complex effects of the drugs on Mycobacterium tuberculosis. We propose a more integrated approach that learns from earlier drug discovery efforts that could help to move drug discovery forward.
Keywords: DprE1, NM4TB, MM4TB, Mycobacterium tuberculosis, PimA
Introduction
It is hard to believe we are still fighting a pathogen that has haunted the human race for thousands of years and yet Mycobacterium tuberculosis, which causes tuberculosis (TB), continues to infect millions of the global population [1,2] and kills ~1.5 million every year [3]. The discovery of antibiotics like streptomycin (discovered in 1944) and the ‘golden age’ of antibiotic drug discovery that followed, which led to isoniazid and pyrazinamide (1952), ethambutol (1961) and rifampicin (1963), remain the cornerstones of therapy to this day. After the approval of rifampicin in 1967 there was a gap of over 40 years until the approval of bedaquiline in 2012. The challenge we have faced since the introduction of these drugs is multidrug resistance (MDR; resistance to at least isoniazid and rifampicin); and occurrence of extensively-drug-resistant (XDR) strains, resistant to at least isoniazid and rifampicin, as well as to any fluoroquinolone and to any of the three second-line injectables (amikacin, capreomycin or kanamycin), thus they are very difficult to treat [5,6][s1]. Combined with the frequent co-infection with HIV, TB treatment represents a tough challenge. There is an urgent need for new therapies and shorter treatments than the usual 6 months for susceptible to over 1 year for drug-resistant TB [s2][7,8]. The current first-line TB drugs were introduced and translated from initial discovery to the clinic in just a couple of years. By contrast, bedaquiline took 16 years from the discovery of a related screening hit to approval [4]. It is apparent something has to change to improve the current situation.
There has been a recent call by Gopal and Dick [9] to go back to look at old TB drugs that are more like fragments. These fragments however tend to be reactive and counter to how drug discovery has evolved in general, which is to find molecules that are generally not reactive and that pass filters, alerts and are not pan-assay interference compounds (PAINS) [10]. From our past analysis, compounds active in vitro against M. tuberculosis tend not to follow these rules [11]. Yet fragments will more readily access M. tuberculosis, perhaps be substrates for enzymes and be metabolized to molecules that in turn target many different proteins and pathways [9]. For example, several of the first-line drugs for TB could be considered fragments (e.g., isoniazid and pyrazinamide) that were developed and have been in use for decades and we are still learning about how they work [9]. A large number of what we would now consider ‘fragment-type’ molecules has been tested in one or more mouse models over the past 70 years, providing a wealth of information (Figure 1). Pyrazinamide (PZA), a key component in shortening TB therapy, was even advanced based on its in vivo activity [12]. The drug is now known to be activated by pyrazinamidase/nicotinamidase PncA to pyrazinoic acid [13], which was proposed to affect membrane transport and energetics [14]. However, several studies suggest various other targets of PZA: the fatty acid synthase (FAS) I system was reported as a primary target of PZA in M. tuberculosis [15], which was later challenged [16]; whereas the ribosomal protein RpsA [17] or the aspartate decarboxylase PanD [18] were recently proposed as additional targets of PZA in M. tuberculosis. Similarly, the old antitubercular para-aminosalicylic acid was relatively recently shown to be a substrate for dihydropteorate synthase, and its active forms then go on to inhibit folate metabolism [19]. This discovery that catalysis rather than inhibition of an enzyme can be an important mechanism for targeting M. tuberculosis presents a new way of thinking about how we can develop drugs for this disease. It has been suggested that no single approach is sufficient for predicting the target or mechanism of an antibiotic [20].
Figure 1.

Partial clustering map of mouse in vivo data showing very small ‘fragments’ tested in mouse (green outline = in vivo active) [71].
The recent EU-funded FP7 collaboration New Medicines for Tuberculosis (NM4TB) was target-based whereas the more-recent More Medicines for Tuberculosis (MM4TB) project used phenotypic screening predominantly with a small degree of target-based screens for targets like gyrase and topoisomerase I. These provide us with examples of projects that consider either target to drug or drug to target approaches [8,21]. The aim of this review is to present our experience with these drug development efforts pointing to specific issues in each, put them into context with the broader picture of M. tuberculosis drug discovery and propose some options for future work.
