

Abstract
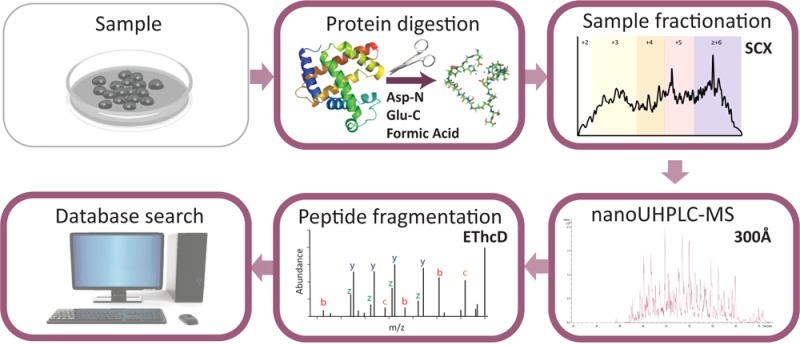
Mass spectrometry (MS)-based proteomics workflows can crudely be classified into two distinct regimes, targeting either relatively small peptides (i.e., 0.7 kDa < Mw < 3.0 kDa) or small to medium sized intact proteins (i.e., 10 kDa < Mw < 30 kDa), respectively, termed bottom-up and top-down proteomics. Recently, a niche has started to be explored covering the analysis of middle-range peptides (i.e., 3.0 kDa < Mw < 10 kDa), aptly termed middle-down proteomics. Although middle-down proteomics can follow, in principle, a modular workflow similar to that of bottom-up proteomics, we hypothesized that each of these modules would benefit from targeted optimization to improve its overall performance in the analysis of middle-range sized peptides. Hence, to generate middle-range sized peptides from cellular lysates, we explored the use of the proteases Asp-N and Glu-C and a nonenzymatic acid induced cleavage. To increase the depth of the proteome, a strong cation exchange (SCX) separation, carefully tuned to improve the separation of longer peptides, combined with reversed phase-liquid chromatography (RP-LC) using columns packed with material possessing a larger pore size, was used. Finally, after evaluating the combination of potentially beneficial MS settings, we also assessed the peptide fragmentation techniques, including higher-energy collision dissociation (HCD), electron-transfer dissociation (ETD), and electron-transfer combined with higher-energy collision dissociation (EThcD), for characterization of middle-range sized peptides. These combined improvements clearly improve the detection and sequence coverage of middle-range peptides and should guide researchers to explore further how middle-down proteomics may lead to an improved proteome coverage, beneficial for, among other things, the enhanced analysis of (co-occurring) post-translational modifications.
At the present time, two quite distinct approaches coexist in mass spectrometry-based proteomics studies: bottom-up, peptide centric approaches and top-down, protein centric approaches. In bottom-up proteomics, proteins are initially enzymatically digested, after which the resulting peptides are typically separated by liquid chromatography (LC) and analyzed by tandem mass spectrometry (MS/MS).1 In proteomics, trypsin represents the “golden standard” for proteases as it generates peptides that are relatively facile to separate by LC and analyze by MS. Therefore, the bottom-up approach is robust and enables high-throughput analysis, allowing the identification and quantification of thousands of proteins from complex lysates. However, sample complexity still imposes a heavy burden on separations prior to MS detection and, especially when using trypsin, a large part of the generated peptides is too small for successful binding to the stationary phases typically used in proteomics. Moreover, peptides smaller than 5 amino acids are typically not analyzed by MS/MS since they provide too little of an analytical value for unambiguous identification. Therefore, quite a portion of tryptic peptides may not be detected or are intentionally ignored.
In top-down proteomics, intact proteins are analyzed,2 which circumvent issues related to peptide-centric proteomics such as the protein inference problem.3 Top-down proteomics provides complete molecular specificity on intact proteins, enabling the analysis of proteoforms4 as intact proteins harbor the entire set of (co-occurring) PTMs.5,6 The main limitations in top-down proteomics are related to the difficulty of efficiently separating proteins and the relatively inefficient formation of fragment ions from larger proteins. Nevertheless, top-down is now becoming feasible, also in a relatively high-throughput manner, albeit mainly limited to proteins with a Mw up to ∼30 kDa. Sequence information on these proteins can be obtained by employing collision-induced dissociation (CID) which selectively fragments the most labile bonds in a protein, which typically provides only limited protein coverage.7 Use of electron capture/transfer dissociation (ECD/ETD)8,9 was shown to improve the sequence coverage. Additionally, ETD preserves most common labile PTMs during fragmentation, facilitating confident PTM site localization. More recent fragmentation methods, such as electron-transfer combined with higher-energy collision dissociation (EThcD) and UV photodissociation, are emerging as efficient fragmentation alternatives for top-down proteomics.10−12
As top-down proteomics typically covers relatively small proteins (10 < Mw < 30 kDa) and trypsin based bottom-up proteomics focuses generally on peptides (0.7 < Mw < 3 kDa), evidently, a gray zone exists between these proteomics approaches. This gap has been filled by a third approach, nowadays, called middle-down proteomics.13−15 This approach also uses protein digestion but aims to yield relatively larger peptides (ideally (far) above 3 kDa).16−19 Middle-down proteomics exhibits particular advantages as the complexity of the digests decreases (as fewer peptides are formed) and may also allow better proteome coverage, including the identification of splice-variants and other isoforms. Longer peptides also increase the probability of detecting multiple co-occurring neighboring PTMs, important to study functionally relevant PTM crosstalk. Most of the pioneering middle-down proteomics studies have been limited to specific applications, for instance, on recombinant monoclonal antibodies,20,21 ubiquitin chains,22 and N-terminal histone chains.23−25 Thus, middle-down proteomics is not yet routinely used for the analysis of full proteomes. We argue that an important bottleneck has been that neither the pipeline developed for bottom-up nor that for top-down analyses is directly applicable and optimal for middle-down proteomics. Therefore, here, we aimed to describe and critically evaluate a workflow optimized for the detection of middle-range sized peptides.
