

Abstract
The Mediator complex-associated cyclin-dependent kinase CDK8 has been implicated in human disease, particularly in colorectal cancer where it has been reported as a putative oncogene. Here we report the discovery of 109 (CCT251921), a potent, selective, and orally bioavailable inhibitor of CDK8 with equipotent affinity for CDK19. We describe a structure-based design approach leading to the discovery of a 3,4,5-trisubstituted-2-aminopyridine series and present the application of physicochemical property analyses to successfully reduce in vivo metabolic clearance, minimize transporter-mediated biliary elimination while maintaining acceptable aqueous solubility. Compound 109 affords the optimal compromise of in vitro biochemical, pharmacokinetic, and physicochemical properties and is suitable for progression to animal models of cancer.
Introduction
The Mediator complex is a multiprotein assembly comprising at least 30 subunits that functions as a regulator of gene transcription in multiple contexts including stem cell function, the immune response, inflammation, cell adhesion, the epithelial to mesenchymal transition and development.1−5 The Mediator complex-associated kinase CDK8 and its paralog CDK19 are cyclin C-dependent enzymes that, along with MED12 and MED13, form the kinase module of the Mediator complex.6,7 CDK8 has been reported to regulate basal transcription by phosphorylation of RNA polymerase II8 and to phosphorylate E2F1, thereby activating WNT signaling.9 Interestingly, CDK8 gene expression correlates with activation of β-catenin, a core transcriptional regulator of canonical WNT signaling, in colon and gastric cancers.10,11 CDK8 gene expression also correlates with increased mortality in colorectal, breast, and ovarian cancers;12 furthermore CDK8 is overexpressed and essential for cell proliferation in melanoma.13 Consistent with these reports, CDK8 is located in a region of chromosome 13 known to undergo copy number gain in ∼60% of colorectal cancers and inducible shRNA-mediated knockdown of CDK8 protein reduces the growth of HT29 and Colo205 colorectal cancer human tumor xenograft animal models harboring CDK8 gene amplification.14 Notably, CDK8 expression transforms NIH3T3 cells into a malignant phenotype whereas a kinase-dead mutant does not, thereby implicating the kinase function of CDK8 in oncogenesis.15 The function and role of CDK19 are less well explored. CDK19 has been reported to form Mediator complexes independent of CDK8; however their context-dependent roles are the subject of ongoing study.7
Previously reported small molecule ligands for CDK8 and its paralog CDK19 have been described in a recent comprehensive review.16 In brief, the steroidal natural product cortistatin A (1) was the first-reported high affinity and selective ligand for CDK8/19 (Chart 1);17 recent disclosures include a patent describing cortistatin A analogs and a report demonstrating potent in vitro and in vivo antileukemic activity of cortistatin A through dual CDK8/19 inhibition.18,19 The marketed kinase inhibitor sorafenib (2) has been cocrystallized with CDK8/cyclin C,20 and subsequently the same group reported a fragment-based approach to CDK8 ligands building from the urea moiety associated with the type II binding mode of sorafenib.21 Type II kinase inhibitors linifanib (3) and ponatinib (4) have also been reported to bind both CDK8 and CDK19.22 Recently, a cell-based HTS campaign seeking inhibitors of p21-activated transcription was reported; this effort led to the discovery of aminoquinazoline-based CDK8/19 ligands, exemplified by senexin B (5) (Chart 1).23 Other series of small molecule CDK8 inhibitors have also been reported in the patent literature.24−26
Chart 1. Small Molecule CDK8/19 Ligands: Cortistatin A (1), Sorafenib (2), Linifanib (3), Ponatinib (4), Senexin B (5), and 6.

We have previously reported the discovery of 6 (CCT251545), a potent, orally bioavailable small molecule inhibitor of WNT signaling from a cell-based pathway screen (Chart 1).27 We identified protein kinases CDK8 and CDK19 as the primary targets of this trisubstituted pyridine series and demonstrated a strong correlation between CDK8 and CDK19 binding affinities in this chemical series.22 Here we describe the medicinal chemistry optimization of 6 to compound 109, a potent, selective, and orally bioavailable inhibitor of CDK8 with equipotent affinity for CDK19 that demonstrates potent cell-based activity together with improved pharmacokinetic and pharmaceutical properties. We demonstrate inhibition of CDK8 function concomitant with reduced proliferation in a human tumor xenograft animal model of colorectal cancer.
Chemistry
The general synthetic method for the preparation of 3-Cl and 3-F substituted pyridine analogues involved initial SNAr displacement at the 4-position of 3-bromo-4,5-dichloropyridine (7) or 3-bromo-4-chloro-5-fluoropyridine (57) to give intermediates 8, 26–36, 58, 59, and 96, which were then subject to Suzuki cross-coupling to give final compounds 17–25, 40–56, 60–63, and 100–105 (Scheme 1 and Tables 2, 3, 4, and 7). The corresponding CF3-substituted pyridines (70, 71, and 72, Table 4) were synthesized by two alternative routes. Compound 71 was prepared by selective copper-mediated trifluoromethylation at the bromo-substituted carbon atom of 8,28 followed by Suzuki coupling at the pyridine chloro substituent. Alternatively, compounds 70 and 72 were prepared by SNAr displacement at the 4-position of 3,5-diiodo-4-chloropyridine 64, followed by copper-mediated trifluoromethylation at one iodo-substituted carbon followed by Suzuki cross-coupling at the other.
Scheme 1. General Synthetic Routes to 3,4,5-Trisubtituted Pyridines.

The asterisk (∗) indicates different solvent and/or slightly different conditions were used for compounds 17, 18, 42, 47, 50, and 52.
Table 2. Introduction of Polarity at the Pyridine C-5 Substituent.

Free base.
TFA salt.
Run in different conditions (see reporter displacement assay in Experimental Section).
In this case the chloro substituent is at C-5 according to nomenclature 8-(3-(3-amino-1H-indazol-6-yl)-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one.
Table 3. Introduction of Polarity at the Pyridine C-4 Substituent.


Free base.
TFA salt.
The chloro substituent is at C-5 according to 5-(5-chloro-4-(1-methyl-1,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide.
The chloro substituent is at C-5 according to 5-(5-chloro-4-(1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide.
The chloro substituent is at C-5 according to 5-(5-chloro-4-(1-(3-methoxypropyl)-1,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide.
The chloro substituent is at C-5 according to 5-(5-chloro-4-(1-(3-(methylsulfonyl)propyl)-1,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide.
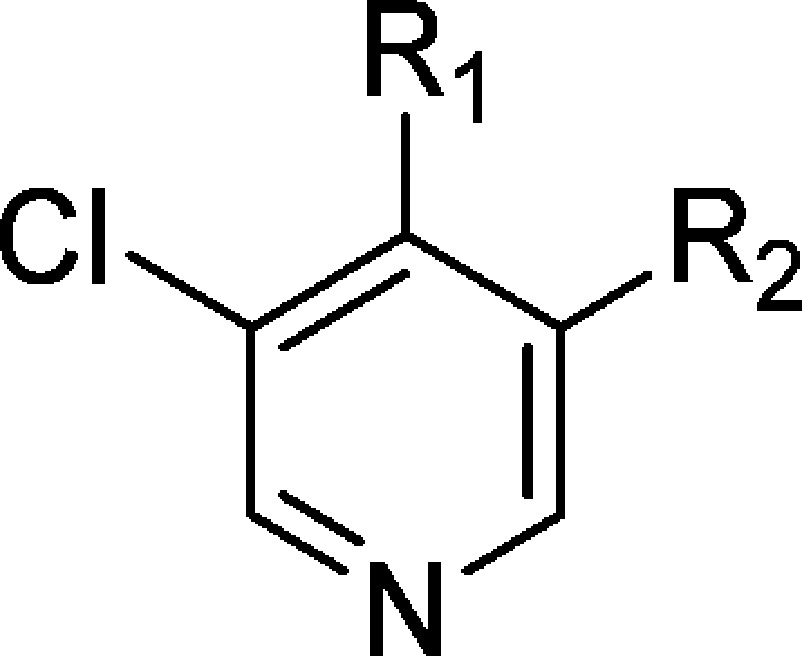
Table 4. Introduction of Chlorine Replacements at the Pyridine C-3 Position.

Free base.
TFA salt.
The CF3 substituent is at C-5 according to 8-(3-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)-5-(trifluoromethyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one.
The CF3 substituent is at C-5 according to 8-(3-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)-5-(trifluoromethyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one.
The CF3 substituent is at C-5 according to 8-(3-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)-5-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one.
Table 7. Attempts To Reduce Efflux in the Pyridine Seriesd.

Free base.
TFA salt.
In this case the chloro substituent is at C-5 according to nomenclature 8-(3-(3-amino-1-methyl-1H-indazol-6-yl)-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one.
M: mouse. R: rat. H: human. log P was calculated using Percepta Batch, version 2015 (www.acdlabs.com). TPSA was calculated in the program MOE (www.chemcomp.com).
2-Aminopyridines (88, 92–95, 108, and 111, Tables 5 and 8) were prepared by SNAr-mediated displacement of the 4-chloro substituent in pyridines 73, 74, and 75 followed by Suzuki cross-coupling (Scheme 2). In the case of 85–87, 89–91, 109, and 110, we found that protection of the primary amine with a PMB group improved both the SNAr displacement and the subsequent palladium-mediated cross-coupling reaction; deprotection was achieved using trifluoroacetic acid at room temperature.
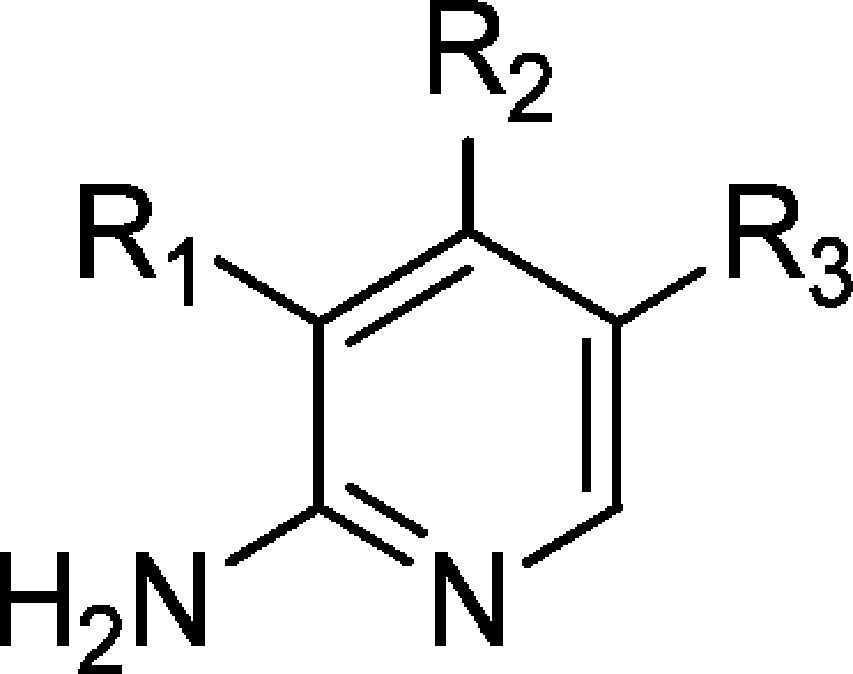
Table 5. 2-Aminopyridine Derivatives.

Free base.
TFA salt.
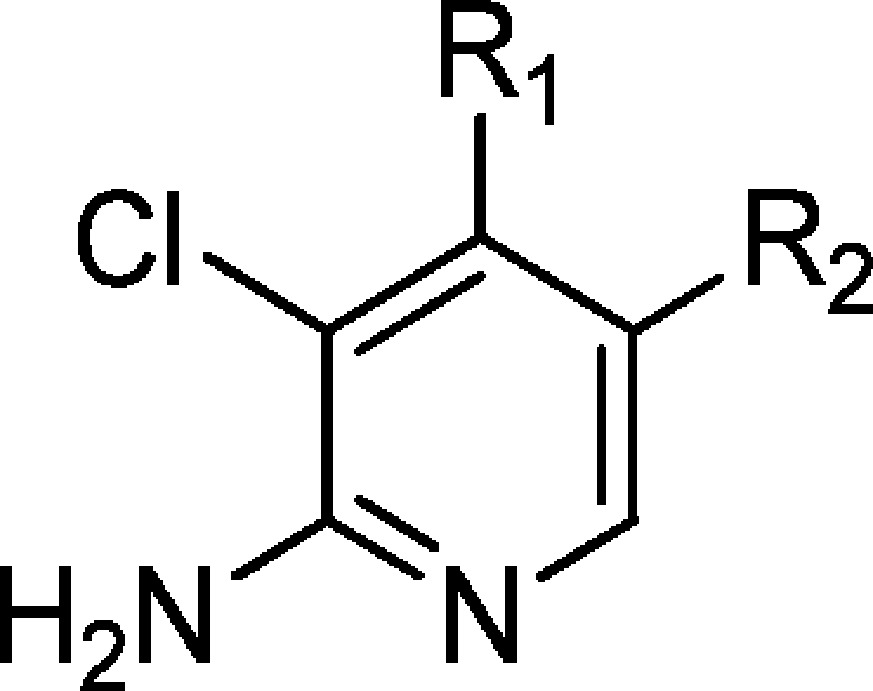
Table 8. Reducing Efflux in the 2-Aminopyridine Seriesd.

free base.
TFA salt.
besylate salt.
M: mouse. R: rat. H: human.
Scheme 2. General Synthetic Routes to 2-Aminopyridine Derivatives.

The asterisk (∗) indicates that KF was added for the synthesis of compounds 76 and 82.
Results and Discussion
Compound 6 is a high affinity ligand for CDK8 and CDK19 (IC50 of 7.2 ± 1.4 and 6.0 ± 1.0 nM, respectively) and demonstrates potent inhibition of WNT-dependent signaling using our previously described inducible luciferase reporter assay in human embryonic kidney cells (HEK293) that contains both estrogen receptor-dishevelled (DVL2) and TCF-luciferase-IRES-GFP constructs (7dF3 IC50 = 5.0 ± 2.0 nM, Table 2);27 it also demonstrates potent activity in LS174T human colorectal carcinoma cells that harbor a reporter-based readout measuring constitutive β-catenin mutation-driven WNT pathway activity (IC50 = 23 ± 11 nM).27 Compound 6 displays moderate in vivo clearance in both mouse and rat; however, high predicted human clearance (∼76% liver blood flow) prevented further progression (Table 1). Furthermore, the volumes of distribution (Vd) across all species tested were low to medium and the aqueous kinetic and thermodynamic solubilities (94 μM and 0.006 mg/mL, respectively) were suboptimal. We therefore turned our attention to lowering the lipophilicity of compound 6 (measured log D = 3.5) with the aim of decreasing the microsomal and in vivo clearance as well as improving aqueous solubility. To increase the Vd, we also attempted the introduction of a weakly basic center.29,30 In the course of improving physicochemical, pharmaceutical, and pharmacokinetic properties, we monitored in vitro biochemical affinity for CDK8 and CDK19 which we found to consistently predict for cell-based activity in both the reporter-based 7dF3 and LS174T assays;22 we also observed consistent SAR in these two reporter-based cellular assays for compounds described in this manuscript (Figure S1 in Supporting Information).
Table 1. Pharmacokinetic Profile of Compound 6a.
| species | Cl (L/h/kg) | LBF (%) | Vd (L/kg) | F (%) | t1/2 (h) |
|---|---|---|---|---|---|
| mouse | 1.87 | 31 | 1.08 | 54 | 0.55 |
| rat | 1.54 | 35 | 1.53 | 88 | 0.97 |
| dog | 0.84 | 33 | 0.74 | 126 | 0.70 |
| human prediction | ∼0.88 | ∼76 | ∼0.85 | ∼70 | ∼0.70 |
Dose: 0.2 mg/kg (iv), 0.5 mg/kg (po).
Upon the basis of our work to identify the molecular targets of 6,22 we knew that extended linear substituents were tolerated at C-5 of the pyridine ring; therefore, we introduced a variety of polar groups on the C-5 phenylpyrazole anticipating that these would enhance metabolic stability and aqueous solubility (Table 2, entries 2–5). Pleasingly, replacement of the pyrazole N1-methyl substituent by hydroxyethyl or 2-hydroxy-2-methylpropyl led to a significant improvement in microsomal stability in all species; furthermore, aqueous solubility also increased (Table 2, entries 2 and 3) while CDK8/19 biochemical affinity and cell-based 7dF3 potency were maintained. Introduction of a basic center resulted in a 4-fold drop in potency in both biochemical and cell-based assays, suggesting that permeability is not responsible for the observed decrease (Table 2, entries 4–6). However, we noticed that compounds such as 17, 18, and 21 (Table 2, entries 2, 3, 6) had reduced microsomal clearance (Clint), which confirmed that the introduction of polarity could be fruitful. Replacement of methylpyrazole by fused 6,5-heterocycles was also investigated. Introduction of 5-substituted-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide (henceforth abbreviated to sultam) improved the microsomal stability of the molecule across all species tested (Table 2, entry 7). Furthermore, the thermodynamic aqueous solubility of 22 was improved by 120-fold; however a 23-fold drop in cell-based potency was also observed. In order to improve cell permeability by lowering H-bond donor count, the N-methyl sultam derivative 23 was prepared (Table 2, entry 8); while this compound was potent in the 7dF3 cell-based assay, both 23 and its regioisomer 24 exhibited significant metabolic instability (Table 2, entries 8 and 9). The aminoindazole 25 gave an acceptable in vitro clearance profile across all species and high aqueous solubility (Table 2, entry 10); however, as for most of the derivatives in Table 2, we observed low oral bioavailability and no improvement of the in vivo mouse clearance compared to 6.
To understand whether the introduction of polarity influenced binding mode, we determined the crystal structure of aminoindazole derivative 25 in complex with the kinase domain of CDK8 and cyclin C (Figure 1, panel A). As previously observed with 6, analog 25 occupies the ATP binding site (Figure 1, panel B).22 All interactions were conserved including a cation−π interaction of the indazole phenyl ring with Arg365 due to insertion of the C-terminal domain of CDK8 into the ATP binding site.22 Notably, binding to the hinge via the pyridine is conserved despite the introduction of a potential alternative 3-aminoindazole hinge-binding motif; pleasingly, the only difference we observe between the binding modes of compounds 6 and 25 is the presence of a DMSO molecule in a small pocket adjacent to 25 (see Figure 1, panel A).