The PimA story: from a promising target to no drug (so far)
Right from the beginning of the NM4TB project a glycosyltransferase PimA (Rv2610) appeared to be a perfect target for rational drug development. It should be noted that this was well before the review by Payne et al., which critically analyzed experience with the target-based screening approach at GlaxoSmithKline (GSK) with other bacteria [22]. PimA attaches a mannosyl residue from GDP-mannose to phosphatidyl-myo-inositol (PI) producing phosphatidyl-myo-inositol monomannoside (PIM1) [23]. By further mannosylations and arabinosylations this metabolic intermediate is converted to prominent mycobacterial glycoconjugates – phosphatidyl-myo-inositol mannosides (Acyl1–2PIM2–6), lipomannan (LM) and lipoarabinomannan (LAM), which serve as important structural elements of the cell envelope [24,25] and also as key players for establishment of the human infection through their interaction with the immune system of the host [26]. In the NM4TB project, PimA was classified as a prioritized ‘category 1 target’ based on essentiality data [23], availability of the protein and its structure [27], convenience of primary (spectrophotometric) and secondary (radiometric) assays and a lack of a human homolog. Thorough optimization of the spectrophotometric assay did not result in its adaptation to high-throughput formats, probably because of problems related to the hydrophobic character of the acceptor substrate PI and/or later-recognized specific properties of the protein [28–30]. Numerous screens were thus performed (Z. Svetlíková, PhD thesis, Comenius University in Bratislava, 2014) in the alternative medium-throughput radiometric assays with PI as an acceptor substrate and purified enzyme [31], or with crude membranes of the Escherichia coli strain producing PimA to satisfy its requirement for membrane association [27]. More than 1000 compounds were examined in these assays that originated from several sources. A set of small molecules was selected from the ChemDiv library (Chemical Diversity, San Diego) using the programs FlexX (Chemical Computing Group, Canada & BioSolveIT, Germany) and ICM (International Coordinate Mechanics, MolSoft L.L.C., USA) based on calculations of their binding affinities to the GDP-Man-binding site of the protein performed by David Giganti and Marcelo Guerin (D. Giganti, PhD thesis, Pasteur Institute, Paris, 2008). Other sets of molecules, provided by our collaborators (György Kéri and János Pató, Vichem, Budapest; Vadim Makarov, Research Center of Biotechnology, Russian Academy of Science, Moscow) reflected structural features of the GDP-Man substrate. In each selection we were able to identify several efficient inhibitors of PimA (>50% inhibition at concentrations <30 μM or 50 μM, depending on the assay), which inhibited the growth of Mycobacterium smegmatis mc2155 with MICs of 9.35–53 μg/ml in the resazurin microtiter assay (REMA). The mechanism of killing caused by these compounds in mycobacteria was examined by radiolabeling of the M. smegmatis mc2155 cultures treated with [14C]-glucose and analysis of the extracted lipids by thin-layer chromatography. This classical biochemistry approach allowed us to focus directly on the expected changes caused by the PimA enzyme inhibitors. The lipid profiles of the drug-treated bacteria should be similar to those obtained by transcriptional silencing of PimA in the conditional mutant of M. tuberculosis, which was prepared in the TetR-Pip off-configuration [s3](i.e., a decrease in PIMs accompanied by accumulation of PI) [32]. Unfortunately, none of the tested compounds showed this phenotype and we concluded that the inhibition of the bacterial growth was not due to PimA targeted by these molecules in mycobacteria (Z. Svetlíková, PhD thesis, Comenius University in Bratislava, 2014). Thus, although PimA is one of only a few targets validated in vivo [32,33], studies to date have so far failed to identify a drug-like inhibitor of PimA inside cells and the project was terminated.
The DprE1 story: from carefully optimized drug to a promiscuous but vulnerable target
Contrary to PimA, DprE1 is an enzyme that was reported as a target of a number of small molecules efficiently killing mycobacteria not only in vitro but also in mouse models of TB (Table 1). The FAD-containing oxidoreductase DprE1 (Rv3790), along with its partner DprE2 (Rv3791), is involved in the conversion of decaprenylphosphoryl-β-d-ribose (DPR) to decaprenylphosphoryl-β-d-arabinose (DPA) [34] – the exclusive donor of arabinofuranose for biosynthesis of fundamental polysaccharides of the mycobacterial cell wall: arabinogalactan and lipoarabinomannan [35]. Its value as a drug target was recognized through a search for the mechanism of action of benzothiazinones (BTZs), sulfur-containing heterocycles, among which derivatives with a nitro group at the 8 position displayed spectacular in vitro, ex vivo and in vivo activities toward mycobacteria. BTZ043, S stereoisomer of 2-[2-methyl-1,4-dioxa-8-azaspiro[4.5]dec-8-yl]-8-nitro-6-(trifluoromethyl)-4H-1,3-benzothiazin-4-one, was selected as a lead compound for further studies [36].
Table 1.
Selected DprEI inhibitors with in vitro and in vivo activity
| Molecule | Name | IC50a | MIC for Mycobacterium tuberculosis | Mouse in vivo | Refs |
|---|---|---|---|---|---|

|
BTZ043 | 0.004 25 μM |
1 ng/ml | Chronic model at 37.5 mg/kg Active | [36,93] |

|
Shirude 3 | 0.01–0.017 μM | 1.56–3.12 μM | Acute and chronic model 100 mg/kg Active | [94] |

|
Shirude 4 | 0.005 | 0.39 μM | Acute and chronic model 100 mg/kg Active | [94] |

|
Chatterji 2 | 0.032 | 0.5–1.56 μM | Chronic model 300 mg/kg Active | [95] |
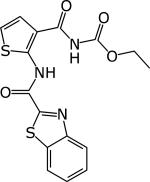
|
TCA1 | 0.19 (μg/ml MIC50) | Tested in chronic and acute model at 100 mg/kg Active | [45] | |
|
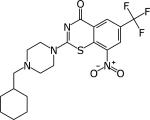
PBZ169 | 0.3 ng/ml ≤0.19 ng/ml |
Chronic model at 50 mg/kg 0.6 log CFU reduction in lung |
[50,93] |
DprE1 activity was established by Amplex Red/horseradish peroxidase assay [43].