The first experimental barrier in the high-throughput middle-down proteomics workflow is the lack of a proteolytic enzyme that can produce controlled populations of middle-range sized peptides (3–10 kDa). A protease cutting at a well-defined peptide length or size would obviously be ideal; however, such an idealase remains to be discovered.26 Some known single-residue specific proteases such as Lys-C,13,27,28 Glu-C,29−31 Asp-N,32 and Lys-N33,34 are thought to produce, when compared to trypsin, a higher proportion of middle-range sized peptides and may thus provide second-best alternatives for middle-down proteomics. Also, some nonenzymatic approaches may be exploited to generate longer peptides. Microwave-accelerated acid hydrolysis, which produces Asp-selective chemical cleavage, had been explored by using a number of acid modifiers.35,36 We evaluated whether the proteases Asp-N and Glu-C or a nonenzymatic acid induced digestion protocol provide middle-range sized peptides. Second, additional analytical issues remain to be optimized for middle-down proteomics, such as the chromatography applied to separate the longer peptides. For this reason, we optimized conditions for sample cleanup and separation of these longer peptides by multidimensional LC. Finally, MS transmission and detection as well as MS/MS fragmentation conditions should be adjusted.37 We fine-tuned the critical MS parameters for improving the detection of middle-range sized peptides. Moreover, we evaluated the performances of ETD, higher-energy collision dissociation (HCD), and EThcD as fragmentation schemes on all the peptide digests obtained to define the best sequencing method for distinct populations of middle-range sized peptides.
Experimental Section
Sample Preparation
HeLa digests were prepared as described previously.38 For preparing the Asp-N and Glu-C digests, the protocols reported by Giansanti et al. were used.39 For the acid induced nonenzymatic digestion, the lysate was diluted to a final concentration of 0.1 μg/μL using a solution of formic acid (FA) (final concentration of 2% FA) and incubated at 100 °C for 1 h. A detailed description of the used protocols can be found in the Supporting Information.
Sample Cleanup and Prefractionation
Following digestion, sample cleanup was performed by using solid-phase extraction (SPE) columns: C18 with a 300 Å pore size. Prior to the MS analysis, samples were fractionated using strong cation exchange (SCX) chromatography. A detailed description of the used methods can be found in the Supporting Information.
LC-MS and LC-MS/MS Set Up
Nano-UHPLC-MS/MS was performed on an Agilent 1290 Infinity System connected to an Orbitrap Fusion. Fused-silica capillary analytical and trap columns were prepared as previously described.40 The UHPLC was equipped with a double frit trapping column and a single frit analytical column. ReprosilPur C18 (3 μm particles, 120 Å pore size 2 cm × 100 μm) was used as a trap column, and Zorbax SB-C18 (1.8 μm particles 80 Å 40 cm × 50 μm) was used for the analytical column. For the 300 Å pore size set up, the used materials were as follows: Zorbax SB-C18 (3.5 μm particles, 300 Å pore size, 2 cm × 100 μm) for the trap column and Zorbax SB-C18 (1.8 μm particles, 300 Å 40 cm × 50 μm) for the analytical column. The column, in both cases, was directly connected to an in-house pulled and gold-coated fused silica needle (with a 5 μm o.d. tip), and a voltage of 2.0 kV was applied. The survey scan range was from 350 to 1500 m/z at a resolution of 60 000 (200 m/z) with an AGC target of 4 ×105. The most intense precursor ions were selected for subsequent fragmentation at Top Speed within a 3 s duty cycle. A resolution of 30 000 (200 m/z) and a maximum injection time of 125 ms were found to be ideal for MS/MS. The AGC target for the MS/MS was set to 1×105. When HCD was used, 35% collision energy (CE) was applied; in the case of EThcD, 40% supplemental activation (SA) was selected and when ETD was used 10% SA was applied. Additionally, charge triggered MS/MS, instead of intensity triggered, was tested for the EThcD charge method.
