

Abstract
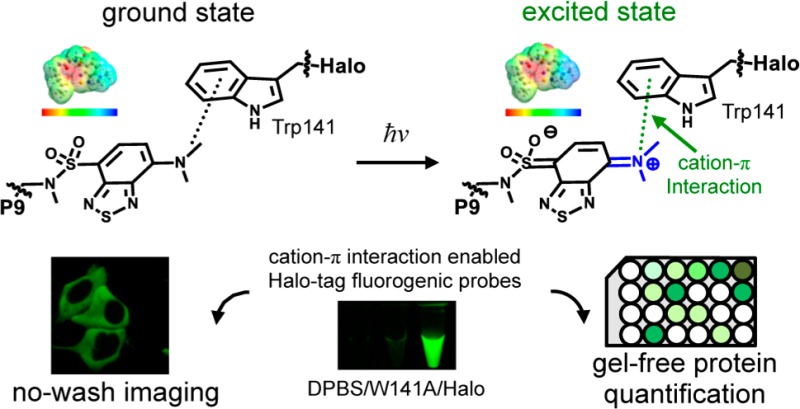
The design of fluorogenic probes for a Halo tag is highly desirable but challenging. Previous work achieved this goal by controlling the chemical switch of spirolactones upon the covalent conjugation between the Halo tag and probes or by incorporating a “channel dye” into the substrate binding tunnel of the Halo tag. In this work, we have developed a novel class of Halo-tag fluorogenic probes that are derived from solvatochromic fluorophores. The optimal probe, harboring a benzothiadiazole scaffold, exhibits a 1000-fold fluorescence enhancement upon reaction with the Halo tag. Structural, computational, and biochemical studies reveal that the benzene ring of a tryptophan residue engages in a cation−π interaction with the dimethylamino electron-donating group of the benzothiadiazole fluorophore in its excited state. We further demonstrate using noncanonical fluorinated tryptophan that the cation−π interaction directly contributes to the fluorogenicity of the benzothiadiazole fluorophore. Mechanistically, this interaction could contribute to the fluorogenicity by promoting the excited-state charge separation and inhibiting the twisting motion of the dimethylamino group, both leading to an enhanced fluorogenicity. Finally, we demonstrate the utility of the probe in no-wash direct imaging of Halo-tagged proteins in live cells. In addition, the fluorogenic nature of the probe enables a gel-free quantification of fusion proteins expressed in mammalian cells, an application that was not possible with previously nonfluorogenic Halo-tag probes. The unique mechanism revealed by this work suggests that incorporation of an excited-state cation−π interaction could be a feasible strategy for enhancing the optical performance of fluorophores and fluorogenic sensors.
Genetically encoded protein tags that can be labeled with synthetic fluorescent molecules are powerful tools for visualizing a protein of interest in live cells.1 Toward this end, numerous genetically encoded fusion protein tags have been developed, including FlAsH tag,2,3 SNAP tag,4 TMP tag,5 BL tag,6 PYP tag,7 ACP/PPTase tag,8 biotin ligase tag,9 sortase tag,10 and Halo tag,11 among others.12,13 A key limitation of this approach is that the introduction of fluorescent molecules to label fusion protein tags typically results in excess unbound probes that yield background fluorescence, requiring extensive washing of samples and limiting time-resolved applications. Fluorogenic “turn-on” probes have been widely used in various applications, represented by redox-responsive fluorescent probes.14−16 These probes eliminate this problem because the chemical probes remain dark until they react with the protein tags. As a result, washing steps are not required to reduce the background and improve the signal-to-noise ratio.
Halo tag11 is a fusion protein tag that is widely used in multiple applications, represented by live cell fluorescence imaging,17 analysis of protein dynamics,18 and probing cellular redox potential perturbation.19 However, fluorogenic probes have been rarely reported for Halo tag. Typical fluorogenic probes for fusion protein tags utilize a fluorescence resonance energy transfer (FRET) quenching mechanism (Figure 1, top box). The fluorescence is unmasked upon covalent attachment of fluorophores and cleavage of the quencher, a chemical reaction that is realized by a surface accessible nucleophile. While this strategy has been successfully implemented in the design of fluorogenic probes for SNAP tag,20,21 TMP tag,22 and BL tag,23 it is ill-suited to Halo tag (Figure 1, top box) because the protein employs a buried nucleophile located at the bottom of a narrow tunnel (Figure S1a) that is not readily accessible to these bulky fluorophore–quencher conjugates. In recent studies, the Johnsson and Hell groups bypassed the narrow tunnel, relying on a spirolactone chemical switch that occurs upon binding to the surface of Halo tag to activate red fluorescence (Figure 1, middle box).17,24−26 These probes exhibit fluorogenicity upon reacting with Halo tag and can enable no-wash super-resolution imaging in live cells. More recently, the Kool group developed a set of red channel fluorophores having twisted internal charge transfer structures that fit into the narrow tunnel and enabled a 27-fold fluorescence increase.27 These probes have been implemented in wash-free imaging of proteins localized in bacterial cells but not in mammalian cells. Although these probes are powerful tools, new fluorogenic probes of different colors and distinct turn-on mechanisms are highly desirable for no-wash live cell imaging using Halo tag.
Figure 1.
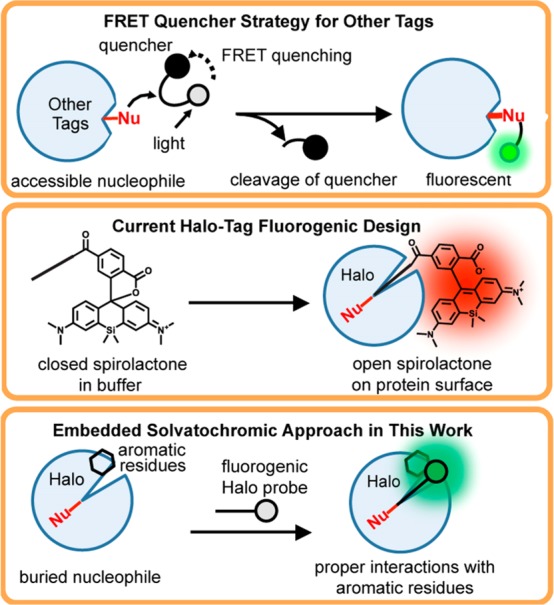
Fluorogenic probe design strategies for fusion protein tags. As shown in the top box, conventional approaches involve a linked FRET quencher that is cleaved when the probe reacts with an accessible nucleophile on the surface. This strategy is not suitable for Halo tag because it contains a deeply buried nucleophile. As shown in the middle box, one current design strategy for fluorogenic probes for Halo tag involves a chemical switch between closed and open forms of a spirolactone group. As shown in the bottom box, an embedded solvatochromic strategy is employed in this work to develop a novel class of fluorogenic probes.
Herein, we report an alternative approach to achieve fluorogenicity, giving rise to a novel class of Halo-tag fluorogenic probes. Solvatochromic fluorophores are molecules with a delocalized aromatic π-system capped with an electron-donating group and an electron-withdrawing group.28,29 They have been extensively utilized in the design of fluorogenic chemical probes for proteins because their quantum yield increases dramatically when they are embedded in hydrophobic protein interiors (Figure 1, bottom box).30−32 Previous studies have demonstrated their general applicability in biological applications, in particular, live cell imaging.33−35 On the basis of these fluorophores, we have developed a novel class of Halo-tag fluorogenic probes of sufficient fluorogenicity for no-wash live cell imaging and gel-free protein quantification. A combination of structural, computational, and biochemical analyses shows that the cation−π interaction between the electron-donating group of the fluorophore in its excited state and a tryptophan residue of the Halo tag contributes significantly to the fluorogenicity of the optimal probe. This unique mechanism of fluorescence turn-on may inform alternative strategies for enhancing the optical performance of fluorophores and fluorogenic sensors.
Methods
Plasmid Construction and Molecular Cloning
For cloning, plasmids were transformed into DH5α Escherichia coli cells. The TDP43 and SOD-1 genes were amplified from the wtTDP43-tdTOMATOHA gene (a gift from Z. Xu, Addgene plasmid 28205) and the pF146 pSOD1WTAcGFP1 gene (a gift from E. Fisher, Addgene plasmid 26407), respectively. These genes were subcloned into a pHTC HaloTag CMV-neo vector by the PIPE cloning method.36 Briefly, a gene of interest and a hosting vector were amplified with PIPE cloning primers and then mixed by being pipetted prior to transformation of DH5α cells for selection and amplification. Halo D106A and W141A mutant plasmids were prepared by site-directed mutagenesis.