The target of BTZs was revealed by two independent genetic approaches, which led to disclosure of DprE1 as the target [36]. First, the cosmids from a M. smegmatis library, conferring resistance to BTZ043, were selected in M. smegmatis and subcloning experiments pinpointed to dprE1. Second, specific point mutations were found in dprE1 genes in the highly resistant mutants of M. smegmatis, Mycobacterium bovis BCG and M. tuberculosis raised against BTZ043. Biochemical studies fully supported DprE1 as a target of BTZs [36]. These included: (i) labeling of BTZ-treated M. smegmatis cultures with [14C] glucose, which clearly showed a decrease in cell wall arabinans compared to control reminiscent of the effects of ethambutol [37]; and (ii) cell-free experiments. Initially, enzymology studies were performed in a crude assay with membrane and cell envelope fractions and 5-phospho-[14C]-ribose diphosphate (P-[14C]-RPP) as a tracer [38], which revealed arrest of conversion of DPR to DPA in the presence of BTZ043. SAR studies showed that the nitro group at position 8 is crucial for the activity of BTZs in whole cells. Accordingly, in the cell-free experiment the amino-derivative BTZ045 was completely inactive [36]. This finding was in agreement with the subsequently confirmed mode-of-action of BTZ043, in which DprE1-bound FADH2 produced by DPR oxidation serves as an electron donor for reduction of NO2 in BTZ043. Conversion of BTZ043 to the reactive nitroso-intermediate, followed by its covalent attachment to the active site cysteine, irreversibly inactivates the enzyme [39,40]. Mutations resulting in replacement of this cysteine to glycine or serine are principal mechanisms of resistance toward BTZs and other nitro-group-containing inhibitors [41]. Later, availability of pure recombinant DprE1 protein from M. smegmatis produced in E. coli and recognition of farnesylphosphoryl ribose (FPR) [42] as a possible substrate surrogate for DprE1 enabled structural studies that led to the clarification of the FAD-dependent mechanism of enzyme inhibition by BTZ043 but also allowed development of ‘clean assays’ employing only the DprE1 enzyme and FPR as the main components [40,43]. These assays take advantage of reduction of FAD accompanying oxidation of FPR substrate during the enzyme cycle. The reaction is then monitored spectrophotometrically using 2,6-dichlorophenolindophenol (DCPIP) as an electron acceptor for reoxidation of FADH2 or, alternatively, in a coupled reaction with Amplex Red and horseradish peroxidase, which react with hydrogen peroxide formed from oxygen serving as an electron acceptor. Interestingly, the amino derivative BTZ045 acted as an efficient noncovalent inhibitor of DprE1 with an IC50 value similar to BTZ043 in these assays [43], although essentially no activity of this compound was observed in the crude assay [36]. These observations are comparable with recent studies by Tiwari et al., who evaluated a BTZ043 analog with the azido-group instead of nitro-group in the 8 position [44]. Although BTZ-N3 was an efficient reversible and noncovalent inhibitor of DprE1 with IC50 9.6 ±0.5 μM in the assay with DCPIP, which is similar to other BTZs [43], examination of its effects in the crude assay did not show any inhibition of DPA production even at a rather high concentration of the compound (100 μg/ml). This is intriguing, because amino- and azido-BTZ analogs have MICs for M. tuberculosis corresponding to 0.5 μg/ml. Nevertheless, failure to show the inhibitory effects in the crude assay is not related to the noncovalent mode of inhibition of the BTZ045 and BTZ-N3, because another noncovalent inhibitor, TCA1 (Table 1), inhibited DPA production efficiently in a dose-dependent manner in this assay [45].
Another version of a clean assay developed and carefully optimized by Batt et al. comprises an E.-coli-produced recombinant DprE1 from M. tuberculosis, FPR and resazurin as substrates [46]. It was used in the search for compounds targeting DprE1 in the GSK publicly available collection (TB set) [47] by an integrated approach employing target-based whole-cell screening with M. bovis BCG overexpressing DprE1 from M. tuberculosis. Out of five compounds conferring increased resistance in this strain compared with the empty vector control, GSK710 was selected for further studies based on the most profound MIC shift between the control and the DprE1-overproducing strain. Interestingly, an enzymology study of the full collection of 177 compounds revealed 29 hits showing >30% inhibition. Their further evaluation showed 11 compounds with IC50 values between 10 and 60 μM, seven compounds between 3 and 10 μM and two compounds <1 μM. Although one of these two compounds was GSK710, these data again point to the issues reported for enzyme target-based screening [22] because, from the spectrum of potent DprE1 inhibitors in the enzyme assay, only one molecule was confirmed to act efficiently on the target in mycobacteria [46].
Soon after its discovery, DprE1 was given an attribute – a magic target [48]. During the following years, when numerous compounds with unrelated structures were discovered from various whole-cell screening campaigns (reviewed by Piton et al. in this issue of Drug Discovery Today), it was attributed a less favorable characteristic (i.e., promiscuous) [8]. However, this feature can be related to accessibility of DprE1 located in the periplasmic space of the mycobacterial cell wall, which makes it particularly vulnerable to the action of the drugs [49]. Nevertheless, despite a variety of compounds targeting DprE1 in mycobacteria, the original BTZ BTZ043 and the optimized next-generation PBTZ169 derivative (Table 1) are the only DprE1 inhibitors in the current TB drug pipeline (http://www.newtbdrugs.org/pipeline.php) that are reported to be in the preclinical phase. The in vivo efficacy data in zebrafish and mouse supports this series as well as combination studies with bedaquiline and pyrazinamide, which were superior to isoniazid, rifampicin and pyrazinamide [50]. In fact, under the iM4TB/Nearmedic partnership, PBTZ169 has already entered early clinical studies in Russia (http://www.nearmedic.ru/en/node/690).
It should be noted that, in contrast to most DprE1 inhibitors, BTZs were not discovered by the classical or modern variants of high-throughput whole-cell screening campaigns of available compound libraries. Quite the opposite, they were produced after careful investigation of the fate of dialkyldithiocarbamates (DDTCs) in mycobacteria and animals. Following the discovery that DDTCs with high antimycobacterial activity are converted to BTZs, a number of their derivatives were synthesized. SAR studies driven by their antimycobacterial activity and favorable in vivo properties (such as plasma protein binding, metabolic stability, etc.) identified BTZ043 as a drug candidate for further studies [36]. We thus believe that the value of the DprE1 target for TB drug development currently lies in the superiority of the original and carefully optimized compound, represented by the lead candidate PBTZ169. Nevertheless, given the high attrition rates at all stages of the TB drug development process, it is reassuring that for DprE1, which appears to be a particularly attractive and vulnerable target, there are a number of inhibitors available, providing opportunities for further backup development.