Data Analysis
The RAW files were processed by using Proteome Discoverer, and the spectra were searched against the UniProt human database. Database searching was performed with Sequest HT and Mascot, and the results were filtered using Percolator41,42 to a peptide and protein FDR < 1%. A detailed description of the data analysis can be found in the Supporting Information.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE43 partner repository with the data set identifier PXD004910, with the following account details:
Username: reviewer09480@ebi.ac.uk
Password: xWhaEOKh
Results and Discussion
Evaluation of Enzymatic Digestion Approaches for the Generation of Middle-Range Sized Peptides
This study was aimed at providing an objective and comprehensive evaluation of the optimization steps required for the generation and analysis by LC-MS/MS of middle-range sized peptides (Figure S-1). We first set out to generate peptide digests of cell lysates, which in principle could yield higher numbers of middle-range sized peptides. Proteases that target single residues, such as Asp-N, Glu-C, and Lys-C, are thought to produce peptides with distinct peptide length distributions compared to trypsin.27,44−46,48 In our initial experiments, we tested Asp-N and Glu-C and digested a HeLa lysate, while a previously published trypsin data set was used as a reference.47 Noteworthy, the median Mw of all identified peptides was found to be around 1.9 kDa for Glu-C and Asp-N, an increase of about 25% compared to 1.5 kDa median Mw for trypsin (Figure S-2, Table S-1). Although this increase in median Mw is significant, ideally we would generate for middle-down proteomics peptides with a median Mw > 3 kDa, and thus, the results obtained were found to be suboptimal. Therefore, we also set out to explore other means to generate peptides from proteins, using a nonenzymatic digestion.
Testing a Nonenzymatic Digestion for the Generation of Middle-Range Sized Peptides
Chemical cleavage methods have been used as an alternative to enzymatic proteolysis to generate peptides from proteins.35 In acid-induced chemical digestion, proteins are incubated at high temperatures in formic acid (FA) diluted solutions.36,49 Diluted FA was reported to cleave proteins mainly at the C-termini of aspartic acid (D).36 We hypothesized that, due to this specificity, FA-induced digestion would potentially yield peptides of middle-sized length.35 To our knowledge, acid-induced chemical digestion has not been generally explored in full complex proteome analysis.50,51 Interestingly, the median Mw of all identified peptides in the FA induced digestion was similar to Asp-N and Glu-C. In addition, the frequency of occurrence of missed cleaved peptides was on par with that for the Glu-C digest data sets (Figure S-2).
Adapting Cleanup and Chromatographic Conditions To Maximize the Retention and Separation of Middle-Range Sized Peptides
Once a considerable population of middle-range sized peptides is generated, it is crucial to avoid their loss in the subsequent steps of the proteomics workflow. The in silicoMw distributions generated based on the Asp-N and Glu-C specificities are 2.39 and 2.37 kDa, respectively. We analyzed experimental data obtained from previous in-depth proteomics studies generated by Asp-N and Glu-C and a much lower average Mw value was found.52,53 We thus hypothesized that this discrepancy can be caused by losses of middle-range sized peptides which can be due to suboptimal separation chemistry being used for sample cleanup, prefractionation, and/or for the choice of unfavorable LC conditions. We thus optimized a range of steps in the proteomics workflow (Figure S-1), prior to MS detection, which were aimed to minimize the losses of middle-range sized peptides. When dealing with these peptides, one of the main aspects to consider is their ability of being adsorbed (mass transfer) and efficiently separated by the porous particles used in the employed chromatographic techniques. A poor choice of pore size will thus result in inadequate retention and loss of resolution. Conventionally, particles with 80–120 Å pore sizes are used for the separation of tryptic peptides. However, a more appropriate pore size for the separation of middle-range sized peptides is 300 Å, as has been previously reported.54 We therefore decided to test the latter pore size material for peptide desalting with solid-phase extraction (SPE) columns, prefractionation, and reversed phase chromatography.
Strong Cation Exchange as a Selective Prefractionation Mode for Middle-Range Sized Peptides
For in depth proteomics studies, typically one or more fractionation steps are included to decrease sample complexity and boost the number of identifications.53,55−57 When evaluating options for multidimensional separation schemes used in proteomics approaches, we prioritized binding and separation of longer peptides with high frequency of basic residues in order to match our digested peptide populations. We, thus, hypothesized that SCX at low pH (typically ∼3) could be appropriate as a fractionation technique. As initially hypothesized, we found a good correlation between the charge and the Mw of the peptides generated by the alternative digestions (Figure S-3a). Interestingly, for middle-down proteomics approaches, the above-described correlation can be exploited by selecting the appropriate fractions containing peptides of higher Mw.