Protein Expression and Purification
E. coli BL21 DE3* competent cells were transformed with pET29b encoding Halo, Halo D106A mutant, or Halo W141A mutant. A single transformant was used to inoculate 30 mL of LB medium containing kanamycin and grown for 16 h while being shaken at 37 °C. Large-scale growth cultures were initiated by the addition of 10 mL of the starter cultures into 1.5 L of freshly prepared LB medium and allowed to grow at 37 °C until the OD600 reached ∼0.8. A final isopropyl β-d-1-thiogalactopyranoside (IPTG) concentration of 0.5 mM was used to induce Halo expression. The cultures were allowed to grow for an additional 5 h or, in the 18 °C overnight. Cells were harvested, resuspended in 20 mL of DPBS, and stored in a −80 °C freezer. Resuspended cells were thawed and lysed by sonication at 4 °C (10 s pulses with 20 s intervening pauses to maintain the low temperature, for a total duration of 15 min; Q500 Sionicator, QSonica) in the presence of a protease inhibitor (1 mM PMSF). Lysed cells were centrifuged for 60 min at 16,000g. The supernatant was collected and loaded onto a 4 mL Bio-Rad NiNTA column and washed with 120 mL of buffer containing 50 mM Tris-HCl (pH 7.5) and 100 mM NaCl. The protein was then eluted by gradient addition of buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 500 mM imidazole over a volume of 64 mL. The Halo-containing fractions were identified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis, pooled, and concentrated. The protein was further purified using a 120 mL HiPrep 16/60 Sephacryl S-200 HR size-exclusion column. The protein-containing fractions were identified by SDS–PAGE gel analysis, pooled, and concentrated. No significant impurities were identified, and Halo purity was estimated to be >98% on the basis of SDS–PAGE.
Quantum Yield Measurements
Quantum yield measurements followed the previously reported protocol.37 Fluorescein was used as the quantum yield (QY) standard (QY of 0.93 in a 0.1 M NaOH aqueous solution). The standard samples were prepared in 0.1 M NaOH. Halo ligands (P4, P8, and P9) were prepared in DMSO as stock solutions. The Halo–ligand conjugate was prepared at a concentration of 16.6 μM. To generate the quantum yields, emission spectra were recorded (Ex. = 435 nm) and the peak area was integrated.
Fluorescence Lifetime Measurements
The Halo–P9 conjugate was prepared by incubating P9 (20 μM) and Halo (50 μM) in PBS buffer. The fluorescence lifetime of the Halo–P9 conjugate was measured by time-correlated single-photon counting (TCSPC) using a FluoroMax 4 fluorimeter (Horiba Scientific). The probe was excited using a 490 nm pulsed laser diode, and fluorescence emission was measured at 530 nm. The fluorescence decay rate was obtained by a fit to an exponential equation using the DAS6 analysis software (Horiba Scientific).
Labeling Kinetics Measurements
The Halo–P9 conjugation reaction was monitored using an Agilent Eclipse fluorescence spectrophotometer with an excitation wavelength of 445 nm and an emission wavelength of 530 nm. The data were corrected for the Halo background in PBS buffer measured under the same conditions. All measurements were averaged from triplicate readings. To follow the conjugation reactions, 0.5 μM P9 was incubated with an excess of purified Halo protein. The time courses were single-exponential and fit to the equation I(t) = I1 + (I0 – I1) × exp(−kobsdt), where I0 is the fluorescence before addition of Halo, I1 is the fluorescence value at infinite time, and kobsd is the observed rate constant. The values of kobsd were plotted as a function of P9 concentration ([P9]) and fit to the equation kobsd = kon[P9], where kon is the rate constant for the conjugation reaction.
Confocal Microscope Imaging
The HEK293T cells were seeded at 25% confluency 24 h prior to transfection in poly-d-lysine-coated 35 mm glass bottom dishes (MatTek Corp.). Cells were grown in DMEM supplemented with 10% FBS and penicillin-streptomycin antibiotics until they reached 50–60% confluency. Transfections of the plasmid encoding the SOD1-Halo or TDP43-Halo conjugate were performed using X-tremeGene 9 DNA transfection reagent (Roche) according to the manufacturer’s instructions. Proteins were expressed for 24 h prior to analyses.
For confocal fluorescence imaging, DMEM was replaced with FluoroBrite DMEM (ThermoFisher) supplemented with 10% FBS, and Hoechst 33342 (0.1 μg/mL), and P9 (2.5 μM) or TMR ligand (2.5 μM). The samples were incubated for 30 min prior to imaging. To wash off unbound TMR ligands, the cells were washed extensively by replacement of the medium with fresh DMEM and incubation for 30 min at 37 °C. The medium was replaced with fresh FluoroBrite DMEM (ThermoFisher) supplemented with 10% FBS prior to imaging for the TMR-washed sample. Confocal images were obtained using an Olympus FluoView FV1000 confocal microscope. The Halo–P9 conjugate fluorescence was visualized using a blue argon (488 nm) laser. Nuclear staining was visualized using a violet laser (405 nm). The TMR ligand was visualized using a green HeNe laser (543 nm).
Determination of the X-ray Structures of Halo and the Halo–P9 Conjugate
Halo [15 mg/mL in 20 mM HEPES (pH 7.5)] was purified as described above and crystallized at room temperature using the sitting-drop vapor diffusion method with 0.1 M sodium chloride, 0.1 M HEPES (pH 7.5), and 1.6 M ammonium sulfate as the precipitating solution. The crystals were mounted on loops and soaked in a well solution supplemented with 25% (v/v) ethylene glycol prior to being flash-frozen in liquid nitrogen. The Halo–P9 conjugate [12 mg/mL in 20 mM HEPES (pH 7.5)] was generated as described and crystallized at room temperature using the hanging-drop vapor diffusion method with 0.2 M ammonium acetate, 0.1 M sodium acetate (pH 4.6), and 20% (w/v) PEG 3000 as the precipitating solution. The crystals were mounted on loops and soaked in a well solution supplemented with 21% (v/v) ethylene glycol prior to being flash-frozen in liquid nitrogen.
Crystallographic data sets were collected at the Life Sciences Collaborative Access Team and GM/CA Collaborative Access Team beamlines at the Advanced Photon Source and processed using HKL2000.38 The Halo structure was determined by molecular replacement with PHASER39 as implemented within the CCP4 software package,40 using the Rhodococcus rhodochrous haloalkane dehalogenase structure (Protein Data Bank entry 1BN7) as the initial search model.41 Phases for the Halo–P9 structure were obtained via the same procedure, but with the coordinates of the apo Halo structure used as the search model. Model building and refinement were performed with Coot42 and Refmac5,43 respectively. Coordinates and geometric restraints for the P9 ligand were generated in JLigand.44 Structure validation and Ramachandran analyses were performed using the Molprobity server.45 Figures were generated with the PyMOL Molecular Graphics software package (Schrödinger LLC).
The asymmetric unit of the apo Halo structure is composed of two Halo monomers related by noncrystallographic symmetry. Residues 3–292 are modeled in each chain, and the final model also includes 437 water molecules, five ethylene glycol molecules, and three chloride ions. Molprobity analysis indicates 97.4% of side chains in favorable rotamer conformations (0.6% of side chains are outliers) and a clash score of 0.43 (100th percentile). The Halo–P9 conjugate also contains two monomers in the asymmetric unit, and the final model consists of residues 3–292 in chain A and 3–297 in chain B, 72 water molecules, two ethylene glycol molecules, two P9 ligands, and two chloride ions. Molprobity analysis of this structure shows 96.3% of rotamers in favorable conformations (0.2% outliers) and a clash score of 1.07 (100th percentile).
Density Functional Theory (DFT)/Time-Dependent DFT Calculation
Computational studies are performed using the wB97xd46 DFT with the 6-31G(d) basis set47 as implemented in Gaussian Development version I.09.18 The polarizable continuum model (PCM)48 is used to implicitly simulate the surrounding protein/water environment. The dielectric constant ε used with PCM is 30 for the low-frequency49 and 1.788 (same as that of water) for the high-frequency dielectric response. The initial geometry is extracted from the Halo–P9 cocrystal, and only atoms from the fluorophore on the P9 unit are relaxed during the optimization. Frequency calculations were performed to verify that the optimized ground-state geometries are true minima on the potential energy surface. In addition, the excited-state geometry has been fully optimized using the time-dependent version of the same DFT functional.