Figure 1.

Crystal structure of 25 in CDK8/cyclin C (panel A, PDB code 5FGK). Overlay of 6 (pink) and 25 (gray) in CDK8/cyclin C (panel B). DMSO is colored cyan.
Although the N-methyl sultam group conferred metabolic instability (Table 2, entry 8), its beneficial effect on the potency was very attractive and we were keen to explore replacement of the spirolactam in combination either with the N-methyl sultam or with our hitherto preferred 4-(1-methyl-1H-pyrazol-4-yl)phenyl substituent at C-5. Consistent with our desire to replace the spirolactam with more polar heterocycles, mass spectrometry-mediated metabolite identification for 6 indicated that the spirolactam was the main site of oxidative metabolism (data not shown). Introduction of a carbamate (Table 3, entry 3) resulted in >100-fold decrease in CDK8 affinity, whereas isomer 41 had a potency profile similar to 6 suggesting that a hydrogen-bond-mediated interaction from the carbamate NH of compound 41 with Asp173 is required for potency (Table 3, entry 4). Compound 42 (Table 3, entry 5), which combines the optimal carbamate isomer and an N-methyl sultam, gave very potent CDK8/19 affinity with improved metabolic stability compared to 6. However, in vivo clearance in mice was higher than that of compound 6. Addition of two methyl groups to block metabolically vulnerable sites in the spirocycle did not lead to improved metabolic stability, and a drop in potency was also observed (Table 3, entry 6). Crystallization of compound 42 (Table 3, entry 5) in CDK8/cyclin C confirmed the carbamate NH hydrogen bond interaction with Asp173 and showed interaction of the carbamate carbonyl group with Lys52 (Figure 2).
Figure 2.
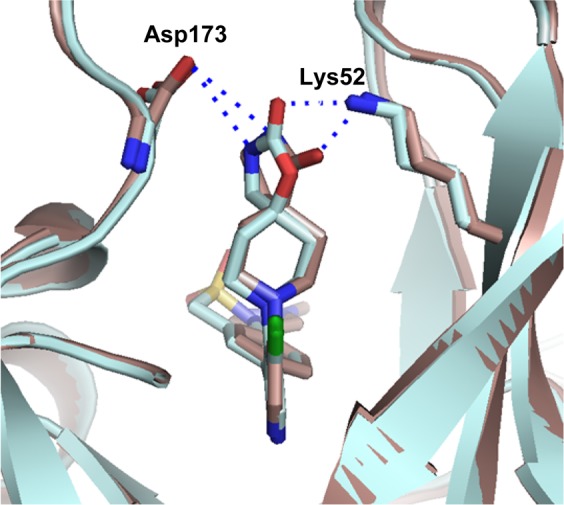
Overlay of lactam 6 (brown) and carbamate 42 (cyan) in CDK8/cyclin C, PDB code 5HBE.
Cognizant of blocking oxidative metabolism on the spirolactam, we prepared additional polar derivatives where the lactam was replaced by imidazolidin-4-one, imidazolidine-2,4-dione, 2-methylimidazol-5-one, and piperazine-2,5-dione (Table 3, entries 7–11). Pleasingly, all derivatives were significantly more stable in microsomal clearance assays; however, translation to cell-based potency was eroded for the imidazolidine-2,4-dione, 2-methylimidazole-5-one, and piperazine-2,5-dione derivatives (Table 3, entries 8–11) and no examples demonstrated significant improvement in in vivo mouse clearance (Table 3, entries 7–11). Surprisingly, the 3-methylpyrazolone derivative 49 (Table 3, entry 12) led to a 13-fold improvement in potency in the 7dF3 assay compared to 23 (Table 3, entry 2) but suffered from poor metabolic stability which was not overcome by exchanging the methyl for a trifluoromethyl group (Table 3, entry 13). Given that the lower dynamic range of our in vitro biochemical assays for CDK8 and CDK19 is approximately 3 nM due to the concentration of enzyme (5 and 6.7 nM, respectively), it is possible that the exquisite cell-based potency for compound 49 is on-target. Introduction of a basic center (compound 51, entry 14) demonstrated that polarity was tolerated at the piperidine C-4 position, but all analogs were metabolically unstable. Unsurprisingly, spiropyrrolidine 52 (Table 3, entry 15) led to a less active compound due to removal of the carbonyl group and loss of interaction with Lys52.
As noted above, a DMSO-filled pocket was observed in the X-ray crystal structure of 25 (Figure 1, panel A), and we postulated that potency could be regained by targeting this region. Addition of a linker on the pyrrolidine nitrogen (Table 3, entries 16–19) led to a 4-fold improvement in potency; however, the metabolic stability of such compounds was poor and, disappointingly, cocrystallization of 54 in CDK8/cyclin C did not demonstrate electron density consistent with binding of the pendant methoxyethyl moiety in the DMSO pocket (Figure 3). Despite its high in vivo clearance, the spirocarbamate 42 (Table 3, entry 5) was attractive from its in vitro profile and we considered spirocarbamate as a potential replacement for the spirolactam in our further optimization.
Figure 3.

Crystal structure of 54 in CDK8/cyclin C (panel A, PDB code 5HBH). Overlay of 25 (gray) with 54 (green) in CDK8/cyclin C (panel B).
Next, we investigated variation of the pyridine C-3 position (Table 4). We had previously demonstrated that small lipophilic residues were tolerated at this position consistent with the protein–ligand crystal structure of 6, 25, and 42 in CDK8/cyclin C where the pyridine C3 substituent is proximal to the lipophilic gatekeeper residue Phe97.27 3-Fluoro derivatives (Table 4, entries 1–4) led to a decrease in both biochemical and cell-based potency consistent with a weaker lipophilic interaction of fluorine compared to chlorine with Phe97.31 Pleasingly, the combination of either spirolactam or spirocarbamate with the N-methyl sultam and C-3 fluoro group resulted in acceptable potency and good solubility; however, both 61 and 63 showed high in vivo clearance in mouse pharmacokinetics (Table 4, entries 2 and 4). Introduction of a C-3 trifluoromethyl group restored potency for spirolactams 70 and 71 (Table 4, entries 5–6); however, they proved metabolically unstable. Carbamate 72, despite being surprisingly less potent than the spirolactam 70, had an acceptable Clint profile and in vivo mouse clearance similar to 6. However, due to the consistently poor aqueous solubility of these CF3 derivatives, we did not progress them further.
The protein–ligand crystal structure of 25 in CDK8/cyclin C demonstrates that the C2-H of the pyridine is proximal to the backbone carbonyl of Asp98 (distance of 2.3 Å, Figure 1), an observation consistent with our previously reported crystal structure of 6 in CDK8/cyclin C.22 We hypothesized that introduction of a hydrogen bond donor at the 2 position of the pyridine could form a favorable interaction with the backbone carbonyl of Asp98. Pleasingly, introduction of a 2-amino group (85, Table 5, entry 1) maintained potency with acceptable metabolic stability and pharmacokinetics, albeit with poor aqueous solubility. At this stage, we decided to introduce the 2-amino functionality into selected optimal compounds from Tables 2, 3, and 4. Similar to 85, all additional 2-aminopyridines (Table 5, entries 2–11) were significantly more metabolically stable than their corresponding pyridine matched pair. For example, the 2-aminopyridine 87 was at least 5-fold more stable in mouse Clint experiments than 25. The presence of the 2-amino functionality also led to lower mouse in vivo clearance; for example, 92 (Table 5, entry 8, Cl = 1.28 L/h/kg) compared to its pyridine matched pair 61 (Table 4, entry 2, Cl = 7.9 L/h/kg). However, we became concerned that reducing in vitro mouse metabolic Clint to <10 μL/min/mg (Table 5, entry 6) did not translate to improved mouse in vivo clearance. Thus, in vitro Clint did not appear to be a sufficient predictor of in vivo clearance for this series. Predicted mouse blood clearance (parallel tube model) was estimated from the Clint data to take into account the polarity of the compounds and their nonspecific protein binding in in vitro clearance assays; however, this method also failed to explain the discrepancy between in vitro and in vivo data (Figure 4).
Figure 4.
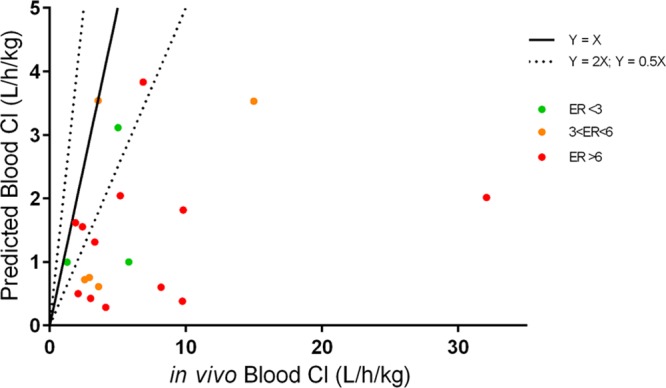
In vitro–in vivo correlation of clearance for compounds from the pyridine and 2-aminopyridine series. X axis is calculated blood clearance from measured plasma clearance in mouse PK. Y axis is predicted blood clearance using the parallel tube model.
We hypothesized that the increased in vivo clearance in comparison with the in vitro predicted clearance may be driven by active transport. Caco-2 assay data confirmed that most compounds had a high efflux ratio (ER) (Table S1). However, it was unclear if the lack of correlation between predicted and observed blood clearance (calculated from measured plasma clearance data) was due to a P-glycoprotein (P-gp) transporter liability (Figure 4). To clarify this matter, we conducted pharmacokinetic evaluation in P-gp knockout (KO) mice for a set of compounds (Table 6). We observed lower in vivo clearance in P-gp KO mice for compounds 42, 48, and 90, while clearance remained unchanged for both 6 and 89 that did not exhibit efflux in the Caco-2 assay. Furthermore, in P-gp KO mice we noted a lower percentage of parent compound cleared unchanged in the feces compared to wild-type mice for all compounds (Table 6). Taken together, these data are consistent with the notion that transporter-mediated hepatic uptake and elimination contribute to the overall clearance of some compounds in this series. However, we noted that even in the P-gp KO mice, compounds 48 and 90, both of which have low Clint, demonstrate moderate in vivo clearance and that compound 48 is still observed in the feces of P-gp KO mice, suggesting that additional nonhepatic elimination mechanisms are also involved.
Table 6. Pharmacokinetic Profile of a Set of Compounds in Wild-Type and P-gp KO Mice.
| wild type
mice |
P-gp KO mice |
|||||
|---|---|---|---|---|---|---|
| ER Caco-2 | mouse Clint (μL/min/mg) | plasma Cl (L/h/kg) | parent compd in feces (%) | plasma Cl (L/h/kg) | parent compd in feces (%) | |
| 6 | 2.5 | 141 | 2.34 | 9.1 | 2.30 | <1 |
| 89 | 2.0 | 18.5 | 2.59 | 17 | 2.84 | <1 |
| 42 | 4.7 | 57 | 4.89 | 18 | 4.00 | <1 |
| 90 | 14.8 | <10 | 4.08 | 20 | 2.85 | <1 |
| 48 | 17.6 | 30 | 4.43 | 67 | 2.22 | 24 |
To understand the contribution of physicochemical parameters to efflux, we conducted a more detailed analysis of the Caco-2 data. Cognizant of the potential for variability in Caco-2 assay data,32 we classified compounds into two large data sets, pyridines (n = 165) and aminopyridines (n = 39), seeking trends to direct our medicinal chemistry design (Figure 5). Analysis of physicochemical property trends in the pyridine series showed that HBD ≤ 2 is necessary but not sufficient to give ER < 3. The probability of compounds in this series exhibiting a low efflux ratio could be further improved by limiting physicochemical properties within the range log P > 2.5, TPSA < 80–100, HBA ≤ 7 with no basic center. Analysis of physicochemical properties versus efflux ratio in a Madin–Darby canine kidney epithelial cell line (MDCK), transfected to express human P-gp (MDR-MDCK), led to broadly similar trends. Analysis of Caco-2 data in the 2-aminopyridine series indicated that physicochemical properties in the range HBD ≤ 3 and TPSA < 100 or HBD ≤ 3 and log P > 3 are necessary for low efflux in this chemical series, and a combination of HBD ≤ 3, TPSA < 100, and log P > 3 is more likely to lead to a low efflux ratio. These observations are consistent with previously published studies demonstrating that a fine balance of physicochemical properties is often required to achieve potency, metabolic stability, and low efflux in a single molecule.33,34
Figure 5.

Physicochemical property analysis results for pyridine and aminopyridine series.
Cognizant of these analyses, we set out to reduce efflux in both the pyridine and 2-aminopyridine series. In the pyridine series, compound 25 (Table 7, entry 1) has an attractive solubility coupled with low Clint and acceptable potency; however, this compound harbors four hydrogen bond donors with an efflux ratio of >1200. Alkylation of the 3-aminoindazole nitrogen in 25 significantly reduced Caco-2 efflux but to the detriment of metabolic stability and potency (Table 7, entries 2–4). Methylation of the hydantoin (Table 7, entry 6) also led to a lower efflux ratio compared to the parent compound 45, again to the detriment of microsomal stability. The potent, metabolically stable, and soluble compound 44 (Table 7, entry 7) bears two hydrogen bond donors and exhibits an efflux ratio of 3.7. Methylation of the N-1 nitrogen of the imidazolidinone ring reduced the efflux ratio (Table 7, entry 8); however, poor microsomal stability was again observed. Replacement of the methyl group by a variety of substituents including trifluoroethyl did not improve the microsomal stability (Table 7, entry 9). As these attempts proved unsuccessful, we turned our attention to the aminopyridine series.
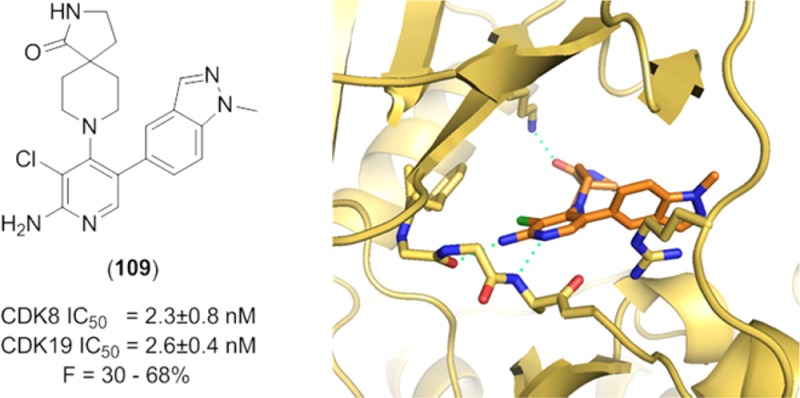
In the 2-aminopyridine series, the presence of additional hydrogen bond donor functionality by virtue of the 2-amino substituent restricted our freedom to modulate physicochemical properties in the remainder of the molecule. Thus, the choice of spirolactam isostere was diminished and we hypothesized that introduction of more lipophilic C5-pyridine substituents may be necessary to stay within our desired physicochemical property range and maximize the chances of low efflux ratios. Thus, we introduced an isopropylpyrazole at C-5 in combination with the spirolactam at C-4 of the 2-aminopyridine scaffold. This tactic led to suboptimal human Clint and an efflux ratio of 7 (Table 8, entry 1). We then combined the spirolactam and spirocarbamate with a methylindazole (Table 8, entries 2 and 3). The spirolactam derivative 109 (measured log D = 2.5) was potent (CDK8 IC50 = 4.9 ± 0.6 nM, CDK19 IC50 = 2.6 ± 0.4 nM with residence times of 53 and 86 min, respectively, in the reporter displacement assay), soluble, and stable with a low efflux ratio and acceptable in vivo mouse pharmacokinetics (Cl = 0.61 L/h/kg, F = 30%, Table 9); however, the carbamate matched pair 110 was less potent and subject to increased efflux. To further improve the metabolic stability, we prepared compound 111 bearing an N-isopropylindazole; however, this compound was less stable in all species. Crystallization of 109 in CDK8/cyclin C (Figure 6) demonstrated a binding mode consistent with those previously observed, and pleasingly, a new hydrogen bond interaction between the exocyclic nitrogen of the 2-aminopyridine scaffold and the backbone carbonyl of Asp98 was observed (3.0 Å).
Table 9. Pharmacokinetic Profile of 109a.
| species | Cl (L/h/kg) | LBF (%) | Vd (L/kg) | F (%) | t1/2 (h) |
|---|---|---|---|---|---|
| mouse | 0.61 | 10 | 0.63 | 30 | 0.78 |
| rat | 1.49 | 34 | 2.0 | 57 | 1.10 |
| dog | 1.07 | 43 | 1.4 | 68 | 0.99 |
| human prediction | ∼0.36 | ∼31 | ∼1.4 | ∼70 | ∼2.7 |
Dose: 0.2 mg/kg (iv), 0.5 mg/kg (po).
Figure 6.
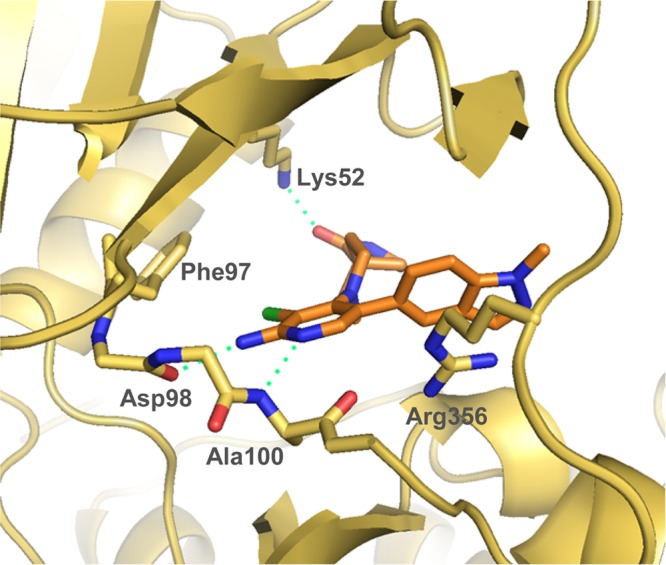
X-ray crystal structure of 109 in CDK8/cyclin C, PDB code 5HBJ.