Optimizing the Pore Size for the Separation of Middle-Range Sized Peptides
We first set up a side-by-side comparison between the conventional reversed phase (RP) C18 material and the 300 Å pore size material, using the same stationary phase chemistry in order to test if the difference in pore size generates a bias against higher Mw peptides. For the comparison, the same amount of Asp-N SCX fractions was injected and identical MS/MS settings were used. We monitored the achieved peak capacity, retention of middle-range sized peptides, the attainable back pressure (see Supporting Information), the overall retention times, and the total number of identifications (Figure S-4a). Only a modest decrease in the overall peak capacity (about 8%) was observed using the 300 Å pore size column. However, when focusing on peptides with Mw > 4 kDa, improved peak widths and increased area under the curves (AUCs) were generally observed (Figure S-4b), ratifying the choice of the 300 Å pore size column for these peptides. The 300 Å pore size column marginally outperformed the conventional RP C18 column in terms of number of identified proteins (4895 vs 4722) and peptides (34 307 vs 32 907). Focusing on peptides identified exclusively with each of the columns, the better performance of the larger pore size material for middle-sized peptides became clearly visible (Figure S-3b). The 300 Å pore size column retained and eluted more efficiently peptides with Mw > 1.5 kDa, confirming our hypothesis that conventional pore size materials typically used for the separation of tryptic peptides negatively affects the retention and separation of middle-range sized peptides.
Tuning MS Settings for Improving the Detection of Middle-Range Sized Peptides
Next, we evaluated several combinations of MS settings, which we thought could be optimized for the detection of middle-range sized peptides. We used the high-field Orbitrap analyzer, which enables higher resolution compared to the previous Orbitrap version at the same transient length and is advantageous for the analysis of larger peptides.58 We evaluated the effects of resolution settings, for both MS and MS/MS scans, on the total number of peptide identifications, particularly the identification success rate of middle-range sized peptides and instrument duty cycle. For this comparison, several Asp-N SCX fractions, containing pools of peptides with distinct charge states, were chosen. We evaluated three methods varying the resolution at both the MS and MS/MS level (Table S-3). By increasing the resolution for MS/MS scans, we obtained a higher number of unique peptides, higher spectral quality (higher XCorr medians), and more middle-range sized peptides for any of the analyzed peptide population (Table S-3, top). Applying higher resolution for MS scans only resulted in a lower number of identified unique peptides, largely due to the lower duty cycle (Table S-3, bottom). Thus, our experiments showed that the chosen higher resolution setting in the MS/MS mode is essential to correctly measure the isotope spacing associated with each ion fragment, improving the identification rates of especially highly charged, longer peptides. Furthermore, we opted to evaluate the effect of longer MS/MS injection times to increase the number of detectable ion fragments and therefore improve peptide sequence coverage. We tested 3 maximum injection times ranging from 75 (typical value for shotgun experiments) to 125 ms with the latter allowing us to increase the sequence fragmentation coverage from 88% to 91% and almost no consequences on the number of unique peptides identified (Table S-4). Optimized method parameters can be found in Table S-5.
Exploring the Benefits of Multiple Fragmentation Methods for the Detection of Middle-Range Sized Peptides
We evaluated the performance of a number of fragmentation methods in identifying middle-range sized peptides from the Asp-N, Glu-C, and FA HeLa digests (Figure 1). In each of these analyses, HCD fragmentation gave the highest number of unique peptides identified, which is an inherent effect due to its shorter duty cycle (twice as many MS/MS scans) compared to ETD and EThcD (Table S-1). However, EThcD showed superior success rates, defined as the number of PSMs divided by the total number of MS/MS acquired. The EThcD success rate was above 55% for both Asp-N and Glu-C, while ETD and HCD success rates were considerably lower, about 36%. Higher score distributions were also obtained for EThcD across the entire Mw range (Figure 1).
Figure 1.

Performance of the fragmentation techniques (ETD, EThcD, and HCD) with respect to peptide Mw in each of the 3 applied digestion protocols. The comparison is based on the number of identified unique peptides as well as on the quality of spectra represented by their XCorr distribution (box plots).
We further calculated the peptide sequence coverages for each digestion method achieved with all the fragmentation methods (Figure 2). The median peptide sequence fragmentation coverage of HCD was 65%, followed by ETD with 82% of coverage. EThcD displayed medians of sequence fragmentation coverage of 95%. This is partly due to the contribution of multiple ion series (c/z and b/y),10 advantageous especially for middle-range sized peptides because they enable unambiguous sequence determination and multiple PTMs identification and site localization.59,60 We focused again on distinct Mw subclasses, peptides displaying a Mw < 2.5 kDa, and peptides with 2.5 < Mw < 4 kDa, and the latter classification contains peptides with Mw > 4 kDa (Figure S-5b–d, respectively). Strikingly, for peptides identified at Mw > 4 kDa, the median peptide sequence coverage obtained by EThcD was constantly around 90%, confirming its unique potential for middle-down proteomics.
Figure 2.
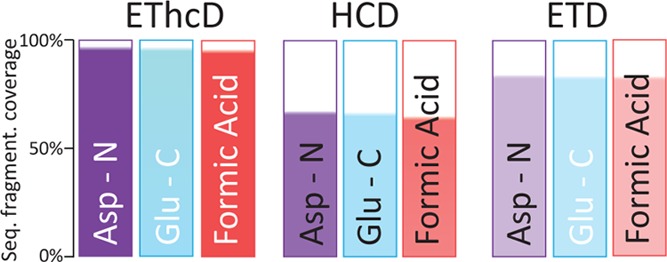
Peptide sequence fragmentation coverage obtained by each fragmentation method in the 3 applied digestion schemes. The median peptide sequence fragmentation coverage was calculated and represented taking into consideration all the SCX fractions for each digestion scheme.