Searching for good drugs and targets
Since the discovery of TMC207 (bedaquiline) and successful identification of its target as ATP-synthase by way of isolation of resistant mutants followed by whole-genome sequencing [51], this method became widely used and represents a standard procedure in the search for the mechanism-of-action of the drugs of interest. However, by its nature this approach frequently does not reveal just the target but also mechanisms responsible for resistance or activation of the drug [52,53]. As pointed out by Lechartier et al. many times, it is not even possible to isolate the resistant mutants, which makes target identification and validation problematic [8]. In such cases transcriptional profiling before and after drug exposure were proposed for shedding light on the effects of the drug in the bacterium [54]. More recently, proteomics and metabolomics approaches proved to be valuable for revealing or confirming drug targets in mycobacteria. Their potential can be exemplified by the study showing that d-Ala–d-Ala-ligase, rather than alanine racemase, is a predominant target of d-cycloserine in mycobacteria [55]; or by a recent report about glutamate racemase (MurI), which was discovered as a primary target of β-chloro-d-alanine and perspective candidate for TB drug development [56]. There have also been several efforts at computational target prediction. For example the hits from M. tuberculosis HTS from GSK were analyzed with two ligand-based methods to predict targets for 776 molecules. The selected targets were then used in a structure-based strategy allowing identification of two molecules identified as inhibitors of DHFR, which acted on this target in mycobacteria as confirmed experimentally [57]. Although “currently, there is poor consensus on the key properties that constitute the ideal drug target for eliminating M. tuberculosis” [58], sophisticated methods were designed for genetic validation of TB drug targets, which include transcriptional silencing but also systems for regulated protein degradation and their combinations. Recently these methods were extensively reviewed by Evans and Mizrahi, who critically highlighted advantages and limitations of these approaches [33]. Major strengths of these state-of-the-art technologies is the possibility to evaluate the targets in vivo, which along with structure-based drug design (SBDD) approaches could [s4]lead to success in obtaining drug candidates. Perhaps, even the value of PimA for drug development could be re-examined by these technologies given the wealth of information about the enzyme and its crucial role in the physiology of mycobacteria.
Looking back on the way forward
Examination of the mechanisms of action of the novel drugs and drug candidates in preclinical and clinical phases listed in the TB Drug Development Pipeline (http://www.newtbdrugs.org/pipeline.php) reveals that, except for three compounds (bedaquiline, the imidazopyridine Q203 and the riminophenazine TBI-166) targeting energy metabolism of mycobacteria, all of the other compounds affect the metabolic pathways, which are considered as highly vulnerable for killing bacteria, because they were chosen by evolution as targets of many natural compounds. These include synthesis of the cell wall (targeted by pretomanid, delamanid, caprazamycin CPZEN-45, dipiperidine SQ609, BTZs PBTZ169 and BTZ043, ethylendiamine SQ109), proteins (sutezolid, spectinamide 1599) or nucleic acids (moxifloxacin, levofloxacin, rifapentine) and represent the same pathways affected by the antibacterial drugs used in a systemic monotherapy [59]. As stated by Silver, “it might be that the ‘good old targets’ are qualitatively different from the crop of all possible novel targets” [59]. Therefore, it might be worth considering that, along with the search for the target by genetic methods, the classical macromolecular synthesis (MMS) assays are applied more frequently in the TB field. It is highly likely that the most straightforward method is the use of radiolabeled precursors for nucleic acids, protein and cell wall and lipid synthesis, as in the ‘golden age’ of antitubercular drug discovery. The advantage is that it is not costly or technically demanding and thus can be performed almost by any laboratory. This approach was recently reported for the novel natural compound teixobactin with an excellent activity against Gram-positive pathogens, including M. tuberculosis [60]. Because it was not possible to obtain mutants of Staphylococcus aureus or M. tuberculosis resistant to teixobactin, its mechanism-of-action was examined by MMS assays in S. aureus. These pointed to peptidoglycan synthesis as a target of the drug. Further studies then specified that the antibiotic acts by binding to the polyprenyl-phosphate-linked precursors of the cell wall. As noted by the authors of this study, teixobactin is an example “how natural products evolved to exploit the inherent weaknesses of bacteria” and, probably, especially among the natural products, more compounds targeting the most vulnerable pathways will be found [60]. This can be exemplified by recent discoveries of pyridomycin, a cell wall inhibitor targeting InhA [61], or griselimycins, which are DNA synthesis inhibitors targeting DnaN [62].
For TB drug candidates MMS assays were recently reported for investigation of the mode-of-action of SQ109 [63] or AU1235 [64], which turned out to be MmpL3 inhibitors. In fact, examination of the effects of the drugs with different cell wall targets in mycobacteria by [14C]-acetate or [14C]-glucose labeling reveals ‘signatures’ in the lipid profiles, which are typical for targets in a specific part of the pathway [i.e., accumulation of trehalose dimycolates (TDM) and trehalose monomycolates (TMM)], for inhibitors of arabinan biosynthesis, lack of TDM and increased TMM for MmpL3 inhibitors, complete loss of TDM and TMM for inhibitors of mycolate production. It should be noted that, for a specific inhibitor of the macromolecular synthesis, there should be a clear distinction in the effects of a particular pathway over the others, because inhibition of all pathways within a narrow concentration range could indicate a nonspecific mechanism of inhibition [65]. Thus, for their use at present, these assays should be carefully optimized using the drugs with known modes of action, taking into consideration the specific effects of the media used for the cultivation of mycobacteria [66]. These kinds of experiments might also be useful with regard to the recent opinion by Gopal and Dick highlighting the fact that some of the key drugs in TB therapy are promiscuous, hitting multiple targets [9]. Although MMS experiments will probably not reveal the precise target of the drug, indication of the activity in a highly vulnerable pathway could warrant further development, including optimization of their properties and their progress to in vivo studies.
Using machine learning to learn from over 70 years of TB research
Indeed, a significant challenge in TB research is identifying compounds that have activity in the mouse models of M. tuberculosis infection [67]. Certainly the mouse differs from the human but it represents a frequently used model for rank ordering compounds for further study [68]. Recent efforts have manually collated historic data for compounds tested in the mouse model of infection [67,69]. Admittedly this represents a combination of data from different strains of mice and acute and chronic mouse models covering over 70 years of data. This resulted in two studies initially with 784 molecules [67] and then, in recent years, the addition of 60 molecules brought this to well over 800 molecules. The compounds were analyzed in terms of their molecular properties and used for generating computational models of in vivo efficacy that were prospectively used to predict other molecules. Our first analysis also enabled us to take a high-level view of the compounds assessed in the mouse over time [4] and the clear gaps in drug discovery. The more-recent curation of 60 additional small molecules with in vivo data published during 2014 and 2015 [69] suggests a general lack of diversity of compounds going in vivo. We have also used multiple computational approaches to analyze these molecules and we showed that models to predict other properties such as mouse liver microsomal half-life (MLM t1/2) and in vitro M. tuberculosis activity incorporating cytotoxicity data could also be used to predict in vivo activity. The creation of this consensus workflow had a positive predicted value (hit rate) >77%. Such computational models can help select antitubercular compounds with desirable in vivo efficacy alongside good MLM t1/2 and in vitro activity [69]. We also found that a new clustering method for data visualization might also help with the assessment of in vivo activity by placing a new molecule of interest in the context of the closest molecules based on molecular descriptors (Figure 1).