In light of its promising profile, compound 109 was further profiled in rat and dog pharmacokinetics (Table 9). Moderate clearance was observed in both species; furthermore, the human pharmacokinetic prediction for 109 was significantly better than for compound 6 (Tables 9 and 1, respectively, ∼31% of liver blood flow for 109 compared to ∼76% for 6). Pleasingly, compound 109 had acceptable aqueous solubility (Table 8, entry 2) and demonstrated minimal activity when tested in a panel of 55 receptors, ion channels, and enzymes at 1 μM (Tables S2 and S3) and in a panel of 279 kinases (Table S4); weak inhibition of CYPs was observed (Table S5). Consistent with the profile of chemical probe 6, compound 109 demonstrated potent inhibition of reporter-based readouts measuring basal WNT pathway activity in human cancer cell lines that have constitutively activated WNT pathway signaling: LS174T (β-catenin mutant), SW480 and Colo205 (APC mutant) or PA-1 human teratocarcinoma cells that are WNT ligand dependent (Table 10).
Table 10. Potency of 6 and 109 versus Reporter-Based Readouts of WNT Pathway Activity in Human Cancer Cell Lines.
| cell line | WNT pathway activation | 6 IC50 (nM) | 109 IC50 (nM) |
|---|---|---|---|
| LS174T | β-catenin mutant | 23 ± 11 | 33 ± 13 |
| SW480 | APC-mutant | 190 ± 30 | 22 ± 2 |
| Colo205 | APC-mutant | 35 ± 3 | 15 ± 1 |
| PA-1 | WNT ligand-dependent | 20 ± 10 | 64 ± 34 |
Compound 109 was then assessed in the in vivo APC-mutant SW620 human colorectal carcinoma xenograft model, treating established tumors in female NCr athymic mice. Mice were treated orally (30 mg/kg q.d.) for 15 days; a 54.2% reduction in tumor weight was observed at day 15 (Figure 7, panels A and B). We monitored inhibition of STAT1SER727 phosphorylation, which we have previously demonstrated to be an in vitro cell-based and in vivo pharmacodynamic biomarker of CDK8 inhibition.22 Reduced STAT1SER727 phosphorylation was maintained for more than 6 h after the last dose (Figure S2) consistent with measured free plasma and tumor exposures that remained above CDK8 IC50 (Figure 7, panel C, Figure S3, and Table S6). In light of its potent and selective profile coupled with good oral pharmacokinetics and duration of in vivo target engagement on oral dosing, compound 109 (CCT251921) has been selected for progression into further preclinical in vivo efficacy and safety studies.
Figure 7.
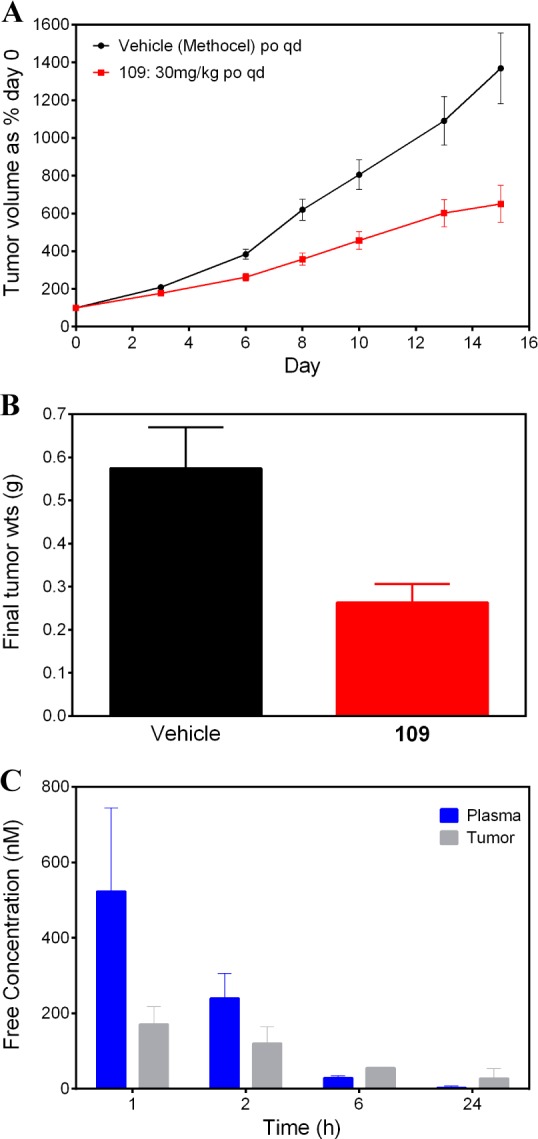
Reduction of (A) tumor volume and (B) tumor weight versus vehicle-treated controls after chronic oral dosing (30 mg/kg q.d.) of 109 to an APC-mutant SW620 human colorectal carcinoma xenograft animal model. (C) Free plasma and free tumor exposure (nM) at 1, 2, 6, and 24 h after the last dose in the same experiment.
Conclusions
Literature evidence9−16 and our own studies22 point to a role for the Mediator complex-associated kinases CDK8 and CDK19 in human disease, particularly in colorectal cancer where CDK8 has been reported as a putative oncogene. CDK19, a paralogue of CDK8, is relatively unexplored, and our previous studies demonstrate that selectivity for CDK8 over CDK19 with a small molecule ligand is likely to be challenging.22 Indeed, in our recently reported discovery of CCT251545 (6), a potent and selective chemical probe for the further exploration of CDK8 and CDK19 pharmacology, we show strong correlation of CDK8 and CDK19 binding affinity.22
Here, we have further optimized the small molecule CDK8/19-selective chemical probe 6 to give compound 109 by improving oral pharmacokinetics and pharmaceutical properties in order to facilitate further in vivo evaluation of CDK8/19 pharmacology and progression into preclinical in vivo efficacy and safety studies. Chemical probe 6, although a high affinity ligand for CDK8 and CDK19, displays moderate in vivo clearance in preclinical species resulting in a clearance prediction to man that may preclude consistent target engagement for extended periods of time; in addition, aqueous solubility was suboptimal and we anticipated that this may limit the maximum absorbable dose. In attempting to reduce oxidative metabolism by reducing lipophilicity while maintaining key interactions within the CDK8 kinase domain, we benefited from detailed knowledge of the binding mode of 6 and analogs through protein–ligand crystal structures in the CDK8 kinase domain. We were able to improve in vitro metabolic stability by introducing a C-2 amino substituent to the pyridine scaffold which reinforced interactions with the kinase hinge region and reduced compound lipophilicity. However, we identified transporter-mediated hepatic uptake as a component of in vivo clearance. In particular, we noted that a significant number of compounds in both the pyridine and 2-aminopyridine series exhibited higher measured in vivo clearance than predicted by experimental in vitro clearance assessment. Through careful correlation of in vitro Caco-2 efflux ratios with physicochemical properties and subsequent medicinal chemistry design within desirable physicochemical property ranges, we were able to identify compounds that demonstrated reduced clearance without increased susceptibility to active hepatic uptake. Compound 109 was identified as the best compromise of in vitro biochemical and pharmacokinetic properties that demonstrated acceptable in vivo pharmacokinetics suitable for progression to in vivo animal models of cancer. Further in vivo evaluation of 109 will be reported in due course.
Experimental Section
Chemistry
Commercially available starting materials, reagents, and dry solvents were used as supplied. Column chromatography was performed on a Biotage SP1 purification system using Thomson or Biotage Flash silica cartridges or on a Companion purification system using Interchim silica cartridges. Preparative TLC was performed on Merck plates. Ion exchange chromatography was performed using acidic Isolute Flash SCX-II columns or basic Isolute Flash NH2 columns. Preparative HPLC was conducted according the following methods. For method A, injections of the sample were made onto a SunFire C18 OBD column (100 Å, 5 μm, 30 mm × 100 mm). Chromatographic separation at room temperature was carried out using Agilent Tehnologies, 1260 Infinity, acetonitrile/water gradient (both modified with 0.1% formic acid) at a flow rate of 50 mL/min. For method B, injections of the sample were made onto a Phenomenex Gemini column (10 μm, 250 mm × 21.2 mm, C18, Phenomenex, Torrance, CA, USA). Chromatographic separation at room temperature was carried out using Gilson GX-281 liquid handler system combined with a Gilson 322 HPLC pump (Gilson, Middleton, WI, USA) over a 15 min gradient elution from 40:60 to 100:0 MeOH/water (both modified with 0.1% formic acid) at a flow rate of 20 mL/min. 1H NMR spectra were recorded on a Bruker Avance 500, Bruker Avance 400, or Avance II 400. Samples were prepared as solutions in a deuterated solvent and referenced to the appropriate internal nondeuterated solvent peak. 13C NMR spectra were recorded at 126 MHz using an internal deuterium lock. The following internal references were used: CDCl3 (δC 77.2), CD3OD (δC 49.0), and DMSO-d6 (δC 39.5); unobserved resonances for quaternary carbon atoms are denoted by “Cq not observed”. LC/MS and HRMS analyses were performed on an Agilent 1200 series HPLC and diode array detector coupled to a 6210 time-of-flight mass spectrometer with dual multimode APCI/ESI source. Analytical separation was carried out according to the following methods. For method A, analytical separation was carried out on a Chromolith Speed ROD column (RP-18e, 50 mm × 4.6 mm) using a flow rate of 2.4 mL/min in a 3.9 min gradient elution with detection at 220 nm. The mobile phase was a mixture of water containing 0.05% formic acid (solvent A) and acetonitrile containing 0.04% formic acid (solvent B). Gradient elution was as follows: 95:5 (A/B) to 0:100 (A/B) over 2.8 min, 0:100 (A/B) for 0.5 min, and then reversion back to 95:5 (A/B) over 0.1 min, finally 95:5 (A/B) for 0.5 min. For method B, analytical separation was carried out on a Chromolith Performance column (RP-18e, 100 mm × 3 mm) using a flow rate of 2.0 mL/min in a 4.8 min gradient elution with detection at 220 nm. The mobile phase was a mixture of water (solvent A) and acetonitrile (solvent B) both containing 0.1% TFA. Gradient elution was as follows: 99:1 (A/B) over 0.2 min, then 99:1 to 0:100 (A/B) over 3.6 min, 0:100 (A/B) for 0.4 min, and then reversion back to 99:1 (A/B) over 0.1 min and finally 99:1 (A/B) for 0.5 min. For method C, analytical separation was carried out at 30 °C on a Merck Purospher STAR column (RP-18e, 30 mm × 4 mm) using a flow rate of 1.5 mL/min in a 4 min gradient elution with detection at 254 nm. The mobile phase was a mixture of MeOH (solvent A) and water (solvent B), both containing 0.1% formic acid. Gradient elution was as follows: 1:9 (A/B) to 9:1 (A/B) over 2.5 min, 9:1 (A/B) for 1 min, and then reversion back to 1:9 (A/B) over 0.3 min, finally 1:9 (A/B) for 0.2 min. For method D, analytical separation was carried out at 30 °C on a Merck Purospher STAR column (RP-18e, 30 mm × 4 mm) using a flow rate of 1.5 mL/min in a 4 min gradient elution with detection at 220 nm. The mobile phase was a mixture of MeOH (solvent A) and water (solvent B), both containing 0.1% formic acid. Gradient elution was as follows: 1:9 (A/B) to 9:1 (A/B) over 2.5 min, 9:1 (A/B) for 1 min, and then reversion back to 1:9 (A/B) over 0.3 min, finally 1:9 (A/B) for 0.2 min. For method E, analytical separation was carried out on XBridge C8 column (50 mm × 4.6 mm, 3.5 μm) using a flow rate of 2.0 mL/min in a 10 min gradient elution with detection at 254 nm. The mobile phase was a mixture of acetonitrile (solvent A) and water (solvent B), both containing 0.1% TFA. Gradient elution was as follows: 5:95 (A/B) to 100:0 (A/B) over 8 min, 100:0 (A/B) for 0.1 min, and then reversion back to 5:95 (A/B) over 0.4 min, finally 5:95 (A/B) for 1.5 min. The following reference masses were used for HRMS analysis: caffeine [M + H]+ 195.087 652; (hexakis(1H,1H,3H-tetrafluoropentoxy)phosphazene [M + H]+ 922.009 798) and hexakis(2,2-difluoroethoxy)phosphazene [M + H]+ 622.028 96 or reserpine [M + H]+ 609.280 657. All compounds submitted for biological testing were determined to be >95% pure by method A, B, C, D, or E unless stated otherwise.
Preparation of Compounds in Table 2, Exemplified by Compounds 23 and 25. 1-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-Dioxide (14)
2-Chlorobenzylsulfonyl chloride (1.86 g, 8.26 mmol) was dissolved in acetone (27 mL), and then ammonium hydroxide (18 mL) was added. The reaction mixture was stirred for 2.5 h at rt, and the solvent was evaporated. The reaction mixture was diluted with EtOAc, and water was added. The two layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated under vacuum. The crude product was purified by Biotage column chromatography (cyclohexane/acetone 90:10 to 60:40) to afford (2-chlorophenyl)methanesulfonamide as a white solid (1.6 g, 94% yield). 1H NMR (500 MHz, CDCl3) δ 7.56–7.53 (m, 1H), 7.47–7.44 (m, 1H), 7.36–7.30 (m, 2H), 4.66 (bs, 2H), 4.57 (s, 2H); LC–MS (method D, ESI, m/z) tR = 1.77 min, parent does not ionize.
(2-Chlorophenyl)methanesulfonamide (450 mg, 2.19 mmol), tris(dibenzylideneacetone)dipalladium (100 mg, 0.109 mmol), 2-di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl (186 mg, 0.438 mmol), and potassium carbonate (605 mg, 4.38 mmol) were loaded in a microwave vial, and THF (8.8 mL) was added. The reaction mixture was stirred at 80 °C in an oil bath for 13 h before being quenched with a sat. aq NH4Cl solution. The solvent was then evaporated and the residue was purified by Biotage column chromatography (cyclohexane/acetone 95:5 to 60:40) to afford 1,3-dihydrobenzo[c]isothiazole 2,2-dioxide as a white solid (296 mg, 80% yield). 1H NMR (500 MHz, CDCl3) δ 7.31–7.26 (m, 1H), 7.26–7.23 (m, 1H), 7.07 (td, J = 7.6, 0.9 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.48 (bs, 1H), 4.39 (s, 2H); LC–MS (method D, ESI, m/z) tR = 1.69 min, parent does not ionize.
To a suspension of 1,3-dihydrobenzo[c]isothiazole 2,2-dioxide (280 mg, 1.66 mmol) and potassium carbonate (229 mg, 1.66 mmol) in DMF (5 mL) was added iodomethane (414 μL, 6.62 mmol). The reaction mixture was stirred for 6 h at rt and was then quenched with a sat. NH4Cl solution. The reaction mixture was concentrated and purified by Biotage column chromatography (cyclohexane/acetone 90:10 to 70:30) to afford 1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide as a white solid (270 mg, 89% yield). 1H NMR (500 MHz, CDCl3) δ 7.37–7.32 (m, 1H), 7.27–7.24 (m, 1H), 7.02 (td, J = 7.6, 1.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 4.34 (s, 2H), 3.14 (s, 3H); LC–MS (method D, ESI, m/z) tR = 2.07 min, parent does not ionize.
1-Methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide (272 mg, 1.49 mmol) was dissolved in DMF (1.5 mL), and N-bromosuccinimide (264 mg, 1.49 mmol) was added. The reaction mixture was stirred at rt for 4 h. After addition of water, the reaction mixture was concentrated. The residue was purified by Biotage column chromatography (cyclohexane/acetone 90:10 to 70:30) to afford 5-bromo-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide as a white solid (330 mg, 85% yield). 1H NMR (500 MHz, CDCl3) δ 7.45–7.41 (m, 1H), 7.37–7.35 (m, 1H), 6.59 (d, J = 8.5 Hz, 1H), 4.30 (s, 2H), 3.09 (s, 3H); LC–MS (method C, ESI, m/z) tR = 2.46 min, parent does not ionize.
5-Bromo-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide (267 mg, 1.02 mmol), bis(pinacolato)diboron (388 mg, 1.53 mmol), potassium acetate (300 mg, 3.06 mmol), and Pd(dppf)Cl2·CH2Cl2 (42 mg, 0.051 mmol) were loaded in a microwave vial, and DME (7.4 mL) was added. The reaction mixture was stirred in an oil bath at 80 °C overnight. The reaction was concentrated and purified by Biotage column chromatography (cyclohexane/acetone 97:3 to 85:15) to afford 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 14 as a white solid (290 mg, 92% yield). 1H NMR (500 MHz, CDCl3) δ 7.80–7.77 (m, 1H), 7.69–7.67 (m, 1H), 6.71 (d, J = 8.0 Hz, 1H), 4.32 (s, 2H), 3.15 (s, 3H), 1.33 (s, 12H); LC–MS (method C, ESI, m/z) tR = 2.82 min, 309/310 (M + H)+.
8-(3-Chloro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (23)
1-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 14 (58 mg, 0.19 mmol), 8-(3-bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one278 (54 mg, 0.16 mmol), and Pd(dppf)Cl2·CH2Cl2 (6.4 mg, 7.9 μmol) were loaded in a microwave vial, and then 0.5 M sodium carbonate in water (440 μL, 0.220 mmol) and acetonitrile (2.8 mL) were added. The reaction was heated at 120 °C for 60 min under microwave irradiation. The solvent was evaporated and the crude material was purified by Biotage column chromatography (DCM/EtOH 99:1 to 85:15) to give 8-(3-chloro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 23 as a white solid (57 mg, 81% yield). 1H NMR (500 MHz, CDCl3) δ 8.42 (s, 1H), 8.17 (s, 1H), 7.29–7.24 (m, 2H), 6.81 (d, J = 8.1 Hz, 1H), 6.36 (s, 1H), 4.48 (s, 2H), 3.31 (t, J = 6.8 Hz, 2H), 3.18 (s, 3H), 3.16–3.10 (m, 2H), 2.81 (t, J = 11.2 Hz, 2H), 1.98 (t, J = 6.8 Hz, 2H), 1.91–1.80 (m, 2H), 1.39–1.33 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 181.6, 153.0, 150.4, 149.9, 141.5, 133.2, 131.0, 130.3, 128.3, 126.6, 118.0, 109.1, 50.8, 47.7, 41.6, 38.8, 32.4, 32.2, 26.6; LC–MS (method C, ESI, m/z) tR = 2.00 min, 447/449 (M + H)+; ESI-HRMS calcd for C21H2435ClN4O3S (M + H)+ 447.1252, found 447.1247.