Next to the peptide sequence coverage obtained by a single fragmentation method, we specifically looked at the performance of each sequencing technique in terms of number of unique peptides identified and XCorr distribution with respect to charge (z) and Mw ranges (Figures 3, S-6, and S-7). The score distributions and therefore the quality of the EThcD spectra are superior in all the studied ranges, while they yielded more identified peptides for the higher charge states (z 5 to 7). Interestingly, EThcD outperformed the other techniques also for lower charged peptides (z 2 to 4). The potential of ETD for the identification of peptides with Mw < 3 kDa, especially for highly charged peptides (z ≥ +5), is clear even though the quality of the spectra is lower than with EThcD fragmentation. Additionally, as expected, the power of HCD for highly charged peptides is diminished compared to +2 and +3 charge states.
Figure 3.

Performance of the peptide fragmentation techniques ETD, EThcD, and HCD for peptides with respect to their Mw and charge states (z). Combined data from Asp-N, Glu-C, and FA HeLa digests whereby the fragmentation parameters for ETD, EThcD, and HCD were optimized. The number of identifications as well as the XCorr distribution (as a measure for spectra quality) are categorized by their z and Mw ranges.
During data dependent MS/MS triggering, the abundance of low mass peptides in the mixture can obscure the precursor selection and thus fragmentation of middle-range sized peptides.62 We showed the correlation between charge state of peptides and their Mw (Figure S-3a) and reasoned that triggering MS/MS based on the highest charge state peptides present in the MS scan would further favor the detection of middle-range sized peptides. We evaluated the performance of this method compared to the usual intensity-based method with Asp-N SCX fractions. Indeed, not only did the peptide Mw median increase by 0.13 kDa by using the charge dependent MS/MS but also we could increase the number of total identified peptides (Figure S-8).
We also evaluated the success rate for each fragmentation technique with respect to Mw of peptide (Figure 4). The analysis confirmed the greater performance of EThcD for middle-down range peptides and of ETD especially for Mw > 6 kDa. However, despite these improved performances for longer peptides, the identification rates seem to drastically decrease at Mw > 4 kDa. One major cause of the decline in efficiency of identification for larger peptides has been related to the need of an increasing ion population required to maintain signal-to-noise for the generated fragment ion species.61 The data suggests our workflow is allowing the transmission of a considerable number of middle range peptides, but MS/MS efficiency is found to limit identification rates.
Figure 4.

Ions selected for MS/MS fragmentation matched to PSMs binned by peptide Mw. The % (in blue) of precursors matched to PSMs is calculated for each Mw bin for all the different fragmentations after merging all data sets from the different digestions. The data clearly indicate the superior performance of EThcD at the higher Mw bins.
Effect of Isotope Deconvolution on Mascot and Sequest for the Identification of Middle-Range Peptides
Triggered by our analysis of success rate with respected to Mw, showing that a great part of the precursors with Mw > 4 kDa is not readily identified, we questioned if we could improve the identification of the high Mw precursors (thus highly charged too) by isotope deconvolution of the MS/MS spectra and use of alternative search engines. Fragmentation spectra of highly charged and high Mw precursors potentially contain multiply charged fragment ions, which are often weighted differently/weaker than singly charged ions by certain search engines.63 We applied the H-Score script to our data,64 searched with the digestion specificity that was dictated by our observation (summarized above, in Figure 5 for Asp-N and similarly in Figure S-9 for the other digestions). We compared the number of unique peptides identified, XCorr distribution, and Mw median for increasing Mw ranges (Figure 5 for Asp-N and Figure S-9 for all data, Table S-6). Our analysis showed that deconvolution of the MS/MS leads to a decrease in XCorr distribution and number of unique peptides identified, while the median of Mw did not significantly increase. Most likely, the decrease in performance of deconvoluted data is due to the cross-correlation algorithm used by Sequest which rewards data rich spectra irrespective of redundancy with respect to fragments present with a range of charge states.65 We thus performed the same comparison, with the well-known, probability-based search engine Mascot. In this case, we obtained an increase of unique peptides identified, Mascot score distributions, and Mw medians of identified peptides for all the digestions and fragmentation techniques (Figure 5 for Asp-N and Figure S-9 for all data, Table S-6). Interestingly, the score distribution was found to be very similar for all 3 fragmentation schemes, perhaps a product of the probability-based scoring. This clearly indicates that Mascot benefits of the isotope deconvolution of multiply charged ions as has been reported previously.63 Nevertheless, the results were comparable to those obtained with Sequest and no deconvolution.
Figure 5.

Effect of deconvolution on Sequest and Mascot searches for the identification of Asp-N middle-range peptides. Specific searches were performed by Sequest HT and Mascot on the Asp-N HeLa digestions data sets for each of the optimized fragmentation methods (ETD, EThcD, and HCD). The number of identifications as well as the XCorr distribution (as a measure for spectra quality) are categorized by their Mw ranges for deconvoluted and nondeconvoluted spectra searched by Sequest HT and Mascot.