Incorporation of 5-Fluorotryptophan into the Halo Protein
5-Fluorotryptophan (5FW, F0896-1G, Sigma-Aldrich) was incorporated into the Halo protein using a tryptophan auxotrophic E. coli strain, W3110(DE3) (a generous gift from the laboratory of S. Benkovic at The Pennsylvania State University) following an established protocol.50 In brief, W3110(DE3) cells were transformed with the pET29b+ plasmid containing the Halo gene and selected on an LB agar plate containing kanamycin (50 mg/L) for single colonies. A 20 mL overnight culture in LB medium containing kanamycin (50 mg/L) at 37 °C was added to 1 L of sterilized defined minimal medium. The minimal medium consists of casamino acids (12 g/L), Na2HPO4·7H2O (12 g/L), KH2PO4 (3 g/L), NH4Cl (1 g/L), NaCl (0.5 g/L), all the pyrimidine and purine bases (1 mM each), folate (1 mM), nicotinic acid (150 μM), thiamin (1 mg/L), riboflavin (150 μM), glucose (2 g/L), FeCl3 (5 mg/L), ZnSO4 (0.4 mg/L), CuSO4 (0.8 mg/L), CoCl2 (0.7 mg/L), MnSO4 (0.5 mg/L), Na2MoO4 (0.7 mg/L), MgSO4 (1 mM), CaCl2 (0.1 mM), kanamycin (50 mg/L), and l-tryptophan (2 mg/L). Cell were grown at 37 °C until they reached a stationary phase because of tryptophan starvation (OD600 ∼ 1.2). IPTG and 5FW were added to final concentrations of 0.5 mM and 100 mg/L, respectively. Halo protein was expressed at 37 °C for 5 h. The cells were harvested, and the 5FW-Halo protein was purified to >95% purity as described for the wild type.
Mass spectrometric analysis was performed on a Waters Q-TOF Premier quadrupole time-of-flight (TOF) mass spectrometer [Waters Corp. (Micromass Ltd.), Manchester, U.K.]. Operation of the mass spectrometer was performed using MassLynx version 4.1 (http://www.waters.com). Samples were introduced into the mass spectrometer using a Waters 2695 high-performance liquid chromatograph. The separation was performed by gradient elution on a Thermo BioBasic C4, 50 mm × 2.1 mm high-performance liquid chromatography column. The mobile phases used were 100% deionized water with 0.1% formic acid (mobile phase A) and 100% acetonitrile (ACN) with 0.1% formic acid (mobile phase B). The sample was injected as received, with an injection volume of 50 μL and a flow rate of 0.2 mL/min. The linear gradient is described in Table 1. Data were acquired during the first 15 min of the run. The nitrogen drying gas temperature was set to 300 °C at a flow rate of 7 L/min. The capillary voltage was 2.8 kV. The mass spectrometer was set to scan from m/z 500 to 2500 in positive ion mode, using electrospray ionization (ESI).
Table 1.
| time (min) | % A (H2O with formic acid) | % B (ACN with formic acid) |
|---|---|---|
| 0.0 | 98.0 | 2.0 |
| 1.0 | 98.0 | 2.0 |
| 2.0 | 5.0 | 95.0 |
| 15.0 | 5.0 | 95.0 |
General Synthetic and Chromatography Methods
Detailed information is provided in the Supporting Information.
Results
Development of Fluorogenic Halo-Tag Probes Containing a Benzoxadiazole Scaffold
To probe whether the Halo-tag protein could accommodate solvatochromic fluorophores in its substrate tunnel, we determined the X-ray crystal structure of the Halo-tag protein to 1.35 Å resolution (PDB: 5UYI, Table S1). Analysis of the structure shows that Halo tag is an ideal target for solvatochromic molecules because the exit of the Halo ligand binding tunnel possesses multiple aromatic residues that could interact favorably with solvatochromic fluorophores (Figure S1b). To test this hypothesis, we chose the benzoxadiazole scaffold as our initial system because it has been shown to exhibit superior performance in fluorogenicity upon binding to proteins32,33,51 and has been developed as fluorogenic probes for the SNAP tag.33 Current nonfluorogenic Halo-tag probes extend the fluorophores (Figure 2a, red) out of the ligand binding pocket via a polyethylene glycol (PEG) linker. To facilitate burial of the probe within the binding tunnel, we evaluated a series of benzoxadiazole-based chromophores (Figure 2a, green) modified with alkane linkers (Figure 2a, R1) that are shorter than the current Halo-tag probes. We anticipated that the shorter linker could enhance fluorescence turn-on by promoting interaction between the fluorophore and the Halo-tag aromatic surface amino acids.
Figure 2.

Fluorogenic benzoxadiazole and benzothiadiazole probes for Halo tag. (a) Current nonfluorogenic probes (top) for Halo tag harbor a six-carbon alkyl chain, a polyethylene glycol (PEG) linker, and a fluorescent fluorophore. The table shows structures of fluorogenic probes for Halo tag evaluated in this work. R1 is a chloroalkane chain of varying length, with and without a sarcosine amide linker. R2 denotes the structure of an electron donating group, which is either a methylamino or dimethylamino substituent. (b) Relative intensity of conjugates of P1–P8 with Halo and (c) its relative labeling kinetics. Halo samples (20 μM) were incubated with P1–P8 (10 μM) for 18 h prior to fluorescence intensity measurements (Ex. = 450 nm, and Em. = 530 nm). The labeling rate was recorded upon mixing 5 μM Halo protein with 0.5 μM ligand (Ex. = 450 nm, and Em. = 530 nm). (d) P9 consists of a six-carbon alkyl chain, a sarcosine amide linker, and a solvatochromic fluorophore based on a benzothiadiazole scaffold. (e) P9 exhibits ∼1000-fold fluorescence enhancement upon reaction with Halo protein (green), compared to P9 in DPBS (red). The probe is weakly fluorescent upon binding to Halo-D106A (black). A solution of the protein (20 μM) was incubated with 0.5 equiv (10 μM) of P9 (green), P8 (yellow), or P4 (blue) in DPBS buffer at 25 °C for 1 h. Fluorescence emission spectra were recorded at 450 nm excitation.
To achieve this goal, we omitted the PEG linker and reduced the length of the alkyl chain to generate probes P1–P3 and P5–P7 with C4, C5, and C6 alkyl linkers, respectively (Figure 2a). Our structural analysis indicated that the short alkyl linkers would likely embed the benzoxadiazole moiety within the internal Halo-tag pockets and allow it to interact with surface aromatic residues to enable fluorescence. On the fluorophore, two different electron-donating groups (Figure 2a, R2) were introduced to enable different electron-donating capacities that could also influence the fluorogenicity of benzoxadiazole (methylamino group for P1–P3 and dimethylamino group for P5–P7 in Figure 2a). Subsequently, we measured the fluorescence of these probes (20 μM) before and after forming covalent conjugation with purified Halo-tag protein (50 μM). For all six probes that we tested, the turn-on fluorescence upon the Halo–probe conjugation was observed to various extents (striped bars in Figure 2b), demonstrating the general applicability of the approach. While the fluorogenicity was moderately influenced by the identity of the electron-donating group, variation of the linker length had the greatest impact on fluorogenicity. Reducing the length of the alkyl linkers (Figure 2a, R1) effectively enhanced the fluorogenicity (striped bars Figure 2b). Series of probes both from P3 to P1 and from P7 to P5 exhibited a positive correlation between the fluorescence intensity and the decrease in the length of the probe linker.
Although the conjugates with shorter linker lengths exhibited desirable increased fluorescence turn-on, they also displayed significant impediments in labeling rate (striped bars in Figure 2c). Series both from P3 to P1 and from P7 to P5 showed the positive correlation between labeling kinetics and longer linkers. Therefore, simply shortening the linker length to embed the fluorophore in the substrate tunnel (Figure S1a) may not be the optimal solution for achieving both adequate fluorogenicity and fast labeling kinetics. We then opted to retain the C6 alkyl chain and introduce additional conformational flexibility by incorporating a terminal sarcosine amide group (P4 and P8). This modification was intended to allow the benzoxadiazole more freedom to access the aromatic residues at the exit of the substrate channel of the Halo-tag protein. As expected, linker extension restored the fluorescence intensity (solid bars in Figure 2b, P4 and P8) while retaining the favorable labeling kinetics (solid bars in Figure 2c, P4 and P8) observed with P3 and P7. With respect to the electron-donating groups on the benzoxadiazole ring, probes with dimethylamino groups (P5–P8) generally exhibited performances better than those of their methylamino counterparts (P1–P4) in terms of both fluorescence intensity and labeling kinetics (Figure 2b,c). Thus, our rational structural optimization resulted in P8 being the optimal probe in terms of maximal fluorogenicity and labeling rate.