(3-Amino-1H-indazol-6-yl)boronic Acid Hydrochloride (16)
In a screw-capped vessel, 6-bromo-1H-indazol-3-amine (95% purity, 500 mg, 2.36 mmol) and 4-(dimethylamino)pyridine (58 mg, 0.47 mmol) were dissolved in THF (10 mL). Di-tert-butyl dicarbonate (2.52 mL, 11.8 mmol) and triethylamine (3.27 mL, 23.6 mmol) were added, and the reaction solution was stirred for 3 days at rt. The reaction mixture was diluted with water (100 mL) and the aqueous layer extracted with EtOAc. The organic layer was washed with water, dried, filtered, and evaporated to dryness to give tert-butyl 3-[bis(tert-butoxycarbonyl)amino]-6-bromoindazole-1-carboxylate (1.42 g, 73% pure, 86% corrected yield) as a colorless oil, which was used without further purification. LC–MS (method B, ESI, m/z) tR = 3.91 min, 534/536 (M + Na)+.
In a screw-capped vessel, tert-butyl 3-[bis(tert-butoxycarbonyl)amino]-6-bromoindazole-1-carboxylate (73% pure, 1.34 g, 1.91 mmol) was dissolved in THF (16 mL). Bis(pinacolato)diboron (486 mg, 1.91 mmol), potassium acetate (375 mg, 3.83 mmol), and Pd(dppf)Cl2·CH2Cl2 (78 mg, 0.096 mmol) were added, and the red reaction mixture was stirred for 15 h at 70 °C. Further bis(pinacolato)diboron (486 mg, 1.91 mmol), potassium acetate (130 mg, 1.33 mmol), and Pd(dppf)Cl2·CH2Cl2 (78 mg, 0.096 mmol) were added, and stirring was continued at 70 °C for additional 4 h. The black reaction mixture was treated with EtOAc, filtered, and evaporated to dryness under reduced pressure. The dark brown residue was purified by flash chromatography (heptane/DCM, gradient) to give tert-butyl 3-[bis(tert-butoxycarbonyl)amino]-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole-1-carboxylate (1.00 g, 90% pure, 84% corrected yield) as a yellow solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 3.94 min, 559/560 (M + H)+.
tert-Butyl 3-[bis(tert-butoxycarbonyl)amino]-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazole-1-carboxylate (90% pure, 1.00 g, 1.61 mmol) was treated with HCl in dioxane (25 mL). The pale yellow solution was stirred at rt for 15 h. The solution was evaporated to dryness and the residue was treated with diethyl ether to obtain a beige solid. The mixture was filtered and the residue was washed with diethyl ether to afford (3-amino-1H-indazol-6-yl)boronic acid hydrochloride 16 (351 mg, 95% pure, 97% yield) as a beige solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 1.34 min, 177/178 (M + H)+.
8-(3-(3-Amino-1H-indazol-6-yl)-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (25)
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one278 (92% pure, 180 mg, 0.480 mmol) and (3-amino-1H-indazol-6-yl)boronic acid hydrochloride 16 (95% pure, 162 mg, 0.720 mmol) were dissolved in acetonitrile (8 mL), and 0.5 M sodium carbonate in water (2.8 mL, 1.4 mmol) and Pd(dppf)Cl2·CH2Cl2 (20 mg, 0.020 mmol) were added. The reaction mixture was stirred for 1 h at 120 °C under microwave irradiation, diluted with acetonitrile (10 mL), filtered and the filtrate evaporated to dryness. The residue was purified by preparative HPLC (method A, 20 min gradient elution from 2:98 to 20:80 acetonitrile/water). The pure fractions were combined and evaporated down to a volume of 20 mL. The solution was neutralized with solid NaHCO3 and extracted with EtOAc (3 × 30 mL). The organic layer was dried over Na2SO4 and evaporated to dryness. The residue was suspended in diethyl ether and filtered under vacuum to afford 8-(3-(3-amino-1H-indazol-6-yl)-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 25 (85 mg, 45% yield) as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 8.45 (s, 1H), 8.25 (s, 1H), 7.63 (dd, J = 8.2, 0.9 Hz, 1H), 7.29 (s, 1H), 6.97 (dd, J = 8.2, 1.4 Hz, 1H), 5.66 (s, 1H), 3.26 (t, J = 6.8 Hz, 2H), 3.21–3.13 (m, 2H), 2.74 (t, J = 11.7 Hz, 2H), 1.97–1.87 (m, 4H), 1.37–1.29 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 181.4, 153.2, 150.9, 149.9, 148.9, 142.5, 137.1, 134.0, 128.2, 121.6, 119.6, 114.2, 110.3, 47.7, 41.5, 38.7, 32.5, 32.2; LC–MS (method C, ESI, m/z) tR = 1.58 min, 397/399 (M + H)+; ESI-HRMS calcd for C20H2235ClN6O (M + H)+ 397.1538, found 397.1522.
Preparation of Compounds in Table 3, Exemplified by Compounds 42 and 54. 1-Oxa-3,8-diazaspiro[4.5]decan-2-one (38)
4-Aminomethyl-1-benzylpiperidin-4-ol (25.0 g, 113 mmol) was suspended in DCM (400 mL), and a solution of triphosgene (32.8 g, 110 mmol) dissolved in DCM (200 mL) was added dropwise while maintaining the temperature between 30 and 35 °C. The solution turned yellow and the mixture was stirred at rt overnight. To the reaction mixture 1 N NaOH solution (200 mL) was added, and the organic phase was separated, washed with water, dried over sodium sulfate, filtered, and evaporated to dryness. The residue was triturated with diethyl ether, the solid obtained was washed with diethyl ether and dried in vacuo to give 13.2 g (82% pure, 39% corrected yield) of 8-benzyl-1-oxa-3,8-diazaspiro[4.5]decan-2-one35 as a yellow solid which was used in the next step without further purification. LC–MS (method B, ESI, m/z) tR = 1.49 min, 247 (M + H)+.
8-Benzyl-1-oxa-3,8-diazaspiro[4.5]decan-2-one (82% pure, 13.2 g, 43.9 mmol) was dissolved in MeOH (60 mL) and THF (60 mL). Pd/C 5% (54.1% H2O, 3.00 g) was added, and the reaction mixture was stirred at rt under hydrogen for 18 h. Since the reaction was incomplete, additional Pd/C 5% (54.1% H2O, 6.00 g) was added and the reaction mixture was stirred at rt for another 18 h. Another portion of palladium was added and the reaction was stirred for 18 h. The catalyst was then filtered off and the filtrate was evaporated to dryness. The residue was triturated with diethyl ether, filtered, and dried in vacuum to yield in 5.39 g (78% yield) of 1-oxa-3,8-diazaspiro[4.5]decan-2-one 38 as a brown solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 0.39 min, 157 (M + H)+.
8-(3-Bromo-5-chloropyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (27)
In a microwave vial 3-bromo-4,5-dichloropyridine277 (2.00 g, 8.80 mmol) was dissolved in NMP (15 mL). 1-Oxa-3,8-diazaspiro[4.5]decan-2-one 38 (1.65 g, 10.6 mmol) and triethylamine (2.44 mL, 17.6 mmol) were added. The reaction was stirred for 1 h at 220 °C under microwave irradiation. The brown reaction solution was treated with 300 mL of water. The beige precipitate was filtered and washed with water and diethyl ether to give 8-(3-bromo-5-chloropyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 27 (2.34 g, 77% yield) as a beige solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 2.01 min, 346/348/350 (M + H)+.
8-(3-Chloro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (42)
8-(3-Bromo-5-chloropyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 27 (2.54 g, 7.33 mmol) was suspended in acetonitrile (60 mL). 1-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 14 (2.72 g, 8.79 mmol), 0.5 M sodium carbonate in water (29.3 mL, 14.6 mmol), and (Pd(dppf)Cl2·CH2Cl2 complex (299 mg, 0.366 mmol) were added. The reaction mixture was stirred at 80 °C for 3 h. After addition of EtOAc and water, the organic layer was separated and washed with water, dried over MgSO4, filtered, and evaporated to dryness. The crude material was purified by flash chromatography (Companion, DCM/MeOH 100:0 to 90:10) to yield in 1.61 g (49% yield) of 8-(3-chloro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 42 as a colorless solid. 1H NMR (500 MHz, DMSO-d6) δ 8.46 (s, 1H), 8.17 (s, 1H), 7.50 (s, 1H), 7.37 (s, 1H), 7.32 (dd, J = 8.2, 1.9 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 4.72 (s, 2H), 3.20 (s, 2H), 3.10 (s, 3H), 2.96–2.82 (m, 4H), 1.77–1.66 (m, 4H); 13C NMR (126 MHz, DMSO-d6) δ 157.7, 152.1, 150.6, 148.8, 141.2, 133.2, 130.2, 130.0, 127.3, 126.0, 118.7, 109.5, 78.7, 50.1, 50.0, 47.1, 35.8, 26.1; LC–MS (method C, ESI, m/z) tR = 1.87 min, 449/451 (M + H)+; ESI-HRMS calcd for C20H2235ClN4O4S (M + H)+ 449.1045, found 449.1032.
8-(3-Bromo-5-chloropyridin-4-yl)-1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decane (36)
To a suspension of 8-boc-1,8-diazaspiro[4.5]decane oxalate (202 mg, 0.839 mmol) in acetonitrile (16.8 mL) were added potassium carbonate (348 mg, 2.52 mmol), potassium iodide (139 mg, 0.839 mmol), and 2-bromoethyl methyl ether (95 μL, 1.0 mmol). The reaction was heated at 80 °C overnight and filtered. The filtrate was concentrated and the crude was purified by Biotage column chromatography (eluting with 1–8% MeOH/aq NH3 (10:1) in DCM) to give tert-butyl 1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decane-8-carboxylate (160 mg, 64% yield) as a colorless oil. 1H NMR (500 MHz, CD3OD) δ 4.11 (br d, J = 14.2 Hz, 2H), 3.53 (t, J = 5.8 Hz, 2H), 3.36 (s, 3H), 3.05–2.67 (m, 6H), 1.89 (s, 4H), 1.67–1.57 (m, 2H), 1.47 (s, 9H), 1.44–1.37 (m, 2H); LC–MS (method C, ESI, m/z) tR = 1.75 min, 299 (M + H)+.
3-Bromo-4,5-dichloropyridine277 (70 mg, 0.31 mmol) and tert-butyl 1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decane-8-carboxylate (120 mg, 0.402 mmol) were introduced in a microwave vial, and then 1-methoxy-2-propanol (773 μL) and triethylamine (130 μL, 0.928 mmol) were added. The reaction mixture was stirred for 2 h at 220 °C under microwave irradiation. The solvent was evaporated and the crude material was purified by Biotage column chromatography (eluting with 2–5% MeOH/aq NH3 (10:1) in DCM) to give 8-(3-bromo-5-chloropyridin-4-yl)-1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decane 36 (86 mg, 72% yield) as a colorless oil. 1H NMR (500 MHz, CD3OD) δ 8.49 (s, 1H), 8.36 (s, 1H), 3.53 (t, J = 6.0 Hz, 2H), 3.45 (td, J = 12.6, 2.3 Hz, 2H), 3.37 (s, 3H), 3.34–3.28 (m, 2H), 2.91 (t, J = 6.6 Hz, 2H), 2.76 (t, J = 6.0 Hz, 2H), 2.00–1.85 (m, 6H), 1.46–1.40 (m, 2H); LC–MS (method C, ESI, m/z) tR = 1.72 min, 388/390/392 (M + H)+.
5-(5-Chloro-4-(1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-Dioxide (54)
8-(3-Bromo-5-chloropyridin-4-yl)-1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decane 36 (27 mg, 0.069 mmol), 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 14 (21 mg, 0.069 mmol), and Pd(dppf)Cl2·CH2Cl2 (2.8 mg, 3.5 μmol) were loaded in a microwave vial, and then acetonitrile (1.2 mL) and 0.5 M sodium carbonate in water (194 μL, 0.0970 mmol) were added. The reaction was stirred at 120 °C for 60 min. The solvents were evaporated and the crude material was purified by Biotage column chromatography (eluting with 1–5% MeOH/aq NH3 (10:1) in DCM) and further purified by SCX-2 column chromatography (loading with DCM:MeOH, elution with 1 N NH3 in MeOH) to afford 5-(5-chloro-4-(1-(2-methoxyethyl)-1,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 54 (14 mg, 41% yield) as a colorless oil. 1H NMR (500 MHz, acetone-d6) δ 8.38 (s, 1H), 8.17 (s, 1H), 7.45 (s, 1H), 7.40–7.37 (m, 1H), 7.05 (d, J = 8.2 Hz, 1H), 4.59 (s, 2H), 3.37 (t, J = 6.5 Hz, 2H), 3.27 (s, 3H), 3.16 (s, 3H), 3.10–3.05 (m, 2H), 2.88–2.73 (m, 4H), 2.60 (t, J = 6.5 Hz, 2H), 1.73–1.59 (m, 6H), 1.16–1.09 (m, 2H); 13C NMR (126 MHz, CD3OD) δ 153.8, 149.6, 148.6, 141.8, 134.2, 130.6, 130.3, 128.0, 126.2, 118.5, 109.0, 71.5, 57.6, 50.8, 49.1, 47.2, 32.3, 31.0, 25.1, 20.0 (Cq and CH2 of the sultam not observed); LC–MS (method C, ESI, m/z) tR = 1.65 min, 491/493 (M + H)+; ESI-HRMS calcd for C24H3235ClN4O3S (M + H)+ 491.1878, found 491.1878.
Preparation of Compounds in Table 4, Exemplified by Compounds 61, 63, and 72. 8-(3-Bromo-5-fluoropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (58)
3-Bromo-4-chloro-5-fluoropyridine 57 (300 mg, 1.43 mmol) and tert-butyl 1-oxo-2,8-diazaspiro[4.5]decane-8-carboxylate (471 mg, 1.85 mmol) were introduced in a microwave vial, and 1-methoxy-2-propanol (3.5 mL) and triethylamine (601 μL, 4.28 mmol) were added. The reaction mixture was stirred for 1 h at 220 °C under microwave irradiation. The solvent was evaporated and the crude material was purified by Biotage column chromatography (DCM/EtOH 98:2 to 92:8) to afford 8-(3-bromo-5-fluoropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 58 (140 mg, 30% yield) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.39 (s, 1H), 8.22 (d, J = 3.6 Hz, 1H), 6.86 (s, 1H), 3.51–3.43 (m, 2H), 3.40–3.35 (m, 2H), 3.24–3.16 (m, 2H), 2.16–2.07 (m, 4H), 1.57–1.50 (m, 2H); 19F NMR (500 MHz, CDCl3) δ −135; LC–MS (method C, ESI, m/z) tR = 2.46 min, 328/330 (M + H)+.
8-(3-Fluoro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (61)
8-(3-Bromo-5-fluoropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 58 (40 mg, 0.12 mmol), 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 14 (49 mg, 0.16 mmol), and Pd(dppf)Cl2·CH2Cl2 (4.98 mg, 6.09 μmol) were loaded in a microwave vial, and acetonitrile (2.1 mL) and 0.5 M sodium carbonate in water (341 μL, 0.171 mmol) were added. The reaction was stirred at 120 °C for 60 min under microwave irradiation. After evaporation of the solvents, the crude material was purified by Biotage column chromatography (DCM/EtOH 98:2 to 95:5) and by SCX-2 column chromatography (loading with DCM/MeOH, elution with 1 N NH3 in MeOH) to give 8-(3-fluoro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 61 (33 mg, 63% yield) as a cream solid. 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 4.2 Hz, 1H), 8.09 (s, 1H), 7.47 (dd, J = 8.2, 1.7 Hz, 1H), 7.44 (d, J = 1.7 Hz, 1H), 6.83 (d, J = 8.2 Hz, 1H), 5.78 (s, 1H), 4.46 (s, 2H), 3.32 (t, J = 6.9 Hz, 2H), 3.26–3.19 (m, 2H), 3.18 (s, 3H), 3.03–2.94 (m, 2H), 2.05 (t, J = 6.8 Hz, 2H), 1.81 (ddd, J = 13.4, 11.3, 4.2 Hz, 2H), 1.36–1.30 (m, 2H); 19F NMR (500 MHz, CDCl3) δ −139; 13C NMR (126 MHz, CDCl3) δ 181.2, 154.6 (d, J = 253.3 Hz), 147.5 (d, J = 3.9 Hz), 143.8, 141.6, 138.1 (d, J = 25.6 Hz), 131.3, 130.3, 130.1, 126.1, 118.1, 109.4, 50.9, 47.5 (d, J = 5.0 Hz), 41.5, 38.7, 32.3, 31.8, 26.6; LC–MS (method C, ESI, m/z) tR = 1.69 min, 431 (M + H)+; ESI-HRMS calcd for C21H24FN4O3S (M + H)+ 431.1548, found 431.1590.
8-(3-Bromo-5-fluoropyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (59)
8-Benzyl-1-oxa-3,8-diaza-spiro[4.5]decan-2-one (7.40 g, 98% pure) was dissolved in MeOH (34 mL), THF (17 mL), and acetic acid (8 mL). Pd/C 5% (54.1% H2O, 4.00 g) was added, and the reaction mixture was stirred at rt under hydrogen for 16 h. The catalyst was then filtered off and the filtrate was evaporated to dryness. The residue was triturated with diethyl ether/diethyl ether, filtered and dried in vacuum to yield 6.16 g (97% yield) of 1-oxa-3,8-diazaspiro[4.5]decan-2-one acetate 38 as a white solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 0.39 min, 157 (M + H)+.