Conclusions
Here, we present a step-by-step tuning of the “standard” bottom-up proteomics workflow with the aim to improve the generation, separation, detection, and identification of middle-down range peptides. We first show that one of the main barriers for the development of middle-down proteomics is the generation of the desired peptides (3.5 kDa < Mw < 10 kDa). A substantial gain in median Mw of the peptides can be made by using Glu-C and Asp-N instead of trypsin, although the median Mw then still only reaches ∼2 kDa. Alternatively, we generated peptides by formic acid-induced digestion. This approach, which had not yet been explored at a proteome-wide scale, provides an alternative for creating peptides, on par in number of achievable peptide and protein identifications with enzyme based approaches, although it suffers from efficiency/sensitivity issues. Interestingly, the average Mw of the peptides generated in the FA digest was found to be similar to that of the Asp-N and Glu-C digests. We next showed that the detection of these longer peptides could be improved by using an optimized SCX separation method and columns packed with larger pore size materials. In testing fragmentation methods, we demonstrate the excellent performance of EThcD for the analysis of middle-range sized peptides, not only presenting the highest identification rate (up to 57% success rate) but also providing higher peptide sequence coverage (up to 95% peptide sequence fragmentation coverage) compared to HCD and ETD fragmentations methods. Furthermore, the Mascot search engine benefited from deconvolution while Sequest showed a minor negative effect. Nevertheless, we also found that the number of effectively identified precursors seems to decrease at higher Mw (>4 kDa) across all fragmentation techniques, suggesting that the MS/MS sequencing may be the major limiting factor in our workflow. Cumulatively, the optimizations made here in peptide generation, separation, detection, fragmentation, and identification expand the number of unambiguous identifications of especially the middle-size range peptides and, therefore, are a major step forward toward an optimized workflow for middle-down proteomics.
Acknowledgments
The authors acknowledge all members of the Heck lab and especially Soenita Goerdayal and Teck Yew Low for technical support and advice. This work was supported by The Netherlands Organisation for Scientific Research (NWO) via the Roadmap Initiative Proteins@Work (project number 184.032.201), the VIDI grant 700.10.429 for S.M., and the TOP-Punt Grant 718.015.003 for A.J.R.H. A.J.R.H. acknowledges additional support through the European Union Horizon 2020 programme FET-OPEN project MSmed, project number 686547.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b03756.
Additional figures and tables containing detailed information on the optimization steps performed in the study (PDF)
Author Contributions
¶ A.C. and F.M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M.-C.; Yates J. R. Chem. Rev. 2013, 113 (4), 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman A. D.; Skinner O. S.; Kelleher N. L. Biochem. Biophys. Res. Commun. 2014, 445 (4), 683–693. 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Aebersold R. Mol. Cell. Proteomics 2005, 4 (10), 1419–1440. 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- Smith L. M.; Kelleher N. L.; Nat. Methods 2013, 10 (3), 186–187. 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuti N.; Kelleher N. L. Nat. Methods 2007, 4 (10), 817–821. 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran J. C.; Zamdborg L.; Ahlf D. R.; Lee J. E.; Catherman A. D.; Durbin K. R.; Tipton J. D.; Vellaichamy A.; Kellie J. F.; Li M.; Wu C.; Sweet S. M. M.; Early B. P.; Siuti N.; LeDuc R. D.; Compton P. D.; Thomas P. M.; Kelleher N. L. Nature 2011, 480 (7376), 254–258. 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki V. H.; Tsaprailis G.; Smith L. L.; Breci L. A. J. Mass Spectrom. 2000, 35 (12), 1399–1406. . [DOI] [PubMed] [Google Scholar]
- Zubarev R. A. Curr. Opin. Biotechnol. 2004, 15 (1), 12–16. 10.1016/j.copbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (26), 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Anal. Chem. 2012, 84 (22), 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Morrison L. J.; Brodbelt J. S. J. Am. Chem. Soc. 2016, 138 (34), 10849–10859. 10.1021/jacs.6b03905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamara S.; Dyachenko A.; Fort K. L.; Makarov A. A.; Scheltema R. A.; Heck A. J. R. J. Am. Chem. Soc. 2016, 138 (34), 10860–10868. 10.1021/jacs.6b05147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes A. J.; Mazur M. T.; Patel H. M.; Walsh C. T.; Kelleher N. L. Proteomics 2001, 1 (8), 927–933. . [DOI] [PubMed] [Google Scholar]