Improvement of Fluorogenicity via a Benzothiadiazole Scaffold, P9
It has been documented that benzothiadiazole is more resistant to photolysis than benzoxadiazole is.52,53 To avoid severe photobleaching in imaging applications, we substituted the benzoxadiazole scaffold in the most optimal probe P8 with a more photolysis-resistant benzothiadiazole moiety, resulting in a new probe P9 (Figure 2d). P9 displayed increased photostability (Figure S2) as well as a striking enhancement (∼1000-fold) in fluorescence intensity upon its covalent conjugation with Halo tag (Figure 2e and Table S2). Importantly, this fluorescence increase was not observed with a Halo D106A mutant11 that cannot form a covalent bond with P9, suggesting that the fluorogenicity is not a consequence of nonspecific binding between P9 and Halo (Figure 2e). In addition, we observed fast labeling kinetics [2.1 × 103 M–1 s–1 (Figure S3)] and a high quantum yield [φ = 0.47 ± 0.03 (Table S2)] upon reaction with Halo. Although this rate is slower than the reported rate constant for commercially available Halo probes (∼2.7 × 106 M–1 s–1),11 it is still within the reported range of rapid-labeling fluorogenic ligands for a variety of other fusion protein tags.20,21,23,32
An Excited-State Cation−π Interaction Identified in the Halo–P9 Conjugate
We next explored the mechanism of the fluorogenicity of P9 in the Halo–P9 conjugate. The benzothiadiazole fluorophore displays significant intrinsic sensitivity to its environment, exhibiting higher fluorescence intensity in nonpolar solvents than in polar solvents. We therefore considered the hypothesis that P9 is bound in a more hydrophobic and fully buried pocket surrounded by a local environment with a low dielectric constant (ε ∼ 6 for the hydrophobic interior of a folded protein vs ε ∼ 80 in aqueous solution; ε is the dielectric constant), leading to a significant fluorescence increase. However, this explanation does not fully address why the fluorescence of the Halo–P9 conjugate was ∼3-fold more intense and exhibited an emission maximal wavelength shorter than that of P9 in all solvents tested (Figure 3b and Figure S4a,b), including benzene and 1,4-dioxane that have low dielectric constants [2.27 and 2.25, respectively (Figure 3b)].
Figure 3.

Cation−π interaction enhances P9 fluorescence. (a) The benzothiadiazole ring of P9 is embedded inside Halo, accommodated by a shift of a loop containing Trp141. The inset at the top right shows that the tertiary N of the dimethylamino group resides close to the geometrical center of the benzene ring of the Trp141 indole (3.8–4.0 Å distance between N and benzene carbons). The inset at the bottom right shows that the dimethylamino group is oriented just slightly twisted in relative to the benzothiadiazole moiety. (b) Fluorescence spectra of the Halo–P9 conjugate (green), W141A–P9 conjugate (orange), P9 in DBPS buffer (red), P9 in 1,4-dioxane (purple), and P9 in benzene (blue). For the Halo–P9 and W141A–P9 conjugates, a solution of the protein (20 μM) was incubated with 0.5 equiv (10 μM) of P9 in DPBS buffer at 25 °C for 1 h. For P9 in different solvents, 10 μM P9 was added in solvents at 25 °C for 1 h. Fluorescence emission spectra were recorded at 450 nm excitation. (c) The dimethylamino group of P9 is thought to take on a positive charge in the excited state and interact with the aromatic group in Trp141 via a cation−π interaction. (d) Fluorescence decay of the Halo–P9 conjugate (green), P9 in DBPS buffer (red), P9 in 1,4-dioxane (purple), and P9 in benzene (blue). P9 (20 μM) was incubated in DPBS buffer, benzene, 1,4-dioxane, or purified Halo protein (50 μM) for 1 h at 25 °C. (e) Electrostatic potential map of Trp141 in the ground state (left), P9 in ground state S0 (middle), and P9 in excited state S3 (right). (f) Geometric model of the cation−π interaction. (g) The left panels shows the simplified geometry of the ground state with θ = 9° and R = 3.8 Å, where the magenta “atom” represents the center of the benzene ring. The right panel shows the simplified geometry at the relaxed excited state with θ = 3° and R = 4.2 Å, exhibiting a canonical cation−π geometry.
To understand the mechanism of fluorescence enhancement, we determined a cocrystal structure of the Halo–P9 conjugate to 1.92 Å resolution (PDB: 5UXZ, Figure S5a,b and Table S1). The benzothiadiazole moiety of P9 was found in a surface-exposed binding site located at the gate of the Halo ligand binding tunnel, making a direct interaction with tryptophan 141 (Figure 3a and Figure S6). Consistent with this observation, the W141A mutation completely eliminated the fluorescence of the P9–Halo conjugate (Figure 3b). Intriguingly, this interaction was not mediated by the commonly expected π–π stacking interaction between the two benzene rings. By contrast, the N atom of the dimethylamino group is positioned close to the geometrical center of the benzene ring, as evidenced by the similar distances between the N atom and the six carbon atoms of this benzene ring (the top right inset of Figure 3a). In addition, the dimethylamino group was identified in a well-defined electron density (Figure S6a,b) and positioned on a plane that was just ∼30° tilted against the benzothiadiazole moiety (the bottom right inset of Figure 3a and Figure S6c). Thus, this interaction is assisted by the benzene ring of Trp141 and the dimethylamino group of P9 in the Halo–P9 conjugate. Given that the Halo–P9 crystal is fluorescent (Figure S5c), we envision that the interaction identified in the structure should contribute to P9’s fluorogenicity for the following reasons.
Solvatochromic fluorophores are known to undergo photoinduced charge transfer in the electronic excited states, rendering the electron-donating end positively charged.54 We hypothesize that the dimethylamino group in P9 becomes positively charged in the excited state and engages in a cation−π interaction55,56 with the benzene ring of Trp141 (Figure 3c). This interaction would not only promote the charge separation of the benzothiadiazole fluorophore in its excited state but also sterically inhibit the rotational motion of the dimethylamino group that leads to a decrease in fluorescence intensity due to the mechanism of a twisted intramolecular charge transfer.57 As a result, P9 conjugated to Halo tag should exhibit a high fluorescence intensity and an extended fluorescence lifetime. Experimentally, we observed both a high quantum yield [0.47 (Table S2)] and a long fluorescence lifetime [τ = 47 ± 3 ns (Figure 3d)] for P9 conjugated to Halo-tag protein, consistent with the suggested mechanism (Figure 3c). By contrast, P9 exhibited a shorter fluorescence lifetime in nonpolar solvents represented by 1,4-dioxane (τ = 14.2 ± 0.4 ns) and benzene (τ = 14.7 ± 0.6 ns) as shown in Figure 3d, consistent with the speculation that the interaction between P9 and Trp141 could be more than simply rendering P9 a nonpolar microenvironment (Figure 3c).
A computational study was performed to test the proposed mechanism in Figure 3c. Using the coordinates generated by the cocrystal structure as a starting point, the geometry of the P9 fluorophore was optimized using the wB97xd/6-31G(d) method in the presence of an implicit solvent model to mimic the protein/water environment (Figure S7a; computational methods are provided in the Supporting Information). To test whether the twist motion of the dimethylamino group on P9 is sterically unfavorable, we performed a dihedral scan on the C–N–C bond angle (black arrow in Figure S7b). The dimethylamino group of P9 had a well-defined electron density (Figure S6a,b) and exhibited an ∼30° tilt relative to the benzothiadiazole ring (Figure S6c). Consistent with this result, we found that the energetically minimal dihedral angle resembled the computationally optimized ground-state geometry that was similar to that of the cocrystal structure. However, the calculated rotational energy barrier (38.4 kcal/mol) was much higher than those that can be traversed in free rotational motion (<5.0 kcal/mol),58 indicating that the dimethylamino group is unlikely to access twisted conformations beyond a narrow dihedral range (Figure S7c). To verify the electrostatic origin of the cation−π interaction, we generated the electrostatic potential maps of P9 in its ground and excited states (Figure 3e). While the dimethylamino group was nearly neutral in the ground state of P9 (green color in Figure 3e), calculation using time-dependent density functional theory (TD-DFT) showed that the dimethylamino group was largely positive upon vertical excitation (blue color in Figure 3e). By contrast, the benzene unit of Trp141 exhibited a negative electrostatic potential in its ground state (red color in Figure 3e) and the partial negative charge remained unchanged during vertical excitation because its orbits did not contribute to the excited state (Figure S8). Thus, the partially positively charged dimethylamino group in its excited state and the partially negatively charged benzene group of Trp141 could effectively form an electrostatically driven cation−π interaction. To provide details of the excited-state cation−π interaction, we further optimized the geometry of the Trp141–P9 complex in the relaxed excited state using the TD-DFT method. Using a geometrical model to quantitatively describe the optimized Trp141–P9 structure (Figure 3f), we found that the nitrogen atom of the dimethylamino group was slightly [θ = 9°, and R = 3.8 Å (Figure 3g and Table S3)] tilted away from the center of the Trp benzene in the ground state (represented by the magenta sphere in Figure 3e). In the relaxed excited state, however, the dimethylamino group aligned well with the benzene center [θ = 3°, and R = 4.2 Å (Figure 3g and Table S4)], exhibiting a signature of a canonical cation−π interaction.55
The Cation−π Interaction Contributes to Fluorescence in the Halo–P9 Conjugate
The structural and computational analyses provided strong evidence that a cation−π interaction exists between Trp141 and P9 upon vertical excitation. To further corroborate whether the excited-state cation−π interaction contributed to the fluorogenicity of P9, we attempted to perturb this interaction using a noncanonical fluorinated tryptophan and tested its effect on the fluorescence of the Halo–P9 conjugate. It is well established that fluorination of a benzene ring reduces its negative charge and compromises the cation−π interaction.59−61 To this end, we incorporated 5-fluorotryptophan [5FW (Figure 4a)] into the Halo protein during its biosynthesis by inducing protein expression in an E. coli tryptophan auxotroph in the presence of excess 5FW.50 Mass spectrometry data showed that 67% of the purified 5FW-Halo protein incorporated 5FW for all nine tryptophan residues (Figure 4b). For the rest of the 5FW-Halo protein, at least seven of the nine tryptophan residues of Halo were substituted with 5FW (Figure 4b). Thus, the high rate of incorporation makes it highly possible that the Trp141 residue is fluorinated in the majority of 5FW-Halo protein in the purified pool. Importantly, the 5FW-Halo protein was folded and functional to an extent similar to that of the wild-type Halo protein, as shown by its P9 labeling kinetics that was indistinguishable from that of wild-type Halo (Figure 4c) and its ability to form a covalent bond with P9 (Figure 4d). However, the fluorescence of P9 was reduced by ∼50% when it was conjugated with 5FW-Halo compared with the wild-type Halo protein (Figure 4e). The fact that the emission maxima remained unchanged with 5FW-Halo strongly suggests that this fluorescence decrease was primarily caused by a compromised cation−π interaction but not a higher dielectric constant of the microenvironment surrounding P9 (Figure 4e). These data are consistent with the notion that fluorination of tryptophan can deactivate the cation−π interaction62 and provides direct evidence of the role of this interaction in the fluorogenicity of P9.