3-Bromo-4-chloro-5-fluoropyridine 57 (656 mg, 3.12 mmol) and 1-oxa-3,8-diazaspiro[4.5]decan-2-one acetate 38 (1.34 g, 6.23 mmol) were introduced in a microwave vial, and 1-methoxy-2-propanol (7.8 mL) and triethylamine (1.34 mL, 9.35 mmol) were added, and the reaction mixture was stirred at 220 °C for 1 h under microwave irradiation. The solvent was evaporated and the crude material was purified by Biotage column chromatography (DCM/EtOH 98:2 to 95:5) to afford 8-(3-bromo-5-fluoropyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 59 (380 mg, 37% yield) as a cream solid. 1H NMR (500 MHz, CDCl3) δ 8.42 (s, 1H), 8.25 (d, J = 3.4 Hz, 1H), 6.20 (s, 1H), 3.58–3.50 (m, 2H), 3.42 (s, 2H), 3.31–3.24 (m, 2H), 2.12–2.06 (m, 2H), 1.99–1.92 (m, 2H); 19F NMR (500 MHz, CDCl3) δ −135; LC–MS (method C, ESI, m/z) tR = 2.32 min, 330/332 (M + H)+.
8-(3-Fluoro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (63)
8-(3-Bromo-5-fluoropyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 59 (50 mg, 0.15 mmol), 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 14 (52 mg, 0.17 mmol), and Pd(dppf)Cl2·CH2Cl2 (6.2 mg, 7.6 μmol) were loaded in a microwave vial, and acetonitrile (2.6 mL) and 0.5 M sodium carbonate in water (424 μL, 0.212 mmol) were added. The reaction mixture was stirred at 120 °C for 60 min. After evaporation of the solvent, the crude material was purified by Biotage column chromatography (DCM/EtOH 98:2 to 95:5) and by SCX-2 column chromatography (loading with DCM/MeOH, elution with 1 N NH3 in MeOH) to afford 8-(3-fluoro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 63 (44 mg, 67% yield) as white solid. 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 4.1 Hz, 1H), 8.08 (s, 1H), 7.39 (dd, J = 8.2, 1.8 Hz, 1H), 7.30 (s, 1H), 6.81 (d, J = 8.2 Hz, 1H), 5.78 (s, 1H), 4.40 (s, 2H), 3.32 (s, 2H), 3.28–3.16 (m, 5H), 3.06–3.03 (m, 2H), 1.92–1.81 (m, 2H), 1.72–1.64 (m, 2H); 19F NMR (500 MHz, CDCl3) δ −139; 13C NMR (126 MHz, CDCl3) δ 158.5, 154.4 (d, J = 253.0 Hz), 147.9 (d, J = 4.0 Hz), 143.1 (d, J = 7.4 Hz), 141.5, 138.0 (d, J = 25.5 Hz), 131.0, 130.3, 130.2, 125.6, 117.9, 109.3, 79.9, 51.3, 50.7, 47.0 (d, J = 4.6 Hz), 36.3, 26.5; LC–MS (method C, ESI, m/z) tR = 1.70 min, 433 (M + H)+; ESI-HRMS calcd for C20H22FN4O4S (M + H)+ 431.1340, found 433.1339.
4-Chloro-3,5-diiodopyridine (64)
In a 3 L three-necked round-bottom flask, 1,4-dihydropyridin-4-one (95% pure, 50 g, 0.50 mol) and N-iodosuccinimide (97% pure, 232 g, 1.00 mmol) were suspended in acetonitrile (1 L). The reaction mixture was heated under reflux for 3 h and then cooled with an ice bath, filtered and the filtrate washed with acetonitrile (150 mL). The light yellow solid was dried at 60 °C under reduced pressure for 15 h to obtain 3,5-diiodopyridin-4-ol36 (165 g, 95% yield) as a light yellow solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 1.34 min, 347 (M + H)+.
In a 3 L three-necked round-bottom flask, 3,5-diiodopyridin-4-ol (150 g, 432 mmol) was suspended in DMF (1 L). This mixture was warmed to 70 °C, and phosphoryl chloride (39.7 mL, 432 mmol) was slowly added in a dropwise manner (caution: slightly exothermic reaction). After completion of the addition of phosphoryl chloride, the reaction mixture was then heated to 95 °C for 30 min. The dark brown mixture was cooled to rt and slowly poured into 6 L of ice–water. A beige precipitate formed. NaHCO3 was added slowly until no more gas formation was observed. The solid was filtered and washed with water (2 L). The residue was suspended in acetonitrile (800 mL) and filtered again. The residue was washed with acetonitrile (100 mL) and dried at 60 °C under reduced pressure for 15 h to give 4-chloro-3,5-diiodopyridine3764 as a yellow solid, which was used without further purification (142 g, 95% pure, 85% corrected yield). LC–MS (method B, ESI, m/z) tR = 3.06 min, 366/368 (M + H)+.
8-(3,5-Diiodopyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (66)
In a 50 mL round-bottom flask, 4-chloro-3,5-diiodopyridine 64 (500 mg, 1.37 mmol) and 1-oxa-3,8-diazaspiro[4.5]decan-2-one acetate 38 (448 mg, 2.05 mmol) were suspended in NMP (10 mL) and triethylamine (570 μL, 4.11 mmol). The reaction mixture was stirred at 240 °C for 3 h and then cooled and poured into water (150 mL). The resulting precipitate was filtered and washed with water (20 mL). The solid was dissolved in DCM (150 mL), the solution was dried over Na2SO4, filtered, and concentrated to dryness. The crude residue was purified by flash column chromatography (Companion, DCM/MeOH 100:0 to 90:10) to give 8-(3,5-diiodopyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (196 mg, 30% yield) 66 as a light brown solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 2.26 min, 486 (M + H)+.
8-(3-Iodo-5-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (68)
Into a 10 mL Schlenck, silver(I) fluoride (57 mg, 0.45 mmol) was loaded and DMF (3 mL) and (trifluoromethyl)trimethylsilane (81 μL, 0.54 mmol) were added at rt, and the resulting brown suspension was stirred for 15 min at rt. Then finely powdered copper (43 mg, 0.67 mmol) was added and the resulting dark red suspension was stirred for 1.5h at rt. The reaction mixture turned green, and a silver precipitate formed on the vessel wall. 8-(3,5-Diiodopyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 66 (196 mg, 0.403 mmol) was added, and the suspension was stirred at 90 °C for 3 h. The green suspension was diluted with DMF (10 mL) and filtered over Celite. The filtrate was evaporated to dryness. The crude residue was dissolved in DCM (10 mL) and treated with diethyl ether (35 mL). The resulting light brown precipitate was filtered and washed with diethyl ether (10 mL), The filtrate was evaporated to dryness to yield in 8-(3-iodo-5-trifluoromethylpyridin-4-yl)-1-oxa-3,8-diaza-spiro[4.5]decan-2-one 68 as a yellow oil (78 mg, 71% pure, 32% corrected yield) which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 2.45 min, 428 (M + H)+.
8-(3-(4-(1-Methyl-1H-pyrazol-4-yl)phenyl)-5-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 2,2,2-Trifluoroacetate (72)
To 1-methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-pyrazole2737 (126 mg, 0.390 mmol) and Pd(dppf)Cl2·CH2Cl2 (5 mg, 0.01 mmol) was added a solution of 8-(3-iodo-5-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 68 (71% pure, 78 mg, 0.13 mmol) in acetonitrile (4 mL) and 0.5 M sodium carbonate solution (780 μL, 0.390 mmol). The reaction mixture was stirred at 120 °C for 1 h under microwave irradiation and diluted with acetonitrile (5 mL), filtered and the filtrate concentrated. The crude product was purified by preparative HPLC (method A, 20 min gradient elution from 2:98 to 25:75 acetonitrile/water to give 8-(3-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)-5-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 2,2,2-trifluoroacetate 72 (18 mg, 24% yield) as a colorless solid. 1H NMR (500 MHz, DMSO-d6) δ 8.82 (s, 1H), 8.53 (s, 1H), 8.24 (d, J = 0.8 Hz, 1H), 7.96 (d, J = 0.8 Hz, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.45 (s, 1H), 7.40 (d, J = 8.2 Hz, 2H), 3.88 (s, 3H), 3.13 (s, 2H), 2.96–2.84 (m, 4H), 1.69–1.56 (m, 4H); 19F NMR (500 MHz, DMSO-d6, TFA salt) δ −74.5; 13C NMR (126 MHz, DMSO-d6, free base) δ 157.7, 155.9, 155.3, 147.7 (d, J = 7.0 Hz), 136.5, 136.2, 134.1, 132.5, 130.0, 128.2, 124.9, 124.0 (q, J = 273 Hz), 121.3, 78.5, 50.0, 48.2, 38.8, 35.5; LC–MS (method C, ESI, m/z) tR = 2.64 min, 458 (M + H)+; ESI-HRMS calcd for C23H23F3N5O2 (M + H)+ 458.1798, found 458.1785.
Preparation of Compounds in Table 5, exemplified by Compounds 86, 92, and 95. 5-Bromo-3,4-dichloropyridin-2-amine (73)
N-Bromosuccinimide (10.9 g, 61.3 mmol) was added to a solution of 4-chloro-2-aminopyridine (7.50 g, 58.3 mmol) in dry acetonitrile (130 mL) at rt under a nitrogen atmosphere. The yellow solution was stirred for 3 h before the mixture was concentrated in vacuo. The resulting crude product was purified by Biotage column chromatography (cyclohexane/EtOAc 5:1 to 1:1) to give 5-bromo-4-chloropyridin-2-amine38 as a yellow solid (10.0 g, 83% yield). 1H NMR (500 MHz, CDCl3) δ 8.16 (s, 1H), 6.62 (s, 1H), 4.57 (s, 2H); LC–MS (method C, ESI, m/z) tR = 2.04 min, 207/209/211 (M + H)+.
N-Chlorosuccinimide (6.11 g, 45.8 mmol) was added portionwise to a solution of 5-bromo-4-chloropyridin-2-amine (10.0 g, 48.2 mmol) in dry acetonitrile (180 mL) at rt under a nitrogen atmosphere. The reaction mixture was heated to 95 °C for 3 h and then cooled to rt. The resultant solid was filtered off and washed with acetonitrile to give the desired compound as a light brown solid. The filtrate was concentrated to 50 mL, and the resulting precipitate was filtered and the resultant solid collected. The two batches of solid material were combined and recrystallized from hot acetonitrile to give two batches of 5-bromo-3,4-dichloropyridin-2-amine 73 (P1, 6.8 g; P2, 2.0 g; 75% yield) which were used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 5.06 (s, 2H); LC–MS (method C, ESI, m/z) tR = 2.93 min, 241/243/245 (M + H)+.
5-Bromo-3,4-dichloro-N,N-bis(4-methoxybenzyl)pyridin-2-amine (81)
A solution of 5-bromo-3,4-dichloropyridin-2-amine 73 (2.37 g, 4.91 mmol) in dry DMF (50 mL) at 0 °C was treated portionwise with sodium hydride (60% in mineral oil) (0.775 g, 18.6 mmol) and the mixture stirred at 0 °C for 15 min before a solution of p-methoxybenzyl chloride (2.10 mL, 15.5 mmol) in dry DMF (1 mL) was added. The mixture was stirred at rt for 2 h before sat. NH4Cl solution and EtOAc were added. The aqueous layer was extracted three times with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The resulting brown residue was purified by Biotage column chromatography (cyclohexane/DCM 100:0 to 75:25) to give 5-bromo-3,4-dichloro-N,N-bis(4-methoxybenzyl)pyridin-2-amine 81 (2.4 g, 80% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.26 (s, 1H), 7.20 (d, J = 8.6 Hz, 4H), 6.85 (d, J = 8.6 Hz, 4H), 4.42 (s, 4H), 3.81 (s, 6H); LC–MS (method C, ESI, m/z) tR = 3.51 min, 361/363/365 (M + H-PMB)+.
8-(2-(Bis(4-Methoxybenzyl)amino)-5-bromo-3-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (82)
5-Bromo-3,4-dichloro-N,N-bis(4-methoxybenzyl)pyridin-2-amine 81 (5.00 g, 10.4 mmol), 8-boc-2,8-diaza-spiro-[4.5]decan-1-one (2.64 g, 10.4 mmol), and potassium fluoride (1.21 g, 20.7 mmol) were loaded in a microwave vial. Triethylamine (4.00 mL, 31.1 mmol) and NMP (25 mL) were added, and the light brown solution was degassed with argon. The reaction mixture was stirred at 220 °C for 1 h under microwave irradiation. After the addition of water, the aqueous layer was extracted twice with EtOAc and the organic layer was washed with water, dried over MgSO4, and concentrated under vacuum. The resulting brown oil was purified by Biotage column chromatography (DCM/EtOH, 97:3 to 90:10) to give 8-(2-(bis(4-methoxybenzyl)amino)-5-bromo-3-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 82 (3.0 g, 54% yield) as a light brown solid. 1H NMR (500 MHz, CDCl3) δ 8.15 (br s, 1H), 7.19 (d, J = 8.7 Hz, 4H), 6.84 (d, J = 8.7 Hz, 4H), 5.62 (s, 1H), 4.38 (s, 4H), 3.80 (s, 6H), 3.42–3.32 (m, 6H), 2.25–2.15 (m, 4H), 1.68–1.46 (m, 2H); LC–MS (method C, ESI, m/z) tR = 3.68 min, 599/601 (M + H)+.
8-(2-Amino-3-chloro-5-(4-(1-(2-hydroxy-2-methylpropyl)-1H-pyrazol-4-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (86)
8-(2-(Bis(4-Methoxybenzyl)amino)-5-bromo-3-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 82 (203 mg, 0.339 mmol) and 2-methyl-1-(4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-pyrazol-1-yl)propan-2-ol 9 (73% pure, 190 mg, 0.407 mmol) were dissolved in acetonitrile (5 mL). 0.5 M aqueous sodium carbonate (1.40 mL, 0.678 mmol) and Pd(dppf)Cl2·CH2Cl2 (25 mg, 0.034 mmol) were added. The reaction mixture was stirred at 120 °C for 1 h under microwave irradiation, and the solvent was evaporated. The crude residue was purified by flash chromatography (n-heptane/EtOAc, gradient and then with DCM/MeOH, gradient) to give 8-(2-(bis(4-methoxybenzyl)amino)-3-chloro-5-(4-(1-(2-hydroxy-2-methylpropyl)-1H-pyrazol-4-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (81 mg, 60% pure, 19% corrected yield) as a light-brown solid which was used directly in the next step. LC–MS (method A, ESI, m/z) tR = 2.59 min, 735/737 (M + H)+.
8-(2-(Bis(4-methoxybenzyl)amino)-3-chloro-5-(4-(1-(2-hydroxy-2-methylpropyl)-1H-pyrazol-4-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (60% pure, 81 mg, 0.066 mmol) was dissolved in TFA (1 mL) and stirred at rt overnight. The crude mixture was concentrated, and the residue was treated with 1 N NaOH. The mixture was extracted with EtOAc, and the organic layer was dried over Na2SO4, filtered, and concentrated. The crude product was dissolved in acetonitrile, and the resultant solid precipitate was filtered and dried in vacuo to afford 8-(2-amino-3-chloro-5-(4-(1-(2-hydroxy-2-methylpropyl)-1H-pyrazol-4-yl)phenyl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 86 (31 mg, 94% yield) as a beige solid. 1H NMR (500 MHz, DMSO-d6) δ 8.12 (s, 1H), 7.91 (s, 1H), 7.64 (s, 1H), 7.62 (d, J = 8.2 Hz, 2H), 7.51 (s, 1H), 7.23 (d, J = 8.2 Hz, 2H), 6.13 (s, 2H), 4.75 (s, 1H), 4.04 (s, 2H), 3.10 (t, J = 6.8 Hz, 2H), 3.01–2.93 (m, 2H), 2.74–2.61 (m, 2H), 1.82 (t, J = 6.8 Hz, 2H), 1.76–1.65 (m, 2H), 1.25–1.18 (m, 2H), 1.10 (s, 6H); 13C NMR (126 MHz, DMSO-d6) δ 180.0, 156.3, 153.3, 147.7, 135.8, 135.7, 131.1, 129.6, 128.1, 124.7, 123.6, 121.1, 108.6, 69.3, 62.2, 47.4, 41.4, 37.8, 31.8, 30.4, 27.3; LC–MS (method C, ESI, m/z) tR = 1.99 min, 495/497 (M + H)+; ESI-HRMS calcd for C26H3235ClN6O2 (M + H)+ 495.2270, found 495.2256.
4,5-Dichloro-3-fluoropyridin-2-amine (74)
To a solution of LDA (2 M, 7.97 mL, 15.9 mmol) in THF (31 mL) at −78 °C was added 5-chloro-3-fluoropyridin-2-amine (934 mg, 6.37 mmol) in solution in THF (9 mL). After 50 min at −78 °C, hexachloroethane (1.44 mL, 12.8 mmol) in THF (9 mL) was added. The reaction mixture was stirred for 40 min and then quenched with NH4Cl and extracted with DCM. The crude material was purified by Biotage column chromatography (DCM 100%) to afford 4,5-dichloro-3-fluoropyridin-2-amine 74 (950 mg, 82% yield) as a cream solid. 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 1.0 Hz, 1H), 4.74 (s, 2H); 19F NMR (500 MHz, CDCl3) δ −137; LC–MS (method C, ESI, m/z) tR = 2.66 min, 181/183/185 (M + H)+.
8-(2-Amino-5-chloro-3-fluoropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (77)
4,5-Dichloro-3-fluoropyridin-2-amine 74 (100 mg, 0.553 mmol) and tert-butyl 1-oxo-2,8-diazaspiro[4.5]decane-8-carboxylate (211 mg, 0.829 mmol) were introduced in a microwave vial, and NMP (1.4 mL) and triethylamine (233 μL, 1.66 mmol) were added. The reaction mixture was stirred for 2 × 1 h at 220 °C under microwave irradiation. The reaction mixture was concentrated and the crude material was purified by Biotage column chromatography (DCM/EtOH 98:2 to 94:6) to afford 8-(2-amino-5-chloro-3-fluoropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 77 (105 mg, 64% yield) as a cream solid. 1H NMR (500 MHz, DMSO-d6) δ 7.67 (s, 1H), 7.59 (s, 1H), 6.17 (s, 2H), 3.35–3.27 (m, 2H), 3.19 (t, J = 6.8 Hz, 2H), 3.12–3.05 (m, 2H), 2.01 (t, J = 6.8 Hz, 2H), 1.79 (td, J = 12.4, 4.2 Hz, 2H), 1.44–1.38 (m, 2H); 19F NMR (500 MHz, DMSO-d6) δ −149; LC–MS (method C, ESI, m/z) tR = 1.73 min, 299/301 (M + H)+.
