

Abstract
Background: We present a 6 year old boy with type I Gaucher treated from 16 months with ERT, developing focal Gaucheroma in the liver at 3.5 years.
Case: The subject presented at 13 months of age with anaemia, thrombocytopenia and hepatosplenomegaly. Gaucher disease was confirmed by leucocyte enzyme assay. A homozygous change: c.1193G>A (p.Arg398Gln) in the GBA gene was identified. He had normal neurology with normal saccades. Imiglucerase was administered at 60 IU/kg/fortnight from 15 months as per Australian regulations with good clinical response. At 3.5 years hepatic ultrasound demonstrated a nodular cystic lesion measuring 7 × 5.3 × 5.1 cm in the right lobe of liver, confirmed on MRI. Biopsy demonstrated acellular hyaline necrosis, portal–portal bridging fibrosis and nodules of Gaucher cells. Cystic fluid comprised necrotic debris and Gaucher cells. Further evaluation over 18 months including repeat MRI, biopsy, alpha-fetoprotein monitoring and whole-body FDG-Pet scan demonstrate no malignancy.
Conclusion: GD is the most common lysosomal storage disorder. The aetiology, natural history and optimal management strategy of rare Gaucheroma in paediatric cases has not been defined particularly in regards to malignancy risk.
Introduction
Gaucher disease (GD) is the commonest lysosomal storage disorder and is caused by deficiency of lysosomal glucocerebrosidase resulting in accumulation of undegraded glucosylceramide and other glycolipids in the reticuloendothelial system (Brady et al. 1965). Phenotypically GD presents as a clinical spectrum ranging from an asymptomatic to a perinatal lethal disorder. It has been traditionally classified into three types – GD 1 (non-neuropathic form), GD 2 (acute neuropathic form) and GD 3 (chronic neuropathic form). GD1 is the commonest type comprising of 95% of all cases (Charrow et al. 2000) and although considered non-neuropathic, peripheral neuropathy (Biegstraaten et al. 2010) and Parkinson’s disease (Tayebi et al. 2001) have been documented in this subset. Most of the patients with GD1 are symptomatic in childhood (Charrow et al. 2004) and often present with hepatosplenomegaly, haematological manifestations (anaemia and thrombocytopenia) or bone disease. Early onset correlates well with severe disease progression (Kaplan et al. 2006). GD2 typically presents within 2 years of age with a rapidly progressive course resulting in death by 2–4 years of age. In GD 3 patients usually survive till third or fourth decade. The current recommendation for use of enzyme replacement therapy (ERT) in GD1 has shown to help significantly with anaemia, thrombocytopenia, bone pain and bone crises (Weinreb et al. 2002; Anderson et al. 2014). The possibility of localised deposition of Gaucher cells in various organs has been described in adults. Such a well-delineated lesion has been termed Gaucheroma historically, although there does not seem to be a very clear definition outlined so far in literature. Radiological follow-up of splenic lesions has been documented in the paediatric cases earlier with most showing complete radiological recovery with ERT (Chippington et al. 2008). However increased risk of malignancies especially haematological is well documented in adult patients with GD (Mistry et al. 2013). The development of solid tumours in adults with type 1 GD has also been reported in the lung, bone, spleen, breast, prostate, colon, brain and liver (Xu et al. 2005): all of them being organs that harbour Gaucher cells. Specifically looking at liver – 3 cases of hepatocellular carcinoma in Gaucher disease have been reported so far (Xu et al. 2005), all in adults. Multiple lesions on radiological examination and HbsAg positivity were present in the case reported in this paper. The other patient included in the review (Xu et al. 2005, patient described in reference 5) had a singe echodense hepatic lesion with metastatic peritoneal deposits having had a previous normal AFP and being HbsAg negative. Thus there are do not appear to be clear diagnostic indicators for predicting HCC in patients with GD. To our knowledge there are no cases of a hepatic Gaucheroma described in a paediatric population. What significance this holds in terms of malignancy risk remains unexplored as well. We thus report a case of Gaucheroma noted in a 3.5-year-old boy after being on ERT for nearly 2 years and his follow-up till 6 years of age.