Figure 4.
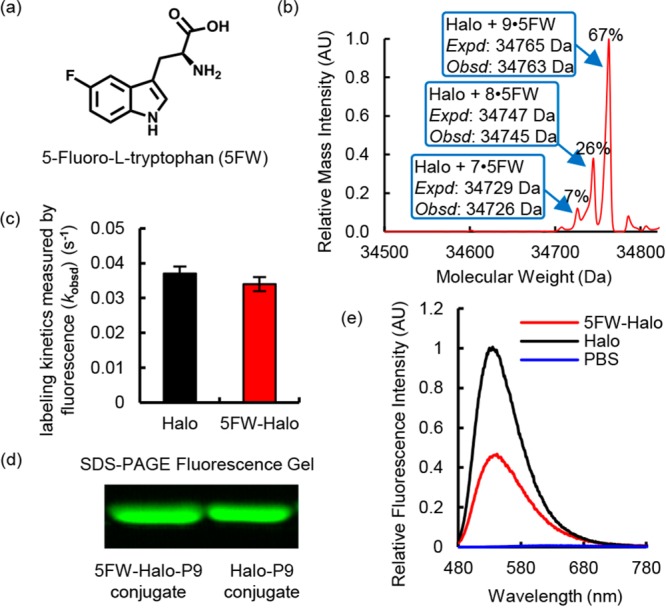
Incorporation of 5-fluorotryptophan (5FW) reduced the fluorescence intensity of the P9–Halo conjugate. (a) Molecular structure of 5FW. (b) Mass spectroscopic evidence of incorporation of 5FW into the Halo protein (5FW-Halo). (c) 5FW-Halo reacts with P9 at an observed rate similar to that observed with wild-type Halo. The reaction mixture contained 10 μM P9 and 30 μM Halo or 5FW-Halo in DPBS buffer. The labeling reaction was monitored at 450 nm excitation and 530 nm emission at 25 °C. (d) 5FW-Halo can be covalently labeled with P9, like wild-type Halo. The labeling reaction was performed using 10 μM P9 and 30 μM Halo or 5FW-Halo for 10 min in DPBS buffer at 25 °C. The Halo–P9 conjugates were visualized on an SDS–PAGE gel using the Bio-Rad Gel Doc EZ imager. (e) The 5FW-Halo–P9 conjugate (red) exhibits an ∼50% decrease in fluorescence intensity, compared to that of the wild-type Halo–P9 conjugate (black). The samples contain 30 μM protein incubated with 10 μM P9 in DPBS buffer at 25 °C for 1 h. Fluorescence emission spectra were recorded at 450 nm excitation.
P9 Enables No-Wash Live Cell Imaging
We next evaluated P9 in live cells for no-wash imaging of Halo-tagged superoxide dismutase (SOD1), transiently expressed in the cytosol of HEK293T cells. The labeling reaction was performed by incubating P9 (2.5 μM) with HEK293T cells expressing SOD1-Halo protein for 30 min in complete DMEM (Figure S9). This condition not only ensured fast labeling of SOD1-Halo protein in cells but also enabled selective detection of the protein in transfected cells via confocal microscopy without additional washing steps (Figure 5a, left panel, green). As a control, fluorescence was not detected in HEK293T cells that had not been transfected with the fusion protein (Figure S10). The P9 probe outperforms the nonfluorogenic Halo-tag TMR ligand (Figure 5a, middle panel) in which extensive washing is necessary to selectively stain the transfected cells (Figure 5a, right panel). A similar no-wash imaging experiment was performed in E. coli, further demonstrating that P9 can permeate a variety of cell membrane and wall structures (Figure S11). To demonstrate no-wash imaging of proteins in subcellular organelles, we used P9 to stain Halo-tagged nuclear TAR DNA binding protein 43 (TDP43-Halo) in HEK293T cells. Without washing, TDP43-Halo (Figure 4b, left panel, green) was selectively visualized in the nucleus (Figure 4b, left panel, blue). By contrast, the Halo-tag TMR ligand required an additional wash step to achieve similar results (Figure 4b, middle and right panels). In both cases, fluorescence SDS–PAGE gels confirmed the selective labeling of SOD1-Halo and TDP43-Halo, indicating the imaging fluorescence primarily arises from the labeled SOD1-Halo and TDP43-Halo (Figure S12). Fluorescence emission spectra were measured using both P9-treated nontransfected HEK293T cells and P9-treated Halo-transfected HEK293T cells to further demonstrate the selective fluorescence response in live cells (Figure S13).
Figure 5.
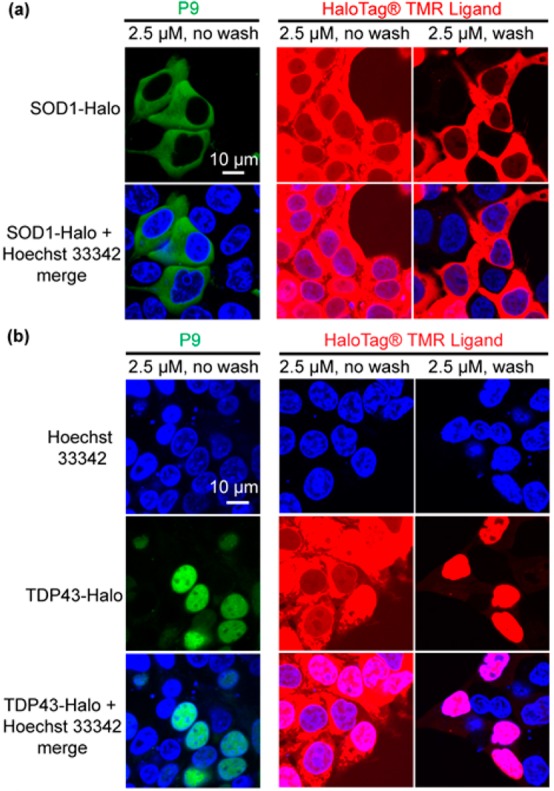
No-wash imaging of Halo-tagged proteins in live cells using P9. (a) No-wash imaging of Halo-tagged cytosolic superoxide dismutase (SOD1) in HEK293T cells after labeling with P9. SOD1-Halo in the cytoplasm of transfected cells can be selectively imaged by P9 (2.5 μM) without a wash step (left lane). The Halo-tag TMR ligand (2.5 μM) exhibits a high fluorescence background without inclusion of washing steps (middle lane). The background can be eliminated by rinsing the cell with fresh medium (right lane). (b) No-wash imaging of Halo-tagged nuclear TAR DNA binding protein 43 (TDP43) in HEK293T cells using P9. TDP43-Halo is visible in the nuclei of transfected cells prior to washing using P9, whereas TDP43-Halo cannot be visualized clearly under the same conditions using TMR ligand (middle lane). HEK293T cells were transiently transfected with SOD1-Halo or TDP43-Halo for 24 h in 35 mm poly-d-lysine-coated glass bottom dishes. P9 and the Halo-tag TMR ligand (2.5 μM) were directly dissolved in the medium. Confocal images were taken after incubation for 30 min at 37 °C. The TMR-ligand-treated samples were washed further and incubated in fresh DMEM for an additional 30 min at 37 °C prior to confocal imaging. Hoechst 33342 is a nuclear staining dye.