- Wu S.; Kim J.; Hancock W. S.; Karger B. J. Proteome Res. 2005, 4 (4), 1155–1170. 10.1021/pr050113n. [DOI] [PubMed] [Google Scholar]
- Garcia B. A. J. Am. Soc. Mass Spectrom. 2010, 21 (2), 193–202. 10.1016/j.jasms.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Wu S.-L.; Kim J.; Bandle R. W.; Liotta L.; Petricoin E.; Karger B. L. Mol. Cell. Proteomics 2006, 5 (9), 1610–1627. 10.1074/mcp.M600105-MCP200. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Wu S.-L.; Kim J.; Karger B. L. J. Chromatogr. A 2007, 1154 (1–2), 295–307. 10.1016/j.chroma.2007.03.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Tran J. C.; Zamdborg L.; Durbin K. R.; Li M.; Ahlf D. R.; Early B. P.; Thomas P. M.; Sweedler J. V.; Kelleher N. L. Nat. Methods 2012, 9 (8), 822–824. 10.1038/nmeth.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskay Ü. A.; Lobas A. A.; Srzentić K.; Gorshkov M. V.; Tsybin Y. O. J. Proteome Res. 2013, 12 (12), 5558–5569. 10.1021/pr400522h. [DOI] [PubMed] [Google Scholar]
- Fornelli L.; Ayoub D.; Aizikov K.; Beck A.; Tsybin Y. O. Anal. Chem. 2014, 86 (6), 3005–3012. 10.1021/ac4036857. [DOI] [PubMed] [Google Scholar]
- Srzentić K.; Fornelli L.; Laskay Ü. A.; Monod M.; Beck A.; Ayoub D.; Tsybin Y. O. Anal. Chem. 2014, 86 (19), 9945–9953. 10.1021/ac502766n. [DOI] [PubMed] [Google Scholar]
- Valkevich E. M.; Sanchez N. A.; Ge Y.; Strieter E. R. Biochemistry 2014, 53 (30), 4979–4989. 10.1021/bi5006305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia B. A.; Siuti N.; Thomas C. E.; Mizzen C. A.; Kelleher N. L. Int. J. Mass Spectrom. 2007, 259 (1–3), 184–196. 10.1016/j.ijms.2006.07.022. [DOI] [Google Scholar]
- Moradian A.; Kalli A.; Sweredoski M. J.; Hess S. Proteomics 2014, 14 (4–5), 489–497. 10.1002/pmic.201300256. [DOI] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Zheng Y.; Smith R. D.; Kelleher N. L. Anal. Chem. 2012, 84 (10), 4271–4276. 10.1021/ac300612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiatsiani L.; Heck A. J. R. FEBS J. 2015, 282 (14), 2612–2626. 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- Wu S.-L.; Kim J.; Hancock W. S.; Karger B. J. Proteome Res. 2005, 4 (4), 1155–1170. 10.1021/pr050113n. [DOI] [PubMed] [Google Scholar]
- Boyne M. T.; Garcia B. A.; Li M.; Zamdborg L.; Wenger C. D.; Babai S.; Kelleher N. L. J. Proteome Res. 2009, 8 (1), 374–379. 10.1021/pr800635m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S.; Schwämmle V.; Ruminowicz C.; Hansen T. A.; Wu X.; Helin K.; Jensen O. N. Proteomics 2014, 14 (19), 2200–2211. 10.1002/pmic.201400084. [DOI] [PubMed] [Google Scholar]
- Sidoli S.; Lin S.; Karch K. R.; Garcia B. A. Anal. Chem. 2015, 87 (6), 3129–3133. 10.1021/acs.analchem.5b00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S. D.; Ueberheide B. M.; Liu Y.; Tackett A. J.; Diaz R. L.; Shabanowitz J.; Chait B. T.; Hunt D. F.; Allis C. D. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (7), 2086–2091. 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney D. L.; Wenger C. D.; Coon J. J. J. Proteome Res. 2010, 9 (3), 1323–1329. 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taouatas N.; Drugan M. M.; Heck A. J. R.; Mohammed S. Nat. Methods 2008, 5 (5), 405–407. 10.1038/nmeth.1204. [DOI] [PubMed] [Google Scholar]
- Hohmann L.; Sherwood C.; Eastham A.; Peterson A.; Eng J. K.; Eddes J. S.; Shteynberg D.; Martin D. B. J. Proteome Res. 2009, 8 (3), 1415–1422. 10.1021/pr800774h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J.; Lohnes K.; Wynne C.; Wang Y.; Edwards N.; Fenselau C. J. Proteome Res. 2010, 9 (8), 3886–3890. 10.1021/pr1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A.; Sowder R. C.; Henderson L. E.; Moore S. P.; Garfinkel D. J.; Fisher R. J. Anal. Chem. 2001, 73 (22), 5395–5402. 10.1021/ac010619z. [DOI] [PubMed] [Google Scholar]
- Di Palma S.; Hennrich M. L.; Heck A. J. R.; Mohammed S. J. Proteomics 2012, 75 (13), 3791–3813. 10.1016/j.jprot.2012.04.033. [DOI] [PubMed] [Google Scholar]