History
A 13-month-old male infant 4th child to consanguineous parents of Lebanese background presented with abdominal distension for 2 months. His development was normal and he was in good health otherwise. There was no significant family history of note. On examination he had liver palpable 7 cm below the right costal margin and spleen was palpable 15 cm along its long axis below the left costal margin. He also had anaemia (Hb 74 g/L, ref 105–138) and thrombocytopenia (Plat 83 × 10 9/l, ref 150–600) noted with mild elevation of liver enzymes (AST 72 μ/L, ref 10–50; GGT 51 μ/L ref 15–45). Systemic examination including saccadic eye movements was normal. Malignancy was suspected and hence bone marrow biopsy was performed, demonstrating several foamy histiocytes on the aspirate. Leucocyte enzyme assay confirmed decreased beta glucocerebrosidase activity (<10 pmol/min/mg protein (ref 600–3,200)) and serum chitotriosidase level was markedly elevated at 11,700 nmol/h/mL (ref 3.6–78) in keeping with GD. At 15 months of age he was commenced on ERT with Imiglucerase at 60 IU/kg/fortnightly. Genetic testing subsequently showed a homozygous change: c.1193G>A (p.Arg398Gln) in the GBA gene. At 3.5 years a routine hepatic ultrasound demonstrated a nodular cystic lesion in the right lobe of liver. This measured 7 × 5.3 × 5.1 cm in size and was confirmed on MRI (Fig. 1). Biopsy demonstrated hyaline necrosis, portal–portal bridging fibrosis and nodules of Gaucher cells (Fig. 2). Alpha fetoprotein was 2 kIU/l (ref range 0–6). Cystic fluid comprised necrotic debris and Gaucher cells. Further evaluation over 18 months including repeat MRI (Fig. 3), biopsy, serial alpha-fetoprotein monitoring and whole-body FDG-Pet scan demonstrate no evidence of malignancy. The patient remained stable throughout and his serum chitotriosidase levels have also shown a steady decline with treatment with the last level being 2,400 nmol/h/mL at 4 year 4 months of age, indicative of positive response to ERT. Subsequently the reference laboratory has been testing plasma glucosylsphingosine levels and our patients have ranged from 230 to 330 (ref <10 nmol/L).
Fig. 1.
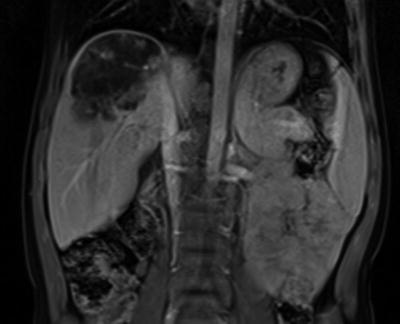
Large subcapsular heterogeneous lesion 7 × 5.3 × 5.1 cm in the right lobe of liver with multiple satellite lesions
Fig. 2.
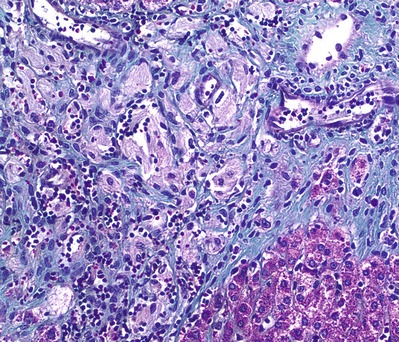
Histopathology of liver biopsy with Masson Trichrome staining shows a nodule of Gaucher histiocytes (light pink) surrounded by fibrosis (green) and lymphocytes. Normal hepatocytes (magenta) are at the bottom right
Fig. 3.
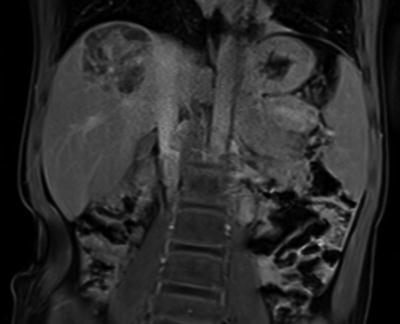
Interval reduction in right lobar hepatic mass, 4.6 × 3.8 × 5.1 cm with diffuse dystrophic calcification within the mass
Discussion
Management of GD in terms of ERT, dose and therapeutic goals is well established based on previous studies (Weinreb et al. 2002; Pastores et al. 2004; Anderson et al. 2014). Hepatic Gaucheroma has only been reported in a 23-year-old adult as a differential for focal nodular hyperplasia (Poll 2009). It is unclear if this adult was receiving ERT and if any follow-up was documented. We know that haematological and solid organ malignancies occur in adults with Gaucher disease. Although the exact aetiology of increased incidence of malignancies in type 1 is unknown, it is believed that accumulated glucocerebroside stimulates the immune system predisposing to haematological carcinogenesis and damage to the immunosurveillance system of T lymphocytes may contributes to solid organ tumours (Shiran et al. 1993; Bertram et al. 2003).
The exact approach to management of a solid organ lesion remains unclear, more so in paediatric population. Serum chitotriosidase level was trending downwards when this lesion was detected. In our patient, liver biopsy was done twice and was in keeping with a benign process. Options include observation, adjunctive or single substrate reduction therapy, resection of primary lesion, partial hepatectomy or liver transplantation. All of these therapeutic options were considered, but none has proven utility in this situation. Funding authorities in Australia do not reimburse doses above 60 IU/kg fortnightly and hence this prescribed dose/kg has been maintained and conservative management of the lesion has continued. Serial alpha-fetoprotein measurements remained normal. Anti-imiglucerase antibody levels were not performed but continuing on the same dose/kg of enzyme resulted in decrease in the size of the lesion with dystrophic calcification within it (Figs. 1 and 3). There were two stellate lesions noted inferior to the main mass which we will follow up on serial imaging. The spectre of looming potential malignancy is a heavy burden for families to bear from early childhood and this has led to huge psychological and emotional impact on the family. Unfortunately with rare disorders having even rarer manifestations it is difficult to explain unquantifiable risks of these findings to the family and for the family to fathom what lies ahead of them.