8-(2-Amino-3-fluoro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (92)
8-(2-Amino-5-chloro-3-fluoropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 7 (20 mg, 0.067 mmol), 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydrobenzo[c]isothiazole 2,2-dioxide 14 (27 mg, 0.087 mmol), and PdCl2(Pcy3)2 (2.5 mg, 3.4 μmol) were loaded in a microwave vial. Acetonitrile (1.1 mL) and 0.5 M sodium carbonate in water (187 μL, 0.0940 mmol) were added, and the reaction mixture was stirred at 150 °C for 30 min under microwave irradiation. After evaporation of the solvent, the crude material was purified by Biotage column chromatography (DCM/EtOH 98:2 to 90:10) to afford 8-(2-amino-3-fluoro-5-(1-methyl-2,2-dioxido-1,3-dihydrobenzo[c]isothiazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 92 (17 mg, 57% yield) as a cream solid. 1H NMR (500 MHz, DMSO-d6) δ 7.54 (s, 1H), 7.48 (s, 1H), 7.45 (d, J = 1.8 Hz, 1H), 7.41 (dd, J = 8.3, 1.8 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 6.02 (s, 2H), 4.67 (s, 2H), 3.12 (t, J = 6.8 Hz, 2H), 3.08–3.01 (m, 2H), 3.06 (s, 3H), 2.89 (t, J = 12.2 Hz, 2H), 1.91 (t, J = 6.8 Hz, 2H), 1.61–1.52 (m, 2H), 1.21–1.15 (m, 2H); 19F NMR (500 MHz, DMSO-d6) δ −153; 13C NMR (126 MHz, DMSO-d6) δ 180.5, 150.4 (d, J = 12.8 Hz), 143.9 (d, J = 5.4 Hz), 142.7 (d, J = 4.3 Hz), 141.1 (d, J = 247 Hz), 140.6, 131.3, 129.5, 126.4, 121.8, 118.7, 109.8, 50.6, 47.5 (d, J = 4.6 Hz), 41.6, 38.3, 32.1, 31.1, 26.6; LC–MS (method C, ESI, m/z) tR = 1.73 min, 446 (M + H)+; ESI-HRMS calcd for C21H25FN5O3S (M + H)+ 446.1657, found 446.1656.
5-Bromo-4-chloro-3-(trifluoromethyl)pyridin-2-amine (75)
3-(Trifluoromethyl)pyridin-2-amine (35.0 g, 0.210 mol) in DMF (350 mL) was cooled to 0–5 °C under a nitrogen atmosphere. N-Bromosuccinimide (38.8 g, 0.210 mol) was added portionwise, and the mixture was stirred at rt for 1 h. The reaction mixture was concentrated, and the light brown viscous liquid residue was triturated with ice-cold water (200 mL). The precipitate was filtered and dried to afford 5-bromo-3-(trifluoromethyl)pyridin-2-amine39 (45.0 g, 86% yield) as an off-white solid that was used directly in the next step.
To a stirred solution of 5-bromo-3-(trifluoromethyl)pyridin-2-amine (25.0 g, 0.100 mol) in THF (250 mL) was added sodium hydride (60% in mineral oil, 7.35 g, 0.180 mol) portionwise at 0–5 °C under a nitrogen atmosphere; the reaction mixture was stirred for 1 h at 0–5 °C. A solution of di-tert-butyl dicarbonate (25.8 mL, 0.110 mol) in THF (50 mL) was added dropwise at 0–5 °C, and stirring was continued for 3 h. Since the reaction was incomplete, sodium hydride (60% in mineral oil, 4.08 g, 0.100 mol) was added portionwise at 0–5 °C and stirring continued for 1 h. A solution of di-tert-butyl dicarbonate (12 mL, 0.050 mol) in THF (20 mL) was added dropwise, and the reaction mixture was stirred for an additional 2 h at 0–5 °C. The mixture was poured onto ice cold water (250 mL) and extracted with ethyl acetate (3 × 150 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, and concentrated under reduced pressure to afford a brown viscous solid. The residue was purified by column chromatography (petroleum ether/EtOAc 100:0 to 94:6) to give tert-butyl (5-bromo-3-(trifluoromethyl)pyridin-2-yl)carbamate (30 g, 74% yield) as an off-white solid that was taken to the next step without further purification.
To a stirred solution of diisoproylamine (13 mL, 0.090 mol) in THF (220 mL) cooled to −70 °C under nitrogen atmosphere was added n-butyllithium (1.6 M solution in hexane, 54 mL, 0.090 mol) dropwise over a period 0.5 h, and the mixture was stirred for additional 30 min at the same temperature. The mixture was slowly warmed to −10 °C and stirred for 20 min. After cooling to −70 °C a solution of (tert-butyl (5-bromo-3-(trifluoromethyl)pyridin-2-yl)carbamate (22 g, 0.040 mol) in THF (100 mL) was added dropwise over a period 20 min at −10 °C. The mixture was stirred for 30 min at −70 °C, then a solution of 1,1,1,2,2,2-hexachloroethane (21 g, 0.090 mol) in THF (100 mL) was added dropwise over a period of 30 min and the reaction mixture stirred for a 1 h at the same temperature. The mixture was slowly warmed to rt and stirred for additional 20 min, poured onto ice-cold water (200 mL), and extracted with EtOAc (3 × 200 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, and concentrated to give the crude product as a brown viscous solid. The crude product was purified by column chromatography (petroleum ether/EtOAc 95:5 to 90:10) to afford tert-butyl (5-bromo-4-chloro-3-(trifluoromethyl)pyridin-2-yl)carbamate (9.5 g, 62% yield) as an off-white solid which was used directly in the next step.
tert-Butyl (5-bromo-4-chloro-3-(trifluoromethyl)pyridin-2-yl)carbamate (9.5 g, 0.020 mol) in 1,4-dioxane (48 mL) was cooled to 0–5 °C. HCl in 1,4-dioxane (4 mol/L, 95 mL) was added dropwise over 20 min. The reaction mixture was stirred at rt for 4 h and then evaporated to give an off-white solid. The residue was taken up in aqueous 10% NaHCO3. The resulting precipitate was filtered off and dried in vacuo. The crude product was purified by column chromatography (petroleum ether/EtOAc 100:0 to 90:10) to afford 5-bromo-4-chloro-3-(trifluoromethyl)pyridin-2-amine 75 (4.85 g, 70% yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.41 (s, 1H), 6.95 (s, 2H); LC–MS (method E, ESI, m/z) tR = 4.38 min, 275/277/279 (M + H)+.
8-(2-Amino-5-bromo-3-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one (80)
5-Bromo-4-chloro-3-(trifluoromethyl)pyridin-2-amine 75 (300 mg, 1.09 mmol) and 1-oxa-3,8-diazaspiro[4.5]decan-2-one 38 (340 mg, 2.18 mmol) were dissolved in NMP (3 mL), and triethylamine (0.453 mL, 3.27 mmol) was added. The reaction mixture was stirred for 2 h at 220 °C under microwave irradiation. The resultant black reaction mixture was poured into 60 mL of water. The brown precipitate was filtered, washed with water and diethyl ether, and then dried to give 8-(2-amino-5-bromo-3-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 80 (280 mg, 89% pure, 58% corrected yield) as a pale brown solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 1.83 min, 395/397 (M + H)+.
8-(2-Amino-5-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)-3-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 2,2,2-Trifluoroacetate (95)
8-(2-Amino-5-bromo-3-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 80 (89% pure, 140 mg, 0.315 mmol) was suspended in acetonitrile (10 mL). 1-Methyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-1H-pyrazole 37 (81% pure, 166 mg, 0.473 mmol), 0.5 M aqueous sodium carbonate (1.26 mL, 0.631 mmol), and Pd(dppf)Cl2·CH2Cl2 (13 mg, 0.016 mmol) were added. The reaction mixture was stirred at 120 °C for 1 h under microwave irradiation. After addition of EtOAc, the mixture was filtered and the filtrate was concentrated. The brown oily residue was purified by preparative HPLC (method A, gradient elution from 2:98 to 30:70 acetonitrile/water in 20 min) to afford 8-(2-amino-5-(4-(1-methyl-1H-pyrazol-4-yl)phenyl)-3-(trifluoromethyl)pyridin-4-yl)-1-oxa-3,8-diazaspiro[4.5]decan-2-one 2,2,2-trifluoroacetate 95 (46 mg, 25% yield) as a white fluffy solid. 1H NMR (500 MHz, DMSO-d6) δ 8.20 (d, J = 0.8 Hz, 1H), 7.92 (d, J = 0.8 Hz, 1H), 7.76 (s, 1H), 7.66 (d, J = 8.2 Hz, 2H), 7.48 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 3.87 (s, 3H), 3.13 (s, 2H), 3.12–3.01 (m, 4H), 1.61–1.45 (m, 4H); 13C NMR (126 MHz, DMSO-d6) δ 160.8, 157.6, 154.2, 144.0, 136.2, 133.1, 132.3, 130.1, 128.1, 124.9, 124.0, 121.3, 78.1, 49.8, 48.1, 38.7, 34.5 (1 Cq not observed); LC–MS (method C, ESI, m/z) tR = 2.05 min, 473 (M + H)+; ESI-HRMS calcd for C23H24F3N6O2 (M + H)+ 473.1907, found 473.1893.
Preparation of Compounds in Table 7, Exemplified by Compounds 100, 101, and 102. 3-(Methylamino)-1H-indazol-6-yl)boronic Acid (97)
6-Bromo-1H-indazol-3-amine (95% pure, 1.00 g, 4.48 mmol) was dissolved in pyridine (20 mL) and the mixture cooled to 0 °C; phenyl chloroformate (0.620 mL, 4.93 mmol) was added dropwise. and the reaction mixture was stirred at 0 °C for 4 h. After addition of DCM (50 mL) and water (50 mL), the layers were separated and the organic layer was washed with brine, dried over Na2SO4, and evaporated to dryness. The residue was purified by flash chromatography (n-heptane/EtOAc, gradient) and by preparative HPLC (method A, gradient elution from 0:100 to 30:70 over 3 min and to 60:40 over 17 min acetonitrile/water) to give first the desired phenyl (6-bromo-1H-indazol-3-yl)carbamate40 (54 mg, 3% yield) as a white solid, which was used directly in the next step, and second undesired phenyl 3-amino-6-bromo-1H-indazole-1-carboxylate (346 mg) as a white solid. LC–MS (method B, ESI, m/z) tR = 2.62 min, 332/334 (M + H)+.
Phenyl (6-bromo-1H-indazol-3-yl)carbamate (54 mg, 0.16 mmol) was dissolved in THF (3 mL), and lithium aluminum hydride solution 1.0 M in THF (325 μL, 0.330 mmol) was added dropwise. The solution turned from colorless to yellow. The mixture was stirred at rt for 2 h and then diluted with water (2 mL) and THF (8 mL). The suspension was filtered over Celite and the mother liquor was evaporated to dryness to give 6-bromo-N-methyl-1H-indazol-3-amine40 (41 mg, 89% pure, 99% corrected yield) as a pale brown solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 1.95 min, 226/228 (M + H)+.
6-Bromo-N-methyl-1H-indazol-3-amine (89% pure, 41 mg, 0.16 mmol), bis(pinacolato)diboron (82 mg, 0.32 mmol), potassium acetate (48 mg, 0.48 mmol), and Pd(dppf)Cl2·CH2Cl2 (13 mg, 0.020 mmol) were loaded in a microwave vial, and acetonitrile was added (4 mL). The reaction mixture was stirred for 1 h at 120 °C under microwave irradiation and then diluted with acetonitrile (10 mL), filtered and the filtrate evaporated to dryness. The crude residue 97 (230 mg, 13% pure, 100% corrected yield) was used in the next step without purification. LC–MS (method B, ESI, m/z) tR = 1.79 min, 191/192 (M + H)+.
8-(3-Chloro-5-(3-(methylamino)-1H-indazol-6-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (100)
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one278 (70.3 mg, 0.20 mmol), (3-(methylamino)-1H-indazol-6-yl)boronic acid 97 (13% pure, 230 mg, 0.160 mmol) were loaded in a microwave vial, and acetonitrile (4 mL), 0.5 M aqueous sodium carbonate (1.23 mL, 0.610 mmol) and Pd(dppf)Cl2·CH2Cl2 (8 mg, 0.01 mmol) were added. The reaction mixture was stirred for 1 h at 120 °C under microwave irradiation. After dilution with acetonitrile (5 mL), the mixture was filtered and the filtrate was concentrated. The crude product was purified by preparative HPLC (method A, 20 min gradient elution from 2:98 to 20:80 acetonitrile/water) to give 8-(3-chloro-5-(3-(methylamino)-1H-indazol-6-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 100 (13 mg, 12% yield) as a white solid. 1H NMR (500 MHz, CD3OD) δ 8.56 (s, 1H), 8.28 (s, 1H), 7.89 (dd, J = 8.4, 0.8 Hz, 1H), 7.41–7.37 (m, 1H), 7.07 (dd, J = 8.4, 1.4 Hz, 1H), 3.41–3.35 (m, 2H), 3.27 (t, J = 6.9 Hz, 2H), 3.06 (s, 3H), 2.97–2.89 (m, 2H), 2.00 (t, J = 6.9 Hz, 2H), 1.92–1.84 (m, 2H), 1.41 (d, J = 13.3 Hz, 2H); LC–MS (method C, ESI, m/z) tR = 1.74 min, 411/413 (M + H)+; ESI-HRMS calcd for C21H2435ClN6O (M + H)+ 411.1695, found 411.1680.
tert-Butyl N-tert-Butoxycarbonyl-N-[1-methyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazol-3-yl]carbamate (98)
6-Bromo-1-methyl-1H-indazol-3-amine (500 mg, 2.21 mmol) and N,N-dimethylpyridin-4-amine (54 mg, 0.44 mmol) were dissolved in DCM (15 mL). Triethylamine (1.74 mL, 13.3 mmol) and di-tert-butyl dicarbonate (1.42 mL, 6.64 mmol) were were added, and the reaction solution was stirred overnight at rt. After addition of water and DCM, the layers were separated and the aqueous layer was extracted with DCM. The organic layer was washed with water, dried, filtered, and evaporated to dryness. The brown solid was purified by flash-chromatography (Companion, n-heptane/EtOAc gradient) to give tert-butyl N-(6-bromo-1-methyl-indazol-3-yl)-N-tert-butoxycarbonylcarbamate (150 mg, 16% yield) as a colorless solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 3.29 min, 448/450 (M + Na)+.
tert-Butyl N-(6-bromo-1-methylindazol-3-yl)-N-tert-butoxycarbonylcarbamate (150 mg, 0.352 mmol) was dissolved in THF (5 mL). Bis(pinacolato)diboron (179 mg, 0.704 mmol), potassium acetate (104 mg, 1.06 mmol), and Pd(dppf)Cl2·CH2Cl2 (29 mg, 0.035 mmol) were added, and the red reaction mixture was stirred overnight at 70 °C. The resultant black reaction mixture was treated with EtOAc, filtered and the filtrate evaporated under vacuum. The dark brown residue was purified by flash chromatography (Companion, heptane/DCM gradient) to give a mixture of [3-[bis(tert-butoxycarbonyl)amino]-1-methyl-indazol-6-yl]boronic acid and tert-butyl N-tert-butoxycarbonyl-N-[1-methyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazol-3-yl]carbamate (2:3 ratio, 182 mg, quant) as a pale yellow solid 98 which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 2.59 min, 413/414 (M + Na)+ and tR = 3.49 min, 495/496 (M + Na)+.
8-(3-(3-Amino-1-methyl-1H-indazol-6-yl)-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 2,2,2-Trifluoroacetate (101)
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one278 (84.4 mg, 0.25 mmol) was suspended in acetonitrile (2 mL). A mixture of tert-butyl N-tert-butoxycarbonyl-N-[1-methyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)indazol-3-yl]carbamate and [3-[bis(tert-butoxycarbonyl)amino]-1-methyl-indazol-6-yl]boronic acid 98 (3:2 ratio, 174 mg, ∼0.368 mmol), 0.5 M aqueous sodium carbonate (982 uL, 0.491 mmol), and Pd(dppf)Cl2·CH2Cl2 (18 mg, 0.025 mmol) were added. The reaction mixture was stirred for 1 h at 120 °C. After addition of EtOAc, the mixture was filtered and the filtrate was concentrated. The brown residue was purified by flash chromatography (Companion, DCM/MeOH gradient) to give tert-butyl (6-(5-chloro-4-(1-oxo-2,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1H-indazol-3-yl)carbamate (47 mg, 71% pure, 27% corrected yield) as a colorless solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 2.19 min, 511/513 (M + H)+.
tert-Butyl (6-(5-chloro-4-(1-oxo-2,8-diazaspiro[4.5]decan-8-yl)pyridin-3-yl)-1-methyl-1H-indazol-3-yl)carbamate (71% pure, 47 mg, 0.066 mmol) was dissolved in 5 N hydrogen chloride in dioxane (5 mL) and MeOH (1 mL). The pale brown solution was stirred at rt for 4 h, and the solvents were evaporated. The residue was purified by preparative HPLC (method A, 20 min gradient elution from 2:98 to 25:75 acetonitrile/water) to give 8-(3-(3-amino-1-methyl-1H-indazol-6-yl)-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 2,2,2-trifluoroacetate 101 (15 mg, 43% yield) as a yellow solid. 1H NMR (500 MHz, CD3OD) δ 8.66 (s, 1H), 8.35 (s, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.47 (s, 1H), 7.04 (d, J = 8.4 Hz, 1H), 3.89 (s, 3H), 3.55–3.47 (m, 2H), 3.27 (t, J = 6.9 Hz, 2H), 3.02–2.95 (m, 2H), 2.01 (t, J = 6.9 Hz, 2H), 1.95–1.87 (m, 2H), 1.50–1.42 (m, 2H); 13C NMR (126 MHz, CD3OD) δ 183.1, 159.7, 148.3, 144.4, 143.3, 143.1, 136.6, 134.7, 127.8, 122.5, 121.2, 115.5, 110.6, 49.8, 43.1, 39.7, 35.1, 33.4, 32.6; LC–MS (method C, ESI, m/z) tR = 1.86 min, 411/413 (M + H)+; ESI-HRMS calcd for C21H2435ClN6O (M + H)+ 411.1695, found 411.1684.