P9 Enables a Facile Gel-Free Protein Quantification Using the Cell Lysate
Another desirable application of fluorogenic probes is a gel-free quantification of protein levels in the cell lysate. Conventional approaches to determine the protein concentration in the cell lysate require multistep immunoblotting. Chemical tags (SNAP tag, Halo tag, BL tag, etc.) simplify this process by exploiting tag-associated fluorescent probes to quantify the band intensity on a fluorescence SDS–PAGE gel (Figure S14a). Because fluorogenic probes like P9 emit only upon conjugation with their fusion protein target, the concentration of the protein of interest can be quantified via fluorescence intensity readings taken directly from the cell lysate (Figure 6a). Such application was first demonstrated using SNAP tag and its fluorogenic probe, reporting a lower limit of detection of 25 nM.21 To evaluate P9 in this context, we expressed Halo protein in HEK293T cells and prepared lysed samples for fluorescence detection. A fluorescence standard curve was generated by addition of purified Halo protein standards to nontransfected lysate (Figure 6b, black dots and curve, R2 of 0.9995), establishing a lower limit of detection at 6.25 nM Halo (Figure 6b, inset). Fluorescence signals were recorded with a plate reader after incubation with P9 (500 nM). The average concentration of Halo protein in the test samples was determined to be 61.1 ± 4.9 nM via a fit of the relative fluorescence intensity in the standard curve with a 10-fold signal-to-noise ratio (Figure 6b, green triangle and diamond, and Figure 6c). Incubation of 2-fold diluted test samples with P9 (500 nM) yielded a measured value of 30.7 ± 4.9 nM Halo (Figure 6c), indicating the robustness of this assay. Conventional fluorescence PAGE analysis with a nonfluorogenic Halo-tag TMR ligand was performed using the same lysate sample to validate the accuracy of this assay (Figure S14b). A comparable average concentration was measured, 52.6 ± 10.1 and 23.7 ± 0.9 nM for the lysate and its 2-fold diluent, respectively (Figure S14c), providing further support for the reliability of gel-free quantification of protein concentration via the Halo–P9 conjugate.
Figure 6.
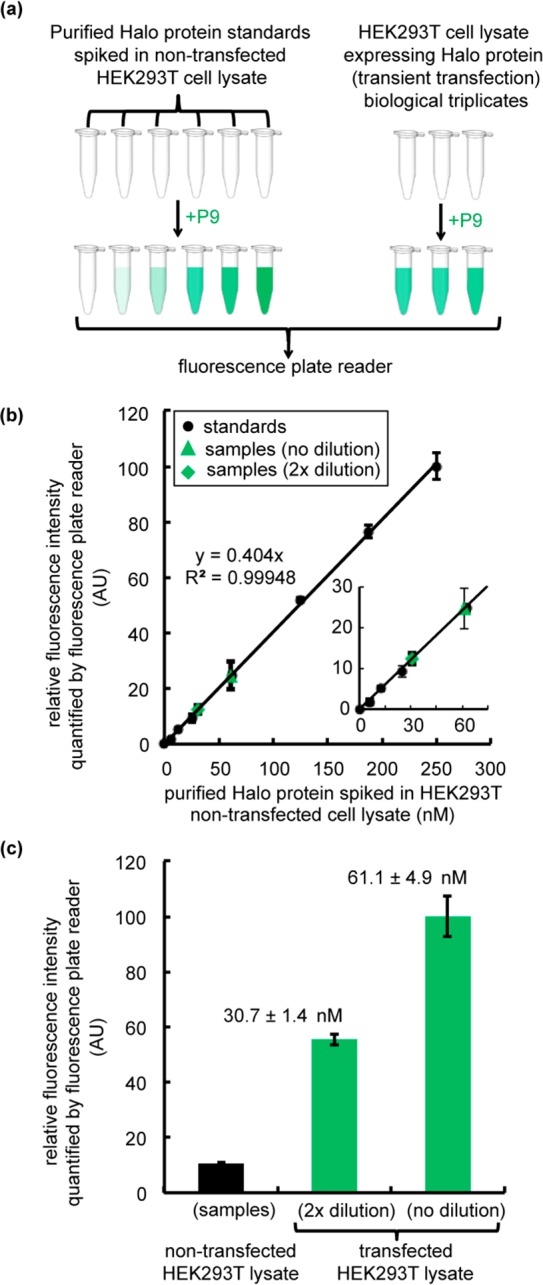
Gel-free quantification of Halo protein expression levels in transiently transfected HEK293T cell lysates. (a) Flowchart describing the procedure for measuring Halo protein expression levels in transfected cell lysates via P9 fluorescence. The workflow involves preparation of Halo standards at concentrations ranging from 6.25 to 250 nM and generation of a standard curve via addition of purified Halo protein to the nontransfected HEK293T cell lysate (0.2 mg/mL). Halo protein was transiently transfected in HEK293T cells for 36 h to generate the test samples. Cells were harvested and lysed by sonication. Lysates were prepared at 1× (0.2 mg/mL) and 2× (0.1 mg/mL) dilutions. To determine the concentration of Halo protein in transfected samples, the standard samples and the test samples were incubated with P9 (500 nM) for 2 h. Fluorescence intensities were recorded in a 96-well plate using a Tecan M1000Pro plate reader (Ex. = 450 nm, and Em. = 530 nm). (b) Standard curve for purified Halo protein in the nontransfected lysate (black dots) compared to the fluorescence intensity of samples containing Halo protein (green triangles and diamonds) in the transfected lysate. Green triangles denote transfected samples measured without dilution (1×), and green diamonds correspond to the 2×-diluted samples. The inset shows a close-up of the low concentration range. (c) Relative fluorescence intensity of nontransfected and transfected lysates treated with P9 (500 nM). All experiments were performed in biological triplicate. Error bars show the standard deviation of three measurements. The concentration of Halo in the transfected sample was determined by fitting the standard data points to a linear function as shown in panel b.
Discussion
Traditionally, the polarity of the surrounding environment has been the primary determinant of the quantum yield and emission maximum of push–pull fluorophores.63 To achieve a fluorescence increase, reducing the polarity of the solvent or fluorophore binding site has been the dominant strategy in optimizing these molecules for use in fluorogenic bioconjugation reactions. By contrast, our work provides evidence of an alternative mechanism for potent activation of a solvatochromic push–pull fluorophore in a biological environment. We show that a strong fluorescence increase can be achieved in a protein–fluorophore conjugate via a cation−π interaction formed between the benzene ring of an aromatic residue and an electron-donating group of the fluorophore that becomes positively charged in its excited state. Cation−π interactions have been extensively described in biological systems, largely in molecular recognition, protein stability, and catalysis.55,56,64−66 The Halo–P9 conjugate is an example of a new function wherein this important molecular interaction is utilized to enable the fluorogenicity of a push–pull fluorophore in a biological context and in a complex cellular milieu.
Positively charged electron-donating groups of fluorophores in the excited states, such as the dimethylamino group, comprise a common structural feature of all classes of fluorophores.63 Solvatochromic push–pull fluorophores, in particular, possess such a positively charged electron-donating group in their excited states (Figure S15), indicating that the cation−π interaction observed here could have broad implications for the design of new fluorogenic biosensors and the improvement of existing solvatochromic fluorophore bioconjugates. In support of this proposal, several recent reports observed spectral tuning when the ion pair−π interactions were utilized to affect the polarized excited state of a solvatochromic fluorophore.67,68 These findings and our work encourage us to speculate that introduction of a cation−π interaction into the excited states of a fluorophore could enhance its optical performance in a wide range of applications, including novel sensor development, high-resolution imaging, and synthesis of optical materials.
Acknowledgments
We thank Anthony Pedley for the HEK293T cell line and technical assistance, Michelle Spiering and Stephen J. Benkovic for providing tryptophan auxotrophic E. coli strain W3110(DE3), Tae-Hee Lee for assistance with fluorescence decay kinetics measurements, the Penn State Microscopy and Cytometry Facility for confocal image acquisition, James R. Miller at the Penn State Proteomics and Mass Spectrometry Core Facility for mass spectrometry data, and Paul S. Cremer and Stephen J. Benkovic for critical reading of the manuscript. We gratefully acknowledge access to resources provided by the Penn State X-ray Crystallography Facility and the Advanced Photon Source, the latter a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor (Grant 085P1000817). Computations were facilitated via the use of advanced computational, storage, and networking infrastructure provided by the Hyak supercomputer system and funded by the STF at the University of Washington.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00056.