Conclusion
Hepatic Gaucheroma is a rare entity in children. We believe this is the first case described in paediatric population. The lesion in our patient was picked up incidentally on routine screening. Our patient has shown features of decrease in size of the lesion with continuation of ERT with no features of malignancy established both pathologically and based on serum markers. More such cases with longitudinal follow-up need to be reported to establish the natural course of these rare lesions.
Compliance with Ethics Guidelines
Conflict of Interest Statement
All authors, Sophy Korula, Penny Owens, Amanda Charlton and Kaustuv Bhattacharya, declare that they have no conflict of interest.
Informed consent was obtained from the patients parents and permission from institutional ethics committee has also been obtained as per the hospital guidelines.
Contributors
Manuscript writing – SK.
Manuscript editing – KB, AC.
Treatment, diagnosis and management of patient – KB, PO, AC, SK.
Footnotes
Competing interests: None declared
Contributor Information
Sophy Korula, Email: jsophyhr@yahoo.co.in.
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Anderson LJ, Henley W, Wyatt KM, Nikolaou V, Waldek S, Hughes DA, Pastores GM, et al. Long-term effectiveness of enzyme replacement therapy in children with Gaucher disease: results from the NCS-LSD cohort study. J Inherit Metab Dis. 2014;37:961–968. doi: 10.1007/s10545-014-9693-8. [DOI] [PubMed] [Google Scholar]
- Bertram HC, Eldibany M, Padgett J, Dragon LH. Splenic lymphoma arising in a patient with Gaucher disease. A case report and review of the literature. Arch Pathol Lab Med. 2003;127(5):e242–e245. doi: 10.5858/2003-127-e242-SLAIAP. [DOI] [PubMed] [Google Scholar]
- Biegstraaten M, Mengel E, Maródi L, Petakov M, Niederau C, Giraldo P, Hughes D, et al. Peripheral neuropathy in adult type 1 Gaucher disease: a 2-year prospective observational study. Brain. 2010;133(10):2909–2919. doi: 10.1093/brain/awq198. [DOI] [PubMed] [Google Scholar]
- Brady RO, Kanfer JN, Shapiro D. Metabolism of Glucocerebrosides. II. Evidence of an enzymatic deficiency in Gauchers disease. Biochem Biophys Res Commun. 1965;18:221–225. doi: 10.1016/0006-291X(65)90743-6. [DOI] [PubMed] [Google Scholar]
- Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160(18):2835–2843. doi: 10.1001/archinte.160.18.2835. [DOI] [PubMed] [Google Scholar]
- Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Prakash-Cheng A, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr. 2004;144(1):112–120. doi: 10.1016/j.jpeds.2003.10.067. [DOI] [PubMed] [Google Scholar]
- Chippington S, McHugh K, Vellodi A. Splenic nodules in paediatric Gaucher disease treated by enzyme replacement therapy. Pediatr Radiol. 2008;38:657–660. doi: 10.1007/s00247-008-0811-3. [DOI] [PubMed] [Google Scholar]
- Mistry PK, Taddei, T, vom Dahl S, Rosenbloom BE (2013) Gaucher disease and malignancy: a model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncog 18(3):235–246 [DOI] [PMC free article] [PubMed]
- Kaplan P, Andersson HC, Kacena KA, Yee JD (2006) The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med 160:603–608 [DOI] [PubMed]
- Pastores GM, Weinreb NJ, Aerts H, Generoso A, Cox TM, Giralt M, Grabowski GA et al (2004) Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 41(Suppl 5):4–14 [DOI] [PubMed]
- Poll LW, Vom Dahl S (2009) Hepatic Gaucheroma mimicking focal nodular hyperplasia. Hepatology 50(3):985–986 [DOI] [PubMed]
- Shiran A, Brenner B, Laor A, Tatarsky I (1993) Increased risk of cancer in patients with Gaucher disease. Cancer 72(1):219–224 [DOI] [PubMed]
- Tayebi N, Callahan M, Madike V, Stubblefield BK, Orvisky E, Krasnewich D, Fillano JJ, et al. Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol Genet Metab. 2001;73(4):313–321. doi: 10.1006/mgme.2001.3201. [DOI] [PubMed] [Google Scholar]
- Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G et al (2002) Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med 113(2):112–119 [DOI] [PubMed]
- Xu R, Mistry P, McKenna G, Emre S, Schiano T, Bu-Ghanim M, Levi G, et al. Hepatocellular carcinoma in type 1 Gaucher disease: a case report with review of the literature. Semin Liver Dis. 2005;25(2):226–229. doi: 10.1055/s-2005-871201. [DOI] [PubMed] [Google Scholar]