- Helbig A. O.; Gauci S.; Raijmakers R.; Van Breukelen B.; Slijper M.; Mohammed S.; Heck A. J. R. Mol. Cell. Proteomics 2010, 9 (5), 928–939. 10.1074/mcp.M900463-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giansanti P.; Tsiatsiani L.; Yew Low T.; Heck A. J. R. Nat. Protoc. 2016, 11, 993. 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- Cristobal A.; Hennrich M. L.; Giansanti P.; Goerdayal S. S.; Heck A. J. R.; Mohammed S. Analyst 2012, 137 (15), 3541–3548. 10.1039/c2an35445d. [DOI] [PubMed] [Google Scholar]
- Käll L.; Canterbury J. D.; Weston J.; Noble W. S.; MacCoss M. J. Nat. Methods 2007, 4 (11), 923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Spivak M.; Weston J.; Bottou L.; Käll L.; Noble W. S. J. Proteome Res. 2009, 8 (7), 3737–3745. 10.1021/pr801109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; Xu Q.-W.; Wang R.; Hermjakob H. Nucleic Acids Res. 2016, 44 (D1), D447–56. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina H.; Horn D. M.; Tang N.; Mathivanan S.; Pandey A. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (7), 2199–2204. 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCoss M. J.; McDonald W. H.; Saraf A.; Sadygov R.; Clark J. M.; Tasto J. J.; Gould K. L.; Wolters D.; Washburn M.; Weiss A.; Clark J. I.; Yates J. R. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (12), 7900–7905. 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi A.; Huttenhower C.; Geer L. Y.; Coon J. J.; Syka J. E. P.; Bai D. L.; Shabanowitz J.; Burke D. J.; Troyanskaya O. G.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (7), 2193–2198. 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N.; Wisniewski J. R.; Geiger T.; Cox J.; Kircher M.; Kelso J.; Pääbo S.; Mann M. Mol. Syst. Biol. 2011, 7, 548. 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapeau G. R. Methods Enzymol. 1977, 47, 189–191. 10.1016/0076-6879(77)47023-X. [DOI] [PubMed] [Google Scholar]
- Inglis A. S. Methods Enzymol. 1983, 91, 324–332. 10.1016/S0076-6879(83)91030-3. [DOI] [PubMed] [Google Scholar]
- Cannon J. R.; Edwards N. J.; Fenselau C. J. Mass Spectrom. 2013, 48 (3), 340–343. 10.1002/jms.3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A.; Mataj A.; Yang Y.; Boysen R. I.; Bowden D. K.; Hearn M. T. W. Anal. Chem. 2010, 82 (21), 8922–8930. 10.1021/ac1017106. [DOI] [PubMed] [Google Scholar]
- Guo X.; Trudgian D. C.; Lemoff A.; Yadavalli S.; Mirzaei H. Mol. Cell. Proteomics 2014, 13 (6), 1573–1584. 10.1074/mcp.M113.035170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Zougman A.; Mann M. J. Proteome Res. 2009, 8 (12), 5674–5678. 10.1021/pr900748n. [DOI] [PubMed] [Google Scholar]
- Pearson J. D.; Lin N. T.; Regnier F. E. Anal. Biochem. 1982, 124 (1), 217–230. 10.1016/0003-2697(82)90240-8. [DOI] [PubMed] [Google Scholar]
- Batth T. S.; Olsen J. V. Methods Mol. Biol. 2016, 1355, 179–192. 10.1007/978-1-4939-3049-4_12. [DOI] [PubMed] [Google Scholar]
- Mohammed S.; Heck A. J. Curr. Opin. Biotechnol. 2011, 22 (1), 9–16. 10.1016/j.copbio.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Di Palma S.; Mohammed S.; Heck A. J. R. Nat. Protoc. 2012, 7 (11), 2041–2055. 10.1038/nprot.2012.124. [DOI] [PubMed] [Google Scholar]
- Scheltema R. A.; Hauschild J.-P.; Lange O.; Hornburg D.; Denisov E.; Damoc E.; Kuehn A.; Makarov A.; Mann M. Mol. Cell. Proteomics 2014, 13 (12), 3698–3708. 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Zhou H.; Taus T.; Altelaar A. F.; Mechtler K.; Heck A. J.; Mohammed S. J. Proteome Res. 2013, 12 (3), 1520–1525. 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Maarten Altelaar A. F.; Van Den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Anal. Chem. 2012, 84 (22), 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Mullen C.; Weisbrod C. R.; Sharma S.; Senko M. W.; Zabrouskov V.; Westphall M. S.; Syka J. E. P.; Coon J. J. J. Am. Soc. Mass Spectrom. 2016, 27 (3), 520–531. 10.1007/s13361-015-1306-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J. R.; Edwards N. J.; Fenselau C. J. Mass Spectrom. 2013, 48 (3), 340–343. 10.1002/jms.3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; Hennrich M. L.; Nolting D.; Zeller M.; Griep-Raming J.; Heck A. J. R.; Mohammed S. J. Proteome Res. 2011, 10 (5), 2377–2388. 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Mathieson T.; Becher I.; Bantscheff M. J. Proteome Res. 2010, 9 (11), 5511–5516. 10.1021/pr1006813. [DOI] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. J. Am. Soc. Mass Spectrom. 1994, 5 (11), 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.