Supporting figures and materials and methods (PDF)
This work has been supported by the Burroughs Wellcome Fund Career Award at the Scientific Interface (X.Z.), a Paul Berg Early Career Professorship (X.Z.), a Lloyd and Dottie Huck Early Career Award (X.Z.), National Institutes of Health Grant GM100011 (A.K.B.), the Searle Scholars Program (A.K.B.), and National Science Foundation Grant CHE-1565520 (X.L.).
The authors declare no competing financial interest.
Supplementary Material
References
- Jing C. R.; Cornish V. W. (2011) Chemical Tags for Labeling Proteins Inside Living Cells. Acc. Chem. Res. 44, 784–792. 10.1021/ar200099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin B. A.; Adams S. R.; Tsien R. Y. (1998) Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269–272. 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- Adams S. R.; Campbell R. E.; Gross L. A.; Martin B. R.; Walkup G. K.; Yao Y.; Llopis J.; Tsien R. Y. (2002) New biarsenical Ligands and tetracysteine motifs for protein labeling in vitro and in vivo: Synthesis and biological applications. J. Am. Chem. Soc. 124, 6063–6076. 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- Keppler A.; Gendreizig S.; Gronemeyer T.; Pick H.; Vogel H.; Johnsson K. (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89. 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- Miller L. W.; Sable J.; Goelet P.; Sheetz M. P.; Cornish V. W. (2004) Methotrexate conjugates: a molecular in vivo protein tag. Angew. Chem., Int. Ed. 43, 1672–1675. 10.1002/anie.200352852. [DOI] [PubMed] [Google Scholar]
- Mizukami S.; Hori Y.; Kikuchi K. (2014) Small-Molecule-Based Protein-Labeling Technology in Live Cell Studies: Probe-Design Concepts and Applications. Acc. Chem. Res. 47, 247–256. 10.1021/ar400135f. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Ueno H.; Mizukami S.; Kikuchi K. (2009) Photoactive yellow protein-based protein labeling system with turn-on fluorescence intensity. J. Am. Chem. Soc. 131, 16610–16611. 10.1021/ja904800k. [DOI] [PubMed] [Google Scholar]
- George N.; Pick H.; Vogel H.; Johnsson N.; Johnsson K. (2004) Specific labeling of cell surface proteins with chemically diverse compounds. J. Am. Chem. Soc. 126, 8896–8897. 10.1021/ja048396s. [DOI] [PubMed] [Google Scholar]
- Chen I.; Howarth M.; Lin W. Y.; Ting A. Y. (2005) Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99–104. 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- Theile C. S.; Witte M. D.; Blom A. E. M.; Kundrat L.; Ploegh H. L.; Guimaraes C. P. (2013) Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 8, 1800–1807. 10.1038/nprot.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Marks K. M.; Braun P. D.; Nolan G. P. (2004) A general approach for chemical labeling and rapid, spatially controlled protein inactivation. Proc. Natl. Acad. Sci. U. S. A. 101, 9982–9987. 10.1073/pnas.0401609101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zhang X.; Tan Y. L.; Bhabha G.; Ekiert D. C.; Kipnis Y.; Bjelic S.; Baker D.; Kelly J. W. (2014) De novo-designed enzymes as small-molecule-regulated fluorescence imaging tags and fluorescent reporters. J. Am. Chem. Soc. 136, 13102–13105. 10.1021/ja5056356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F. B. A.; Li P.; Li G. Y.; Zhao G. J.; Chu T. S.; Han K. L. (2011) A Near-IR Reversible Fluorescent Probe Modulated by Selenium for Monitoring Peroxynitrite and Imaging in Living Cells. J. Am. Chem. Soc. 133, 11030–11033. 10.1021/ja202582x. [DOI] [PubMed] [Google Scholar]
- Yu F. B.; Li P.; Wang B. S.; Han K. L. (2013) Reversible Near-Infrared Fluorescent Probe Introducing Tellurium to Mimetic Glutathione Peroxidase for Monitoring the Redox Cycles between Peroxynitrite and Glutathione in Vivo. J. Am. Chem. Soc. 135, 7674–7680. 10.1021/ja401360a. [DOI] [PubMed] [Google Scholar]
- Lou Z. R.; Li P.; Han K. L. (2015) Redox-Responsive Fluorescent Probes with Different Design Strategies. Acc. Chem. Res. 48, 1358–1368. 10.1021/acs.accounts.5b00009. [DOI] [PubMed] [Google Scholar]
- Lukinavicius G.; Umezawa K.; Olivier N.; Honigmann A.; Yang G. Y.; Plass T.; Mueller V.; Reymond L.; Corrêa I. R. Jr.; Luo Z. G.; Schultz C.; Lemke E. A.; Heppenstall P.; Eggeling C.; Manley S.; Johnsson K. (2013) A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 5, 132–139. 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- Neklesa T. K.; Tae H. S.; Schneekloth A. R.; Stulberg M. J.; Corson T. W.; Sundberg T. B.; Raina K.; Holley S. A.; Crews C. M. (2011) Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 7, 538–543. 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. Y.; Haegele J. A.; Disare M. T.; Lin Q. S.; Aye Y. (2015) A Generalizable Platform for Interrogating Target- and Signal-Specific Consequences of Electrophilic Modifications in Redox-Dependent Cell Signaling. J. Am. Chem. Soc. 137, 6232–6244. 10.1021/ja5132648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T.; Johnsson K.; Okuno H.; Bito H.; Inoue T.; Nagano T.; Urano Y. (2011) Real-Time Measurements of Protein Dynamics Using Fluorescence Activation-Coupled Protein Labeling Method. J. Am. Chem. Soc. 133, 6745–6751. 10.1021/ja200225m. [DOI] [PubMed] [Google Scholar]
- Sun X. L.; Zhang A. H.; Baker B.; Sun L.; Howard A.; Buswell J.; Maurel D.; Masharina A.; Johnsson K.; Noren C. J.; Xu M. Q.; Correa I. R. (2011) Development of SNAP-Tag Fluorogenic Probes for Wash-Free Fluorescence Imaging. ChemBioChem 12, 2217–2226. 10.1002/cbic.201100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing C.; Cornish V. W. (2013) A fluorogenic TMP-tag for high signal-to-background intracellular live cell imaging. ACS Chem. Biol. 8, 1704–1712. 10.1021/cb300657r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukami S.; Watanabe S.; Akimoto Y.; Kikuchi K. (2012) No-wash protein labeling with designed fluorogenic probes and application to real-time pulse-chase analysis. J. Am. Chem. Soc. 134, 1623–1629. 10.1021/ja208290f. [DOI] [PubMed] [Google Scholar]
- Butkevich A. N.; Mitronova G. Y.; Sidenstein S. C.; Klocke J. L.; Kamin D.; Meineke D. N.; D’Este E.; Kraemer P. T.; Danzl J. G.; Belov V. N.; Hell S. W. (2016) Fluorescent Rhodamines and Fluorogenic Carbopyronines for Super-Resolution STED Microscopy in Living Cells. Angew. Chem., Int. Ed. 55, 3290–3294. 10.1002/anie.201511018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G.; Reymond L.; Umezawa K.; Sallin O.; D’Este E.; Gottfert F.; Ta H.; Hell S. W.; Urano Y.; Johnsson K. (2016) Fluorogenic Probes for Multicolor Imaging in Living Cells. J. Am. Chem. Soc. 138, 9365–9368. 10.1021/jacs.6b04782. [DOI] [PubMed] [Google Scholar]
- Xue L.; Karpenko I. A.; Hiblot J.; Johnsson K. (2015) Imaging and manipulating proteins in live cells through covalent labeling. Nat. Chem. Biol. 11, 917–923. 10.1038/nchembio.1959. [DOI] [PubMed] [Google Scholar]
- Clark S. A.; Singh V.; Vega Mendoza D.; Margolin W.; Kool E. T. (2016) Light-Up ″Channel Dyes″ for Haloalkane-Based Protein Labeling in Vitro and in Bacterial Cells. Bioconjugate Chem. 27, 2839–2843. 10.1021/acs.bioconjchem.6b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti E.; Kocsis L. S.; Brummond K. M. (2012) Synthesis and Photophysical Properties of a Series of Cyclopenta[b]naphthalene Solvatochromic Fluorophores. J. Am. Chem. Soc. 134, 12418–12421. 10.1021/ja3055029. [DOI] [PubMed] [Google Scholar]
- Brummond K. M.; Kocsis L. S. (2015) Intramolecular Didehydro-Diels-Alder Reaction and Its Impact on the Structure-Function Properties of Environmentally Sensitive Fluorophores. Acc. Chem. Res. 48, 2320–2329. 10.1021/acs.accounts.5b00126. [DOI] [PubMed] [Google Scholar]