N,N,1-Trimethyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazol-3-amine (99)
4-Bromo-2-fluorobenzonitrile (1.0 g, 5.0 mmol) was dissolved in ethanol (11 mL), and methylhydrazine (0.756 mL, 14.4 mmol) was added. The reaction mixture was stirred under microwave irradiation at 120 °C for 50 min. The reaction was quenched with water, and the product was extracted with EtOAc three times. The combined organic layers were dried over MgSO4 and concentrated to give 6-bromo-1-methyl-1H-indazol-3-amine41 (940 mg, 83% yield) as a brown solid. 1H NMR (500 MHz, CDCl3) δ 7.47 (dd, J = 8.7, 0.7 Hz, 1H), 7.38 (dd, J = 1.5, 0.7 Hz, 1H), 7.13 (dd, J = 8.7, 1.5 Hz, 1H), 3.80 (s, 3H); LC–MS (method C, ESI, m/z) tR = 2.50 min, 226/228 (M + H)+.
To 6-bromo-1-methyl-1H-indazol-3-amine (736 mg, 3.26 mmol) in formic acid (3.7 mL, 98 mmol) was added formaldehyde 37% in water (897 μL, 32.6 mmol). The reaction mixture was heated at 100 °C for 2 h and concentrated. The crude material was purified by Biotage column chromatography (cyclohexane/EtOAc 95:5 to 82:18) to afford 6-bromo-N,N,1-trimethyl-1H-indazol-3-amine (85 mg, 10% yield) as a brown solid. 1H NMR (500 MHz, CDCl3) δ 7.60 (dd, J = 8.7, 0.6 Hz, 1H), 7.37 (dd, J = 1.5, 0.6 Hz, 1H), 7.06 (dd, J = 8.7, 1.5 Hz, 1H), 3.82 (s, 3H), 3.07 (s, 6H); LC–MS (method C, ESI, m/z) tR = 3.11 min, 254/256 (M + H)+.
6-Bromo-N,N,1-trimethyl-1H-indazol-3-amine (100 mg, 0.394 mmol), bis(pinacolato)diboron (150 mg, 0.590 mmol), potassium acetate (116 mg, 1.18 mmol), and Pd(dppf)Cl2·CH2Cl2 (16 mg, 0.020 mmol) were loaded in a flask, and DME (2.8 mL) was added. The reaction mixture was heated at 80 °C overnight and concentrated. The crude material was purified by Biotage column chromatography (cylohexane/EtOAc 95:5 to 70:30) to give N,N,1-trimethyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazol-3-amine 99 (100 mg, 84% yield). 1H NMR (500 MHz, CDCl3) δ 7.75 (dd, J = 8.1, 0.9 Hz, 1H), 7.72 (s, 1H), 7.39 (d, J = 8.1 Hz, 1H), 3.91 (s, 3H), 3.09 (s, 6H), 1.38 (s, 12H); LC–MS (method C, ESI, m/z) tR = 3.19 min, 301/302 (M + H)+.
8-(3-Chloro-5-(3-(dimethylamino)-1-methyl-1H-indazol-6-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (102)
8-(3-Bromo-5-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one278 (13 mg, 0.036 mmol), N,N,1-trimethyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazol-3-amine 99 (12 mg, 0.040 mmol), and Pd(dppf)Cl2·CH2Cl2 (1.5 mg, 1.8 μmol) were loaded in a microwave vial, and acetonitrile (624 μL) and 0.5 M sodium carbonate in water (101 μL, 0.0510 mmol) were added. The reaction was stirred at 120 °C for 60 min under microwave irradiation. The solvent was evaporated, and the crude material was purified by Biotage column chromatography (DCM/EtOH 98:2 to 91:9) and then by preparative HPLC (method B). Fractions were concentrated under vacuum and the product was solubilized in DCM and filtered on a basic Isolute flash NH2 column to afford 8-(3-chloro-5-(3-(dimethylamino)-1-methyl-1H-indazol-6-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 102 (8 mg, 50% yield). 1H NMR (500 MHz, CDCl3) δ 8.46 (s, 1H), 8.27 (s, 1H), 7.82 (dd, J = 8.4, 0.8 Hz, 1H), 7.13 (dd, J = 1.4, 0.8 Hz, 1H), 6.88 (dd, J = 8.4, 1.4 Hz, 1H), 5.68 (s, 1H), 3.92 (s, 3H), 3.28–3.23 (m, 2H), 3.21–3.15 (m, 2H), 3.13 (s, 6H), 2.71 (t, J = 11.8 Hz, 2H), 2.00–1.90 (m, 4H), 1.36–1.29 (m, 2H); LC–MS (method C, ESI, m/z) tR = 2.40 min, 439/441 (M + H)+; ESI-HRMS calcd for C23H2835ClN6O (M + H)+ 439.2008, found 439.2004.
Preparation of Compounds in Table 8, Exemplified by Compound 109. 8-(2-Amino-3-chloro-5-(1-methyl-1H-indazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (109)
Into a 500 mL screw cap jar, 8-(2-(bis(4-methoxybenzyl)amino)-5-bromo-3-chloropyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 82 (82% pure, 13.4 g, 18.4 mmol), 1-methylindazole-5-boronic acid 39 (97% pure, 4.99 g, 27.5 mmol), and Pd(dppf)Cl2·CH2Cl2 (749 mg, 0.920 mmol) were weighed in and suspended in acetonitrile (300 mL) and sodium carbonate solution (0.5 M, 73.4 mL, 36.7 mmol). The reaction mixture was stirred at 80 °C overnight. The mixture was diluted with acetonitrile (200 mL), filtered, and evaporated to dryness. The crude residue was purified by flash chromatography (DCM/MeOH, gradient). The isolated product was suspended in acetonitrile (100 mL) and treated with diethyl ether (200 mL). The resultant beige precipitate was filtered, washed with diethyl ether (100 mL), and dried under vacuum. The mother liquor was evaporated to 50 mL volume and treated with diethyl ether (100 mL). The resulting precipitate was filtered, washed with diethyl ether (40 mL), and dried under vacuum. The two batches of compound were combined to yield in 8-(2-(bis(4-methoxybenzyl)amino)-3-chloro-5-(1-methyl-1H-indazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (7.05 g, 59% yield) as a colorless solid which was used directly in the next step. LC–MS (method B, ESI, m/z) tR = 2.78 min, 651/653 (M + H)+.
8-(2-(Bis(4-methoxybenzyl)amino)-3-chloro-5-(1-methyl-1H-indazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one (7.05 g, 10.8 mmol) was dissolved in trifluoroacetic acid (70 mL) and the reaction mixture stirred at rt for 4 h. The solution was evaporated to dryness. The crude residue was suspended in DCM and treated with diethyl ether (350 mL). The white precipitate was filtered and washed with diethyl ether (100 mL) and dried under vacuum overnight. The residue was dissolved in DMSO (50 mL) and poured into sodium carbonate solution (0.25 M, 500 mL). The resulting light yellow precipitate was filtered, washed with water (150 mL), and dried at 70 °C under vacuum overnight to give 8-(2-amino-3-chloro-5-(1-methyl-1H-indazol-5-yl)pyridin-4-yl)-2,8-diazaspiro[4.5]decan-1-one 109 (3.34 g, 75% yield) as a light yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.05 (d, J = 1.0 Hz, 1H), 7.69–7.64 (m, 2H), 7.63–7.60 (m, 1H), 7.49 (s, 1H), 7.27 (dd, J = 8.6, 1.6 Hz, 1H), 6.10 (s, 2H), 4.07 (s, 3H), 3.07 (t, J = 6.7 Hz, 2H), 2.98–2.92 (m, 2H), 2.67–2.55 (m, 2H), 1.74 (t, J = 6.7 Hz, 2H), 1.71–1.61 (m, 2H), 1.16 (d, J = 12.6 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ 180.0, 156.2, 153.5, 148.2, 138.7, 132.4, 130.6, 128.4, 124.2, 123.7, 120.3, 109.2, 108.8, 47.3, 41.4, 37.8, 35.5, 31.8, 30.3; LC–MS (method C, ESI, m/z) tR = 1.79 min, 411/413 (M + H)+. ESI-HRMS calcd for C21H2435ClN6O (M + H)+ 411.1695, found 411.1681.
Mouse, Rat, and Human Clint Determination
Microsomes (final concentration 0.5 mg/mL), 50 mM phosphate buffer pH 7.4, and compound (final concentration 1 μM) were added to the assay plate and allowed to preincubate for 5 min at 37 °C. The reaction was initiated by the addition of NADPH (final concentration 1.5 mM), and the plate was shaken at 800 rpm at 37 °C. After 0, 5, 10, 20, and 30 min aliquots were taken and the reaction was stopped using cold acetonitrile. The samples were centrifuged at 4000 rpm for 30 min at 4 °C and analyzed by LC–MS/MS. Four test compounds were pooled for analysis. The in vitro intrinsic clearance was calculated from the rate of compound disappearance.
Caco-2 Papp Determination
Caco-2 cells were maintained in DMEM in an atmosphere of 8.5% CO2. For transport experiments 0.125 × 106 cells/well of were seeded on polycarbonate filter inserts and allowed to grow and differentiate for 14 ± 1 days before the cell monolayers were used for experiments. Drug transport experiments were carried out using a cocktail approach in a four-dimensional setting. Apparent permeability coefficients were determined for A → B and B → A directions with and without the presence of cyclosporine A as a transporter inhibitor. Up to five test items and reference compounds were dissolved in Hank’s balanced salt solution at pH 7.4 to yield a final concentration of 1 μM. The assays were performed in HBSS containing 25 mM HEPES (pH 7.4) in an atmosphere of 5% CO2 at 37 °C. Prior to the study, the monolayers were washed in prewarmed HBSS. At the start of the experiments prewarmed HBSS containing the test items was added to the donor side of the monolayer and HBSS without test items was added to the receiver side. The plates were shaken at 150 rpm at 37 °C during the experiment. After 2 h the Transwell insert containing the monolayer was carefully removed and placed in a new plate and aliquots of both the receiver and donor sides were taken and diluted with an equal volume of acetonitrile containing the internal standard. The mixture was centrifuged and supernatant analyzed by LC–MS/MS. The apparent permeability coefficients (Papp) were calculated using the formula: Papp = [Vrec/(A × C0,donor)](dCrec/dt) × 106 with dCrev/dt being the change in concentration in the receiver compartment with time, Vrec the volume of the sample in the receiver compartment, C0,donor the concentration in the donor compartment at time 0, and A the area of the compartment with the cells.
In Vivo Mouse PK In-Life Phase
Female NMRI mice (n = 5) received either a single intravenous (bolus) injection or single oral administration (by gavage) of the compound in a cocktail preparation. Doses of 0.2 mg/kg (per compound, intravenous administration) and 0.5 mg/kg (per compound, perorally) were given as solutions in DMSO/PEG 200/water. Consecutive blood samples were taken retroorbitally under isofluorane inhalation from n = 3 animals per route of administration after 0.1 (iv only), 0.25 (po only), 0.5, 1, 2, 4, and 6 h and were further processed to obtain plasma. In addition, feces of 2 mice per route of administration were collected over the time interval from 0 to 24 h (pooled for analysis, no blood sampling).
In Vivo Rat PK In-Life Phase
Male Wistar rats (n = 6) received either a single intravenous (bolus) injection or a single oral administration (by gavage) of the compound in a cocktail preparation. Doses of 0.2 mg/kg (pre compound, intravenous administration) and 0.5 mg/kg (per compound, perorally) were given as solutions in DMSO/PEG 200/water. Consecutive blood samples were taken sublingually under isofluorane inhalation from n = 3 animals per route of administration after 0.1 (iv only), 0.25 (po only), 0.5, 1, 2, 4, and 6 h and were further processed to obtain plasma. In addition, feces and urine of 3 rats per route of administration were collected over the time interval from 0 to 24 h (pooled for analysis, no blood sampling).
In Vivo Dog PK In-Life Phase
Female Beagle dogs received either a single intravenous (bolus) injection or an oral administration (by gavage) of the compound in a cocktail preparation. Doses of 0.2 and 0.5 mg/kg were given intravenously and per orally as a solution in DMSO/PEG 200/water, respectively. Consecutive blood samples were taken by puncture of the vena cephalica from n = 2 animals per route of administration (crossover design) predose and after 0.1 (only (iv), 0.25 (only po), 0.5, 1, 2, 4, 6, 24, and 48 h and were further processed to obtain plasma. The Institute of Cancer Research does not use non-rodent species in research and, where this is deemed essential, require ethical approval for use by organizations with whom we collaborate. Pharmacokinetic analysis of compounds 6 and 109 in dogs, necessary for prediction of human pharmacokinetics, was approved by the ICR Ethics Committee. Studies were sponsored and conducted in full compliance with national regulations at an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited site of Merck Biopharma.
Bioanalytics
The concentrations of compound in plasma and feces were quantified using an UPLC method with tandem mass spectrometric detection (LC–MS/MS). The LC–MS system consisted of a Waters Acquity UPLC coupled to an AB Sciex mass spectrometer API 5500 Q-trap. The UPLC separation was carried out on a reversed phase column (Acquity UPLC BEC C8, 1.7 μM, 2.1 mm × 50 mm) using a mobile phase gradient with 0.1% formic acid and acetonitrile as eluents. The detection of the drug was performed using multiple reaction monitoring in the positive ionization mode. Plasma samples were spiked with internal standard, and the analyte was extracted from the matrix using tert-butyl methyl ether (tBME). The organic phase was evaporated to dryness under a stream of nitrogen. The residue was dissolved in acetonitrile/0.1% formic acid for LC–MS/MS analysis. Feces samples were homogenized with 4 times their volume ethanol/water (4:1). Aliquots of the aqueous ethanolic extracts were further diluted with acetonitrile/0.1% formic acid, spiked with internal standard, and directly injected into the system.
Prediction of Human Clearance
Allometric methods used for clearance prediction to human were simple allometry (3-species), allometry with (a) correction by fu, (b) with correction by fu and MLP (maximum life span), (c) with correction by fu and BrW (brain weight); in addition, in vitro to in vivo (iviv) scaling from liver microsomes and hepatocytes was used for clearance prediction (for compounds 6 and 109, iviv from hepatocytes of 0.53 and 0.215 L/h/kg may be included in the mean calculation). For compound 6, the clearance prediction ranged from 1.45 L/h/kg (simple allometry) to 0.485 L/h/kg (BrW). Allometry with correction for fu and MLP was chosen because of the highest R2 value (0.9992) in the linear regression plot; the mean of all methods gives a clearance prediction of 0.88 L/h/kg. For volume allometry, methods included 3-species allometry with correction for fu and human–dog proportionality. Values ranged between 0.7 and 1.0 L/kg; the selected method of choice was allometry with correction by fu (Vss = 0.85 L/kg), which represents roughly the mean of the values. For compound 109 the corresponding values were as follows: clearance prediction; MLP = 0.511 L/h/kg, BrW = 0.343 L/h/kg. Allometry with correction for fu and scaling from liver microsomes and hepatocytes was included; the mean of all methods gave a clearance prediction of 0.36 L/h/kg. For volume allometry, methods included 3 species allometry with correction for and human–dog proportionality. Values ranged between 1.3 and 1.5 L/kg; the mean of all methods gave a volume prediction of 1.4 L/kg.
In Vitro Cell-Based Reporter Assays. 7dF3 Luciferase Reporter Assay27
7dF3 cells were seeded at 5000 per well in 50 μL of Dulbecco’s modified Eagle medium (DMEM) with 4 mM supplemental l-glutamine in 384-well white tissue culture plates (Corning 3570, USA). After incubating overnight at 37 °C/5% CO2, compounds ranging in final concentration from 90 μM to 0.3 nM were added in duplicate to the wells. After 2 h of further incubation as above, β-oestradiol was added to a final concentration of 10 μM. The cells were incubated for 24 h at 37 °C/5% CO2, and then 25 μL of luciferase reagent (SteadyGlo, Promega,USA) was added and mixed. After leaving the plate for 60 min at room temperature, luminescence was read on a plate luminescence reader (TopCount, PerkinElmer, USA). Percentage inhibition for each compound concentration was calculated using total counts (no compound) and low counts (no β-oestradiol) as highs and lows, respectively. IC50 was subsequently determined using a curve-fitting software package in Excel (Excelfit, IDBS, U.K.). Data are reported as average values of at least two determinations with standard deviations unless otherwise noted.
In Vitro Biochemical Assays. CDK8 Lanthascreen Binding Assay
The assay format was a FRET based Lanthascreen binding competition assay. A dye-labeled ATP competitive tracer served as a FRET acceptor upon binding to CDK8. This CDK8 was His-tagged. The FRET donor was a strepavidin-Eu-chelate bound to a biotinylated anti-His antibody. When the tracer is competed by an inhibitor, the respective FRET signal is affected. The CDK8 used for this assay was a protein coexpressed with CycC. The assay procedure for assays in the 384-well plate format (Greiner PS, white, medium binding no. 784075) was the following: 2 μL of CDK8/biotin-anti-His Ab/SA-Eu mix in assay buffer was pipetted into the wells of the microplates. Then 1 μL compounds in 20 mM Hepes buffer/5% DMSO was added. The plates were shaken for 30 s and incubated for 20 min at rt. Then 2 μL of Alexa647 tracer in assay buffer was added. The plates were shaken for 30 s and incubated for 60 min at rt in the dark. Finally the plates were read out on a PerkinElmer Envision 2104 (mode LANCE/TRF, excitation 340 nm, emissions at 615 and 615 nm). For evaluation the emission ratios (615 nm/655 nm) were calculated. These were normalized to data from control wells containing vehicle only or 6 at 30 uM. The assay buffer was 50 mM Hepes, pH 7.5 (Merck no. 1.10110), 10 mM MgCl2 (Merck no. 1.05833), 1 mM EGTA (Merck no. 1.08435), 0.01% Brij-35 (Pierce no. 28316). The final concentrations of the reaction components in 5 μL total assay volume were 1% DMSO (Merck no. 1.02950), 5 nM CDK8 (CDK8/CycC Invitrogen no. PV4402), 2 nM biotin-a-His Ab (Invitrogen no. PV6089), 2 nM SA-europium (Invitrogen no. PV5899), 10 nM Alexa647 tracer (Invitrogen no. PV5592). Data are reported as average values of at least two determinations with standard deviations unless otherwise noted.