Making these data publically accessible is potentially useful for revisiting old compounds that might have failed but which might be amenable to being revived based on what we know now about M. tuberculosis. Indeed, we are increasingly revisiting the past. Examples include the rediscovery of the triazine series of compounds [70] (Figure 2), which followed on from their initial detection in 1969 [71], or oxyphenbutazone [72], which was rediscovered from studies in the 1960s [73] in which patients with TB were given this drug for treatment of pain and fever. Even natural products are not immune to rediscovery because gliotoxin was recently identified in a screen to have antitubercular activity [74] but it had already been described in 1950 [75], and again more recently [76]. We identified this because we had curated over 700 molecules with their potential targets from literature publications in a mobile app called TB Mobile [77,78]. Resources like this [79,80], mouse in vivo compounds [67,69] and literature in vitro screening data from SRI/NIAID [81-83] that has been collated in CDD (http://www.collaborativedrug.com) [11,84] all represent starting points for potential knowledge-based selection of compounds for further follow-up or design. Using the computational models or the data on their own can help in understanding the properties that confer in vitro activity, in vivo activity as well as the multidimensional properties such as cytotoxicity and microsomal stability (Figure 3). However, are we using the historic data to help make better decisions?
Figure 2.

Partial clustering map of mouse in vivo data (green = in vivo active) showing triazines [71].
Figure 3.

Using published data on TB to aid drug discovery, integrating data on Mycobacterium tuberculosis targets, machine learning models using M. tuberculosis in vitro, in vitro data and molecular property predictors. Abbreviation: Mtb, M. tuberculosis.
In 2010 GSK published a set of 14 000 compounds with antimalarial activity [85]. We used these compounds as a testset for two Bayesian models: one was generated with ~220 000 compounds with single-point screening data and a second used ~2200 compounds with dose–response data from the Molecular Libraries Small Molecule Repository (MLSMR) (data from [81]), which had been recently published [84]. These prediction data for M. tuberculosis activity were subsequently published in the CDD public database and provided as supplementary data in a paper analyzing various datasets [86]. In 2013, GSK published the results of phenotypic screening of 2 million compounds, which identified a set of 177 leads [47]. After downloading the flat file from ChEMBL-NTD, the molecules and data were uploaded in to CDD and we looked at overlap of this set with the previous Bayesian model predictions generated by us in 2010 (Figure 4). Fifteen molecules were found to overlap in the datasets. Upon closer examination of the Bayesian model predictions we found that each unique model correctly predicted 11 out of 15 compounds (73.3% correct classification). This illustrates how databases such as CDD can be used as a repository for computational predictions, which can be accessed and compared years later as experimental data becomes accessible.
Figure 4.

Comparison of Bayesian model predictions and experimental data against Mycobacterium tuberculosis for GSK compounds (green = correct prediction, red = incorrect prediction, yellow = M. tuberculosis data).
Clearly, few people are taking advantage of what has been done to assist TB drug discovery. How can we change this? Several examples of drug repurposing could help. The identification of the proton pump inhibitor lansoprazole which is reduced to an active metabolite intracellularly and shown to possess in vivo activity in mouse [87] is a recent example derived from screening FDA-approved drugs in vitro. Target-based computational models have led to approved drug compounds that were not previously considered as antibiotics, with good activity against M. tuberculosis Topo I (e.g., amsacrine, imipramine and norclomipramine) [88,89] and ThyX (idebenone) [90] but with poorer MICs. So this gets to the additional challenge of finding molecules that hit the sweet spot of in vitro activity at the target and in the whole cell, and that is before trying in vivo.
Concluding remarks
Some have discussed taking more of a mechanism-based approach to TB drug development [20] or a pathway-based approach to screening [58]. A recent opinion [91] has also laid out criteria for antituberculous hits and leads to ensure the quality of compounds, in particular focusing on the massive Global Health Innovative Technology Fund's high-throughput screen being undertaken in Japan against M. tuberculosis. It is unclear how this is going to address the still fundamental problem of a poor preclinical and clinical pipeline [7]. Granted there is still the huge leap from in vitro screen to activity in the mouse model but are we setting the standards unacceptably high compared with tuberculosis drug discovery of the past?
We have focused on our experiences with a very small number of targets and methods, however many of the successes and challenges in the MM4TB project will be summarized in other articles in this issue. From our experience there is no guarantee of success using any of the approaches taken and there is certainly a major role for good fortune.
As we have stated, there is considerable information in the public domain on compounds that are active either in vitro or in vivo against M. tuberculosis and increasingly we are finding approved drugs that have some degree of whole-cell and/or target activity. Although repurposing an already approved drug would certainly shave many years off the drug development timeline, for M. tuberculosis it represents a ‘Hail Mary’ approach. The shifts in the TB drug discovery community, whether driven by the failure of big pharma with their target-based screens for other bacterial targets [22] or funding organizations prioritizing whole-cell screens over target-based screens, can be seen as having an influence. What really matters to many is finding compounds active in the mouse acute or chronic model of M. tuberculosis [4].
We propose that future collaborative projects could follow MM4TB and learn from our experiences. These might use a more diverse array of methods that spread the risk and include the following ten examples:
Going back to earlier published in vivo data and finding compounds that failed and trying to fix the flaws caused by poor ADME, improving solubility and or metabolic stability by using computational models and in vitro testing [92].
Use the computational models to identify compounds that could be active in vivo [67,69] from the 1000s of published in vitro hits.