- MacNevin C. J.; Gremyachinskiy D.; Hsu C. W.; Li L.; Rougie M.; Davis T. T.; Hahn K. M. (2013) Environment-Sensing Merocyanine Dyes for Live Cell Imaging Applications. Bioconjugate Chem. 24, 215–223. 10.1021/bc3005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loving G.; Imperiali B. (2008) A versatile amino acid analogue of the solvatochromic fluorophore 4-N,N-dimethylamino-1,8-naphthalimide: A powerful tool for the study of dynamic protein interactions. J. Am. Chem. Soc. 130, 13630–13638. 10.1021/ja804754y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y. D.; Chiang P. Y.; Wang C. W.; Tan K. T. (2013) Environment-sensitive fluorescent turn-on probes targeting hydrophobic ligand-binding domains for selective protein detection. Angew. Chem., Int. Ed. 52, 8124–8128. 10.1002/anie.201302884. [DOI] [PubMed] [Google Scholar]
- Liu T. K.; Hsieh P. Y.; Zhuang Y. D.; Hsia C. Y.; Huang C. L.; Lai H. P.; Lin H. S.; Chen I. C.; Hsu H. Y.; Tan K. T. (2014) A Rapid SNAP-Tag Fluorogenic Probe Based on an Environment-Sensitive Fluorophore for No-Wash Live Cell Imaging. ACS Chem. Biol. 9, 2359–2365. 10.1021/cb500502n. [DOI] [PubMed] [Google Scholar]
- Yu W. T.; Wu T. W.; Huang C. L.; Chen I. C.; Tan K. T. (2016) Protein sensing in living cells by molecular rotor-based fluorescence-switchable chemical probes. Chem. Sci. 7, 301–307. 10.1039/C5SC02808F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y. S.; Gao R. C.; Wu T. W.; Cho C.; Tan K. T. (2016) Fluorescent Probe Encapsulated in SNAP-Tag Protein Cavity To Eliminate Nonspecific Fluorescence and Increase Detection Sensitivity. Bioconjugate Chem. 27, 1872–1879. 10.1021/acs.bioconjchem.6b00290. [DOI] [PubMed] [Google Scholar]
- Klock H. E.; Lesley S. A. (2009) The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 498, 91–103. 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- Wurth C.; Grabolle M.; Pauli J.; Spieles M.; Resch-Genger U. (2013) Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 8, 1535–1550. 10.1038/nprot.2013.087. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Storoni L. C.; Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 61, 458–464. 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J.; Peat T. S.; Richard R.; Kan L.; Swanson P. E.; Affholter J. A.; Holmes I. H.; Schindler J. F.; Unkefer C. J.; Terwilliger T. C. (1999) Haloalkane dehalogenases: Structure of a Rhodococcus enzyme. Biochemistry 38, 16105–16114. 10.1021/bi9913855. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Lebedev A. A.; Young P.; Isupov M. N.; Moroz O. V.; Vagin A. A.; Murshudov G. N. (2012) JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr., Sect. D: Biol. Crystallogr. 68, 431–440. 10.1107/S090744491200251X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai J. D.; Head-Gordon M. (2008) Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Pople J. A.; Binkley J. S. (1984) Self-Consistent Molecular-Orbital Methods 0.25. Supplementary Functions for Gaussian-Basis Sets. J. Chem. Phys. 80, 3265–3269. 10.1063/1.447079. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. (2005) Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Li L.; Li C.; Zhang Z.; Alexov E. (2013) On the ’Dielectric ″Constant″ of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 9, 2126–2136. 10.1021/ct400065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillion P.; Sexton D. J.; Benkovic S. J. (1998) Clamp subunit dissociation dictates bacteriophage T4 DNA polymerase holoenzyme disassembly. Biochemistry 37, 1819–1827. 10.1021/bi972526a. [DOI] [PubMed] [Google Scholar]
- Neto B. A. D.; Lapis A. A. M.; da Silva Júnior E. N.; Dupont J. (2013) 2,1,3-Benzothiadiazole and Derivatives: Synthesis, Properties, Reactions, and Applications in Light Technology of Small Molecules. Eur. J. Org. Chem. 2013, 228–255. 10.1002/ejoc.201201161. [DOI] [Google Scholar]
- Uchiyama S.; Kimura K.; Gota C.; Okabe K.; Kawamoto K.; Inada N.; Yoshihara T.; Tobita S. (2012) Environment-Sensitive Fluorophores with Benzothiadiazole and Benzoselenadiazole Structures as Candidate Components of a Fluorescent Polymeric Thermometer. Chem. - Eur. J. 18, 9552–9563. 10.1002/chem.201200597. [DOI] [PubMed] [Google Scholar]
- Neto B. A. D.; Carvalho P. H. P. R.; Correa J. R. (2015) Benzothiadiazole Derivatives as Fluorescence Imaging Probes: Beyond Classical Scaffolds. Acc. Chem. Res. 48, 1560–1569. 10.1021/ar500468p. [DOI] [PubMed] [Google Scholar]
- Marini A.; Munoz-Losa A.; Biancardi A.; Mennucci B. (2010) What is Solvatochromism?. J. Phys. Chem. B 114, 17128–17135. 10.1021/jp1097487. [DOI] [PubMed] [Google Scholar]
- Ma J. C.; Dougherty D. A. (1997) The cation-π interaction. Chem. Rev. 97, 1303–1324. 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. (2013) The Cation-π Interaction. Acc. Chem. Res. 46, 885–893. 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; English B. P.; Chen J.; Slaughter J. P.; Zhang Z.; Revyakin A.; Patel R.; Macklin J. J.; Normanno D.; Singer R. H.; Lionnet T.; Lavis L. D. (2015) A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 12, 244–250. 243 pages following page 250 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku J.; Lansac Y.; Jang Y. H. (2011) Time-Dependent Density Functional Theory Study on Benzothiadiazole-Based Low-Band-Gap Fused-Ring Copolymers for Organic Solar Cell Applications. J. Phys. Chem. C 115, 21508–21516. 10.1021/jp2062207. [DOI] [Google Scholar]
- Davis M. R.; Dougherty D. A. (2015) Cation-π interactions: computational analyses of the aromatic box motif and the fluorination strategy for experimental evaluation. Phys. Chem. Chem. Phys. 17, 29262–29270. 10.1039/C5CP04668H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C. J.; Gao J. M. (2013) Exploring and Exploiting Polar-π Interactions with Fluorinated Aromatic Amino Acids. Acc. Chem. Res. 46, 907–915. 10.1021/ar300086n. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. (2008) Physical organic chemistry on the brain. J. Org. Chem. 73, 3667–3673. 10.1021/jo8001722. [DOI] [PubMed] [Google Scholar]
- Xiu X. A.; Puskar N. L.; Shanata J. A. P.; Lester H. A.; Dougherty D. A. (2009) Nicotine binding to brain receptors requires a strong cation-π interaction. Nature 458, 534–U510. 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt C. (1994) Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 94, 2319–2358. 10.1021/cr00032a005. [DOI] [Google Scholar]
- Tsou L. K.; Tatko C. D.; Waters M. L. (2002) Simple cation-π interaction between a phenyl ring and a protonated amine stabilizes an α-helix in water. J. Am. Chem. Soc. 124, 14917–14921. 10.1021/ja026721a. [DOI] [PubMed] [Google Scholar]
- Daze K. D.; Hof F. (2013) The Cation-π Interaction at Protein-Protein Interaction Interfaces: Developing and Learning from Synthetic Mimics of Proteins That Bind Methylated Lysines. Acc. Chem. Res. 46, 937–945. 10.1021/ar300072g. [DOI] [PubMed] [Google Scholar]
- Watt M. M.; Collins M. S.; Johnson D. W. (2013) Ion-π Interactions in Ligand Design for Anions and Main Group Cations. Acc. Chem. Res. 46, 955–966. 10.1021/ar300100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa K.; Humbert-Droz M.; Letrun R.; Vauthey E.; Wesolowski T. A.; Sakai N.; Matile S. (2015) Ion Pair-π Interactions. J. Am. Chem. Soc. 137, 11047–11056. 10.1021/jacs.5b05593. [DOI] [PubMed] [Google Scholar]
- Chuard N.; Fujisawa K.; Morelli P.; Saarbach J.; Winssinger N.; Metrangolo P.; Resnati G.; Sakai N.; Matile S. (2016) Activation of Cell-Penetrating Peptides with ionpair-π Interactions and Fluorophiles. J. Am. Chem. Soc. 138, 11264–11271. 10.1021/jacs.6b06253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.