Identify compounds likely to be metabolically activated (e.g., norclomipramine is a metabolite of clomipramine, and only the former is active vs M. tuberculosis topoisomerase I in vitro [89]).
Optimize the delivery of compounds or their formulation to get in vivo activity.
Focus on old well-validated targets and use all available experimental and computational resources (e.g., InhA, DHFR, etc.).
Virtually screen small fragment libraries with the machine learning models (e.g., in vivo and in vitro).
Use NMR to screen fragment libraries versus validated M. tuberculosis targets and use as a starting point for structure-based design.
Get active compounds to in vivo studies faster by using MMS assays and biochemistry for evaluation of the affected pathways.
Mine through all the compounds discarded in NM4TB, MM4TB and possibly other large collaborations and identify those that could be optimized.
Mine through the compounds in NM4TB and MM4TB and predict targets using computational approaches, verify in vitro and use as a starting point for target to drug.
Highlights.
We explain the shift from target-based to phenotypic HTS for TB
We describe EU-funded FP7 collaborations, NM4TB and MM4TB projects
We detail the success with DprE1 and failure with PimA
We suggest how we can learn from earlier in vitro and in vivo efforts and data
We propose future approaches to TB drug discovery
Acknowledgments
We gratefully acknowledge our NM4TB and MM4TB colleagues for their kind support and, in particular, Dr Vadim Makarov and Professor Stewart Cole. S.E. kindly acknowledges the collaboration and discussions with Dr Joel S. Freundlich, Dr Alex M. Clark, Dr Alexander L. Perryman and Dr Robert Reynolds. K.M. would like to thank Dr Jana Korduláková for discussions and for critically reading the manuscript, as well as current and former students for performing the experiments mentioned in this review. The work was supported by grants from the European Community's Sixth and Seventh Framework Programs (grant LHSP-CT-2005-018923, NM4TB Consortium; grant 260872, MM4TB Consortium). K.M. acknowledges current support by the Ministry of Education, Science, Research and Sport of the Slovak Republic (grant VEGA 1/0441/15 and grant 0395/2016 for Slovak/Russian cooperation in science 2015-15075/33841:1-15E0) and the Slovak Research and Development Agency (APVV-15-0515). S.E. acknowledges that the machine learning work described was partially supported by Award Number 9R44TR000942-02 ‘Biocomputation across distributed private datasets to enhance drug discovery’ from the NIH National Center for Advancing Translational Sciences.
We dedicate this review to our dear colleagues who sadly passed away far too soon: Dr Barry Furr (from our Scientific Advisory Board) and Dr György Kéri (Vichem).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Teaser: We describe the shift from a target-based approach to one that uses phenotypic high-throughput screens providing examples of success and failure from recent large-scale FP7 collaborations for tuberculosis drug discovery.
References
- 1.WHO Global Tuberculosis Report. 2013 Available at: http://apps[s5].who.int/iris/bitstream/10665/91355/1/9789241564656_eng.pdf.
- 2.WHO Global Tuberculosis Report. 2014 http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf?ua=1.
- 3.Lonnroth K, et al. Towards tuberculosis elimination: an action framework for low-incidence countries. Eur. Respir. J. 2015;45:928–952. doi: 10.1183/09031936.00214014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekins S, et al. Minding the gaps in tuberculosis research. Drug Discov. Today. 2014;19:1279–1282. doi: 10.1016/j.drudis.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 5.Jakab Z, et al. Consolidated action plan to prevent and combat multidrugand extensively drug-resistant tuberculosis in the WHO European region 2011–2015: cost-effectiveness analysis. Tuberculosis. 2015;95(Suppl. 1):212–216. doi: 10.1016/j.tube.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 6.Zignol M, et al. Drug-resistant tuberculosis in the WHO European region: an analysis of surveillance data. Drug Resist. Updat. 2013;16:108–115. doi: 10.1016/j.drup.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Wong EB, et al. Rising to the challenge: new therapies for tuberculosis. Trends Microbiol. 2013;21:493–501. doi: 10.1016/j.tim.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lechartier B, et al. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 2014;6:158–168. doi: 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gopal P, Dick T. Reactive dirty fragments: implications for tuberculosis drug discovery. Curr. Opin. Microbiol. 2014;21:7–12. doi: 10.1016/j.mib.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 11.Ekins S, et al. Analysis and hit filtering of a very large library of compounds screened against Mycobacterium tuberculosis. Mol. BioSyst. 2010;6:2316–2324. doi: 10.1039/c0mb00104j. [DOI] [PubMed] [Google Scholar]
- 12.Malone L, et al. The effect of pyrazinamide (aldinamide) on experimental tuberculosis in mice. Am. Rev. Tuberc. 1952;65:511–518. [PubMed] [Google Scholar]
- 13.Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996;2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003;52:790–795. doi: 10.1093/jac/dkg446. [DOI] [PubMed] [Google Scholar]
- 15.Zimhony O, et al. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat. Med. 2000;6:1043–1047. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- 16.Boshoff HI, et al. Effects of pyrazinamide on fatty acid synthesis by whole mycobacterial cells and purified fatty acid synthase I. J. Bacteriol. 2002;184:2167–2172. doi: 10.1128/JB.184.8.2167-2172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi W, et al. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science. 2011;333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi W, et al. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerg. Microbes Infect. 2014;3:e58. doi: 10.1038/emi.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakraborty S, et al. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science. 2013;339:88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakraborty S, Rhee KY. Tuberculosis drug development: history and evolution of the mechanism-based paradigm. Cold Spring Harb. Perspect. Med. 2015;5:a021147. doi: 10.1101/cshperspect.a021147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sala C, Hartkoorn RC. Tuberculosis drugs: new candidates and how to find more. Future Microbiol. 2011;6:617–633. doi: 10.2217/fmb.11.46. [DOI] [PubMed] [Google Scholar]
- 22.Payne DJ, et al. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 23.Kordulakova J, et al. Definition of the first mannosylation step in phosphatidylinositol mannoside synthesis. PimA is essential for growth of mycobacteria. J. Biol. Chem. 2002;277:31335–31344. doi: 10.1074/jbc.M204060200. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda T, et al. Critical roles for lipomannan and lipoarabinomannan in cell wall integrity of mycobacteria and pathogenesis of tuberculosis. MBio. 2013;4:e00472–00412. doi: 10.1128/mBio.00472-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bansal-Mutalik R, Nikaido H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc. Natl. Acad. Sci. U. S. A. 2014;111:4958–4963. doi: 10.1073/pnas.1403078111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishra AK, et al. Lipoarabinomannan and related glycoconjugates: structure, biogenesis and role in Mycobacterium tuberculosis physiology and host-pathogen interaction. FEMS Microbiol. Rev. 2011;35:1126–1157. doi: 10.1111/j.1574-6976.2011.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guerin ME, et al. Molecular recognition and interfacial catalysis by the essential phosphatidylinositol mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 2007;282:20705–20714. doi: 10.1074/jbc.M702087200. [DOI] [PubMed] [Google Scholar]
- 28.Giganti D, et al. Conformational plasticity of the essential membrane-associated mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 2013;288:29797–29808. doi: 10.1074/jbc.M113.462705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giganti D, et al. Secondary structure reshuffling modulates glycosyltransferase function at the membrane. Nat. Chem. Biol. 2015;11:16–18. doi: 10.1038/nchembio.1694. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigo-Unzueta A, et al. Molecular basis of membrane association by the phosphatidylinositol mannosyltransferase PimA enzyme from mycobacteria. J. Biol. Chem. 2016;291:13955–13963. doi: 10.1074/jbc.M116.723676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sipos A, et al. Lead selection and characterization of antitubercular compounds using the Nested Chemical Library. Tuberculosis. 2015;95(Suppl. 1):200–206. doi: 10.1016/j.tube.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 32.Boldrin F, et al. The phosphatidyl-myo-inositol mannosyltransferase PimA is essential for Mycobacterium tuberculosis growth in vitro and in vivo. J. Bacteriol. 2014;196:3441–3451. doi: 10.1128/JB.01346-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans JC, Mizrahi V. The application of tetracyclineregulated gene expression systems in the validation of novel drug targets in Mycobacterium tuberculosis. Front. Microbiol. 2015;6:812. doi: 10.3389/fmicb.2015.00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikusova K, et al. Decaprenylphosphoryl arabinofuranose, the donor of the d-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J. Bacteriol. 2005;187:8020–8025. doi: 10.1128/JB.187.23.8020-8025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolucka BA, et al. Recognition of the lipid intermediate for arabinogalactan/arabinomannan biosynthesis and its relation to the mode of action of ethambutol on mycobacteria. J. Biol. Chem. 1994;269:23328–23335. [PubMed] [Google Scholar]
- 36.Makarov V, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikusova K, et al. Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 1995;39:2484–2489. doi: 10.1128/aac.39.11.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scherman MS, et al. Polyprenylphosphate-pentoses in mycobacteria are synthesized from 5-phosphoribose pyrophosphate. J. Biol. Chem. 1996;271:29652–29658. doi: 10.1074/jbc.271.47.29652. [DOI] [PubMed] [Google Scholar]
- 39.Trefzer C, et al. Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl-beta-d-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010;132:13663–13665. doi: 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- 40.Trefzer C, et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-beta-d-ribofuranose 2′-oxidase DprE1. J. Am. Chem. Soc. 2012;134:912–915. doi: 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- 41.Mikusova K, et al. DprE1 –from the discovery to the promising tuberculosis drug target. Curr. Pharm. Des. 2014;20:4379–4403. doi: 10.2174/138161282027140630122724. [DOI] [PubMed] [Google Scholar]
- 42.Liav A, et al. Stereoselectivity in the synthesis of polyprenylphosphoryl β-d-ribofuranoses. Tetrahedron Lett. 2006;47:8781–8783. [Google Scholar]
- 43.Neres J, et al. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci. Transl. Med. 2012;4:150ra121. doi: 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tiwari R, et al. Design, syntheses, and anti-TB activity of 1,3-benzothiazinone azide and click chemistry products inspired by BTZ043. ACS Med. Chem. Lett. 2016;7:266–270. doi: 10.1021/acsmedchemlett.5b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F, et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E2510–2517. doi: 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batt SM, et al. Whole cell target engagement identifies novel inhibitors of Mycobacterium tuberculosis decaprenylphosphoryl-β-d-ribose oxidase. ACS Infectious Diseases. 2015;1:615–626. doi: 10.1021/acsinfecdis.5b00065. [DOI] [PubMed] [Google Scholar]
- 47.Ballell L, et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem. 2013;8:313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manina G, et al. Decaprenylphosphoryl-beta-d-ribose 2′-epimerase from Mycobacterium tuberculosis is a magic drug target. Curr. Med. Chem. 2010;17:3099–3108. doi: 10.2174/092986710791959693. [DOI] [PubMed] [Google Scholar]
- 49.Brecik M, et al. DprE1 is a vulnerable tuberculosis drug target due to its cell wall localization. ACS Chem. Biol. 2015;10:1631–1636. doi: 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- 50.Makarov V, et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014;6:372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andries K, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 52.Ioerger TR, et al. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS One. 2013;8:e75245. doi: 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Albesa-Jove D, et al. Rv2466c mediates the activation of TP053 to kill replicating and non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 2014;9:1567–1575. doi: 10.1021/cb500149m. [DOI] [PubMed] [Google Scholar]
- 54.Boshoff HI, et al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 55.Prosser GA, de Carvalho LP. Metabolomics reveal d-alanine:d-alanine ligase as the target of d-cycloserine in Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2013;4:1233–1237. doi: 10.1021/ml400349n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prosser GA, et al. Glutamate racemase is the primary target of beta-chloro-d-alanine in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2016 doi: 10.1128/AAC.01249-16. doi: 10.1128/AAC.01249-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mugumbate G, et al. Mycobacterial dihydrofolate reductase inhibitors identified using chemogenomic methods and in vitro validation. PLoS One. 2015;10:e0121492. doi: 10.1371/journal.pone.0121492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kana BD, et al. Future target-based drug discovery for tuberculosis? Tuberculosis. 2014;94:551–556. doi: 10.1016/j.tube.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silver LL. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 2007;6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
- 60.Ling LL, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hartkoorn RC, et al. Towards a new tuberculosis drug: pyridomycin –nature's isoniazid. EMBO Mol. Med. 2012;4:1032–1042. doi: 10.1002/emmm.201201689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kling A, et al. Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science. 2015;348:1106–1112. doi: 10.1126/science.aaa4690. [DOI] [PubMed] [Google Scholar]
- 63.Tahlan K, et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012;56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grzegorzewicz AE, et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012;8:334–341. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Silver LL. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pethe K, et al. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun. 2010;1:57. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ekins S, et al. Looking back to the future: predicting in vivo efficacy of small molecules versus Mycobacterium tuberculosis. J. Chem. Inf. Model. 2014;54:1070–1082. doi: 10.1021/ci500077v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Franzblau SG, et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis. 2012;92:453–488. doi: 10.1016/j.tube.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 69.Ekins S, et al. Machine learning model analysis and data visualization with small molecules tested in a mouse model of Mycobacterium tuberculosis infection (2014–2015). J. Chem. Inf. Model. 2016;56:1332–1343. doi: 10.1021/acs.jcim.6b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ekins S, et al. Bayesian models leveraging bioactivity and cytotoxicity information for drug discovery. Chem. Biol. 2013;20:370–378. doi: 10.1016/j.chembiol.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bruhin H, et al. Antituberculosis activity of some nitrofuran derivatives. J. Pharm. Pharmacol. 1969;21:423–433. doi: 10.1111/j.2042-7158.1969.tb08283.x. [DOI] [PubMed] [Google Scholar]
- 72.Gold B, et al. Nonsteroidal anti-inflammatory drug sensitizes Mycobacterium tuberculosis to endogenous and exogenous antimicrobials. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16004–16011. doi: 10.1073/pnas.1214188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoffmann K, Onoz E. Inhibitory effect of oxyphenbutazone against Mycobacterium tuberculosis in vitro]. Arzneimittelforschung. 1969;19:241–242. in German. [PubMed] [Google Scholar]
- 74.Stanley SA, et al. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 2012;7:1377–1384. doi: 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tompsett R, et al. Tuberculostatic activity of blood and urine from animals given gliotoxin. J. Immunol. 1950;65:59–63. [PubMed] [Google Scholar]
- 76.Nicholas GM, et al. Inhibition and kinetics of Mycobacterium tuberculosis and Mycobacterium smegmatis mycothiol-S-conjugate amidase by natural product inhibitors. Bioorg. Med. Chem. 2003;11:601–608. doi: 10.1016/s0968-0896(02)00345-0. [DOI] [PubMed] [Google Scholar]
- 77.Clark AM. TB Mobile. 2013 Available at: https://itunes.apple.com/us/app/tb-mobile/id567461644?mt=8.
- 78.Ekins S, et al. TB Mobile: a mobile app for anti-tuberculosis molecules with known targets. J. Cheminform. 2013;5:13. doi: 10.1186/1758-2946-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clark AM, et al. New target predictions and visualization tools incorporating open source molecular fingerprints for TB Mobile 2.0. J. Cheminform. 2014;6:38. doi: 10.1186/s13321-014-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ekins S, et al. Bayesian models for screening and TB Mobile for target inference with Mycobacterium tuberculosis. Tuberculosis. 2014;94:162–169. doi: 10.1016/j.tube.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ananthan S, et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis. 2009;89:334–353. doi: 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maddry JA, et al. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis. 2009;89:354–363. doi: 10.1016/j.tube.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reynolds RC, et al. High throughput screening of a library based on kinase inhibitor scaffolds against Mycobacterium tuberculosis H37Rv. Tuberculosis. 2012;92:72–83. doi: 10.1016/j.tube.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ekins S, et al. A collaborative database and computational models for tuberculosis drug discovery. Mol. BioSystems. 2010;6:840–851. doi: 10.1039/b917766c. [DOI] [PubMed] [Google Scholar]
- 85.Gamo F-J, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 86.Ekins S, Williams AJ. When pharmaceutical companies publish large datasets: an abundance of riches or fool's gold? Drug Discov. Today. 2010;15:812–815. doi: 10.1016/j.drudis.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 87.Rybniker J, et al. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat. Commun. 2015;6:7659. doi: 10.1038/ncomms8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Godbole AA, et al. Inhibition of Mycobacterium tuberculosis topoisomerase I by m-AMSA, a eukaryotic type II topoisomerase poison. Biochem. Biophys. Res. Commun. 2014;446:916–920. doi: 10.1016/j.bbrc.2014.03.029. [DOI] [PubMed] [Google Scholar]
- 89.Godbole AA, et al. Targeting Mycobacterium tuberculosis topoisomerase I by small-molecule inhibitors. Antimicrob. Agents Chemother. 2015;59:1549–1557. doi: 10.1128/AAC.04516-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Djaout K, et al. Predictive modeling targets thymidylate synthase ThyX in Mycobacterium tuberculosis. Sci. Rep. 2016;6:27792. doi: 10.1038/srep27792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katsuno K, et al. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015;14:751–758. doi: 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- 92.Perryman AL, et al. Predicting mouse liver microsomal stability with ‘pruned’ machine learning models and public data. Pharm. Res. 2015;33:433–449. doi: 10.1007/s11095-015-1800-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Makarov V, et al. The 8-pyrrole-benzothiazinones are noncovalent inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015;59:4446–4452. doi: 10.1128/AAC.00778-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shirude PS, et al. Azaindoles: noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J. Med. Chem. 2013;56:9701–9708. doi: 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- 95.Chatterji M, et al. 1,4-Azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother. 2014;58:5325–5331. doi: 10.1128/AAC.03233-14. [DOI] [PMC free article] [PubMed] [Google Scholar]