Reporter Displacement Assay for CDK8 and CDK19
Interaction of compounds with human CDK8(aa1-464)/cyclin C(aa1-283) and human CDK19(aa1-502)/cyclin C(aa1-283) was analyzed using the reporter displacement assay provided by Proteros Biostructures GmbH.42 In brief, the reporter displacement assay is based on reporter probes that are designed to bind to the site of interest of the target protein. The proximity between reporter and protein results in the emission of an optical signal. Compounds that bind to the same site as the reporter probe displace the probe, causing signal diminution. Reporter displacement is measured over time after addition of compounds at various concentrations. For IC50 determination, percent probe displacement values are calculated for the last time point at which the system has reached equilibrium. For each compound concentration, percent probe displacement values are calculated and plotted against the compound concentration. IC50-like values (corresponding to 50% probe displacement) are calculated using standard fitting algorithms. Data are reported as average values of at least two determinations with standard deviations unless otherwise noted.
Human Tumor Xenograft Efficacy Study
All animal procedures were conducted in accordance with NCRI guidelines43 and U.K. Home Office regulations. Female athymic nude mice Crl:Nu(NCr)-Foxn1nu supplied by Charles River Laboratories were acclimatized for 1 week prior to use. The human colorectal adenocarcinoma cell line, SW620 (ATCC, no. CCL-227), was expanded in culture, and 5 million cells were injected sc into the right flanks of animals (n = 12 per group) under isofluorothane anesthesia. Tumors were established for 7 days when dosing commenced (day 0) and continued for 16 days.
Compound 109 was prepared 24 h prior to administration to mice. The compound was weighed into a glass 30 mL universal tube, and Methocel solution (0.5% Methocel, 0.25% Tween 20 in sterile phosphate buffered saline) was added to provide a final concentration of 3 mg/mL 109. The preparation was agitated at room temperature by magnetic stirrer to bring it into a fine suspension before use. Animals were dosed orally by gavage every 24 h at 0.1 mL per 10 g body weight. Tumors were measured three times weekly by Vernier calipers and body weights recorded. At the end of the study, animals were culled at intervals: 3 control and 3 treated at 1, 2, 6, and 24 h after the final dose. Heparinized blood was collected by cardiac puncture, spun, and plasma snap frozen for analysis of compound exposure. Tumors were excised, weighed and samples snap frozen for compound quantification and PD analyses. The 30 mg/kg q.d. schedule was well tolerated with no significant body weight loss. Tumor growth was significantly inhibited (T/C = 45.8; P = 0.0241 final tumor weights).
Pharmacokinetic Analysis
Plasma and tumor homogenates were measured by LC–MS/MS on a Water TQS following a separation on a Waters Acquity BEH C18 column (50 mm × 2.1 mm; 1.7 μm) with conditions of 0.1% formic acid (mobile phase A) and methanol (mobile phase B). The column was equilibrated at initial condition of 95% A and 5% B, linear gradient over 3 min to 100% B, held over 1 min, followed by linear gradient back to 5% B over 0.1 min, at 0.6 mL/min flow rate. Detection was achieved in positive electrospray ionization mode by multiple reaction monitoring, 412.17 > 376.40 at 33 eV for compound 109, and 299.19 > 91.24 at 33 eV, 299.19 > 177.29 at 26 eV for the internal standard olomoucine. Samples were quantified by external standard calibration (8-point calibration ranging from 2 nM to 50 000 nM). Quality controls were included at 25, 250, 2500, and 7500 nM in duplicate at the beginning and the end of the analytical run.
Tumor Xenograft Processing and Western Blotting
Tumors were excised, immediately snap-frozen in liquid nitrogen, and stored at −80 °C until use. Samples were transferred to MK28 reinforced homogenizing tubes with metal beads (Stretton Scientific), and CDK lysis buffer (50 mM Tris-HCl, pH7.4, 1 mM EDTA, 1% Triton X-100 (v/v), 150 mM NaCl, 1 mM activated sodium orthovanandate, 1 mM PMSF, protease cocktail (Sigma P8340 1:100), and phosphatase inhibitors (Sigma P5726 and P0044 1:50 dilution) were added immediately. The samples were ground using a Precellys 24 at 6000 rpm, 2 × 20 s (Stretton Scientific) and lysates frozen at −80 °C until use. For analysis, samples were debanked and sonicated for 3 min, on ice for 10 min. Tubes were spun at 14 000 rpm at 4 °C for 10 min, and supernatants were collected, aliquoted, and frozen at −80 °C until use. Tumor lysates were diluted 1:5 in lysis buffer and the concentrations determined using a Direct Detect spectrometer (Merck Millipore).
Samples were aliquoted, then boiled in electrophoresis sample buffer and loaded on SDS-4-12% PAGE gels. After transfer to PVDF membranes, blots were blocked at room temperature for 1 h in blocking buffer (5% dry milk in TNT: 1 M Tris-HCL, pH 8, 5 M NaCl, 0.1% Tween 20) and then incubated at 4 °C overnight with the appropriate antibody (Table 11). After washing with TNT, blots were incubated with HRP-conjugated secondary antibodies in blocking buffer at room temperature for 1 h, and bands were visualized using the enhanced chemoluminescence (ECL prime) method.
Table 11. Antibodies Used in Western Blottinga.
| antibody name | source | catalogue no. | dilution | type |
|---|---|---|---|---|
| rabbit anti-Stat1 | Santa Cruz | sc-346 | 1:1000 | primary |
| rabbit anti phospho-Stat1Ser727 | CST | 8826 | 1:2000 | primary |
| mouse anti GAPDH | Abcam | Ab-8245 | 1:10000 | loading control |
| α-mouse IgG HRP-linked | CST | 7076 | 1:10000 | secondary |
| anti-rabbit IgG HRP-linked | CST | 7074 | 1:2500 | secondary |
Membranes were cut and incubated at the same time for phospho-StatSER727 (84, 91 kDa), total Stat1 (84, 91 kDa), and GAPDH (37 kDa).
Acknowledgments
This work was supported by Cancer Research UK [Grant C309/A11566]. We acknowledge NHS funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden. We thank Dr. Amin Mirza, Meirion Richards, and Dr. Maggie Liu for their assistance with NMR, mass spectrometry and HPLC, Gary Nugent and Dr. Christian Herhaus for chemoinformatics support, and Kate Trotman for administrative support. We thank the team of Proteros Biostructures GmbH, Bunsenstrasse 7a, D-82152 Martinsried, Germany, for the reporter displacement assay and in particular Dr. Elisabeth V. Schneider and Dr. Alfred Lammens for the X-ray crystal structures with CDK8/cyclin C.
Glossary
Abbreviations Used
- CDK
cyclin-dependent kinase
- Cl
clearance
- Clint
intrinsic clearance
- ER
efflux ratio
- F
bioavailability
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- kin
kinetic
- LBF
liver blood flow
- t1/2
half-life
- PMB
p-methoxybenzyl
- SNAr
nucleophilic aromatic substitution
- therm
thermodynamic
- TPSA
topological polar surface area
- Vd
volume of distribution
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01685.
Caco-2 data for compounds 6, 17–25, 41, 42, 44–49, 51, 54, 61, 63, 72, 85–95 (Table S1), CEREP profile of 109 (Tables S2 and S3), kinase profile of 109 (Table S4), CYP450 inhibition for 109 (Table S5), plasma and tumor concentration (Table S6), correlation of 7dF3 and LS174T cell-based potency (Figure S1), analyses of phospho-STAT1SER727 (Figure S2), total plasma and tumor concentration (Figure S3), thermodynamic and kinetic solubility protocols, preparation of compounds 9–13, 15, 17–22, 24, 26, 28–35, 40, 41, 43–53, 55, 56, 60, 62, 65, 67, 69–71, 76, 78, 79, 83–85, 87–91, 93, 94, 96, 103–108, 110, 111, 1H NMR spectra for all final compounds, and small molecule X-ray crystallographic data analysis (including Fo – Fc omit maps at 2.5σ cutoff) for compounds 25, 42, 54, and 109 (PDF)
Molecular formula strings (CSV)
Accession Codes
Atomic coordinates and structure factors for the crystal structures of CDK8/cyclin C with compounds 25, 42, 54, and 109 can be accessed using PDB codes 5FGK, 5HBE, 5HBH, and 5HBJ, respectively.
Author Contributions
The manuscript was written with contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): A.M., C.R., S.C., M.S., O.A-P., M.-J.O.-R., C.T., M.V., A.d.H.B., R.B., P.W., T.D., P.A.C., F.I.R., S.A.E., and J.B. are current or former employees of The Institute of Cancer Research, which has a commercial interest in the development of WNT pathway inhibitors. K.S., F.S., M.C., O.P., M.B., P.C., D.M., D.S., R.S., D.W., C.E. and F.R. are current employees of Merck.
Supplementary Material
References
- Allen B. L.; Taatjes D. J. The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsten J. O.; Zhu X.; Gustafsson C. M. The multitalented Mediator complex. Trends Biochem. Sci. 2013, 38, 531–537. 10.1016/j.tibs.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Kim S.; Xu X.; Hecht A.; Boyer T. G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006, 281, 14066–14075. 10.1074/jbc.M602696200. [DOI] [PubMed] [Google Scholar]
- Schiano C.; Casamassimi A.; Vietri M. T.; Rienzo M.; Napoli C. The roles of mediator complex in cardiovascular diseases. Biochim. Biophys. Acta, Gene Regul. Mech. 2014, 1839, 444–451. 10.1016/j.bbagrm.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Schiano C.; Casamassimi A.; Rienzo M.; de Nigris F.; Sommese L.; Napoli C. Involvement of Mediator complex in malignancy. Biochim. Biophys. Acta, Rev. Cancer 2014, 1845, 66–83. 10.1016/j.bbcan.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Galbraith M. D.; Donner A. J.; Espinosa J. M. CDK8: a positive regulator of transcription. Transcription 2010, 1, 4–12. 10.4161/trns.1.1.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui T.; Fukasawa R.; Tanaka A.; Hirose Y.; Ohkuma Y. Identification of target genes for the CDK subunits of the Mediator complex. Genes Cells 2011, 16, 1208–1218. 10.1111/j.1365-2443.2011.01565.x. [DOI] [PubMed] [Google Scholar]
- Rickert P.; Seghezzi W.; Shanahan F.; Cho H.; Lees E. Cyclin C/CDK8 is a novel CTD kinase associated with RNA polymerase II. Oncogene 1996, 12, 2631–2640. [PubMed] [Google Scholar]
- Morris E. J.; Ji J. Y.; Yang F.; Di Stefano L.; Herr A.; Moon N. S.; Kwon E. J.; Haigis K. M.; Naar A. M.; Dyson N. J. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature 2008, 455, 552–556. 10.1038/nature07310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R.; Shima K.; Nosho K.; Irahara N.; Baba Y.; Bojarski E.; Giovannucci E. L.; Hahn W. C.; Fuchs C. S.; Ogino S. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 2010, 126, 2863–2873. 10.1002/ijc.24908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. Y.; Han S. I.; Lim S. C. Roles of cyclin-dependent kinase 8 and beta-catenin in the oncogenesis and progression of gastric adenocarcinoma. Int. J. Oncol. 2011, 38, 1375–1383. 10.3892/ijo.2011.948. [DOI] [PubMed] [Google Scholar]
- Porter D. C.; Liang J.; Kaza V.; Chumanevich A. A.; Altillia S.; Farmaki E.; Chen M.; Schools G. P.; Chatzistamou I.; Pena M. M.; Friedhoff L. T.; Wentland M. P.; Broude E.; Kiaris H.; Roninson I. B.. Targeting the seed and the soil of cancers with selective small-molecule inhibitors of CDK8/19: Chemopotentiating, chemopreventive, anti-invasive and anti-metastatic activities. Presented at the AACR Annual Meeting 2014, San Diego, CA, 2014.
- Kapoor A.; Goldberg M. S.; Cumberland L. K.; Ratnakumar K.; Segura M. F.; Emanuel P. O.; Menendez S.; Vardabasso C.; Leroy G.; Vidal C. I.; Polsky D.; Osman I.; Garcia B. A.; Hernando E.; Bernstein E. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler A. S.; McCleland M. L.; Truong T.; Lau S.; Modrusan Z.; Soukup T. M.; Roose-Girma M.; Blackwood E. M.; Firestein R. CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res. 2012, 72, 2129–2139. 10.1158/0008-5472.CAN-11-3886. [DOI] [PubMed] [Google Scholar]
- Firestein R.; Bass A. J.; Kim S. Y.; Dunn I. F.; Silver S. J.; Guney I.; Freed E.; Ligon A. H.; Vena N.; Ogino S.; Chheda M. G.; Tamayo P.; Finn S.; Shrestha Y.; Boehm J. S.; Jain S.; Bojarski E.; Mermel C.; Barretina J.; Chan J. A.; Baselga J.; Tabernero J.; Root D. E.; Fuchs C. S.; Loda M.; Shivdasani R. A.; Meyerson M.; Hahn W. C. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T.; Mikula M.; Wiklik K.; Brzozka K. CDK8 kinase-An emerging target in targeted cancer therapy. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1617–1629. 10.1016/j.bbapap.2015.05.011. [DOI] [PubMed] [Google Scholar]
- Cee V. J.; Chen D. Y.; Lee M. R.; Nicolaou K. C. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew. Chem., Int. Ed. 2009, 48, 8952–8957. 10.1002/anie.200904778. [DOI] [PubMed] [Google Scholar]
- Shair M. D.Cortistatin analogues and syntheses and uses thereof. WO2015100420 A1, 2015.
- Pelish H. E.; Liau B. B.; Nitulescu II; Tangpeerachaikul A.; Poss Z. C.; Da Silva D. H.; Caruso B. T.; Arefolov A.; Fadeyi O.; Christie A. L.; Du K.; Banka D.; Schneider E. V.; Jestel A.; Zou G.; Si C.; Ebmeier C. C.; Bronson R. T.; Krivtsov A. V.; Myers A. G.; Kohl N. E.; Kung A. L.; Armstrong S. A.; Lemieux M. E.; Taatjes D. J.; Shair M. D. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. 10.1038/nature14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider E. V.; Bottcher J.; Blaesse M.; Neumann L.; Huber R.; Maskos K. The structure of CDK8/CycC implicates specificity in the CDK/cyclin family and reveals interaction with a deep pocket binder. J. Mol. Biol. 2011, 412, 251–266. 10.1016/j.jmb.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Schneider E. V.; Bottcher J.; Huber R.; Maskos K.; Neumann L. Structure-kinetic relationship study of CDK8/CycC specific compounds. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8081–8086. 10.1073/pnas.1305378110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T.; Clarke P. A.; Esdar C.; Waalboer D.; Adeniji-Popoola O.; Ortiz-Ruiz M.; Mallinger A.; Samant R. S.; Czodrowski P.; Musil D.; Schwarz D.; Schneider K.; Stubbs M.; Ewan K.; Fraser E.; TePoele R.; Court W.; Box G.; Valenti M.; de Haven Brandon A.; Gowan S.; Rohdich F.; Raynaud F.; Schneider R.; Poeschke O.; Blaukat A.; Workman P.; Schiemann K.; Eccles S. A.; Wienke D.; Blagg J. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat. Chem. Biol. 2015, 11, 973–980. 10.1038/nchembio.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roninson I. B.; Porter D. C.; Wentland M. P.. CDK8/CDK19 selective inhibitors and their use in anti-metastatic and chemopreventative methods for cancer. WO2013116786 A1, 2013.
- Hu H.; Jiang M.; Jin T.; Niu R.; Wang J.; Yang S.; Yuan T.; Zhou C.; Wang M.;, Zhou Z.. Novel phenyl-pyridine/pyrazine amides for the treatment of cancer. WO2014029726 A1, 2014.
- Rzymski T.; Zarebski A.; Dreas A.; Osowska K.; Kucwaj K.; Fogt J.; Cholody M.; Galezowski M.; Czardybon W.; Horvath R.; Wiklik K.; Milik M.; Brzózka K.. Substituted tricyclic benzimidazoles as kinase inhibitors. WO2014072435 A1, 2014.
- Romero D. L.; Chaudhary D.; Robinson S.; Masse C. E.; Morin M. J.. CDK8 inhibitors and uses thereof. WO2014194245 A3, 2014.
- Mallinger A.; Crumpler S.; Pichowicz M.; Waalboer D.; Stubbs M.; Adeniji-Popoola O.; Wood B.; Smith E.; Thai C.; Henley A. T.; Georgi K.; Court W.; Hobbs S.; Box G.; Ortiz-Ruiz M. J.; Valenti M.; De Haven Brandon A.; TePoele R.; Leuthner B.; Workman P.; Aherne W.; Poeschke O.; Dale T.; Wienke D.; Esdar C.; Rohdich F.; Raynaud F.; Clarke P. A.; Eccles S. A.; Stieber F.; Schiemann K.; Blagg J. Discovery of Potent, Orally Bioavailable, Small-Molecule Inhibitors of WNT Signaling from a Cell-Based Pathway Screen. J. Med. Chem. 2015, 58, 1717–1735. 10.1021/jm501436m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremlev M. M.; Mushta A. I.; Tyrra W.; Yagupolskii Y. L.; Naumann D.; Moller A. Me3SiCF3/AgF/Cu—A new reagents combination for selective trifluoromethylation of various organic halides by trifluoromethylcopper, CuCF3. J. Fluorine Chem. 2012, 133, 67–71. 10.1016/j.jfluchem.2011.07.025. [DOI] [Google Scholar]
- Gleeson M. P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- Charifson P. S.; Walters W. P. Acidic and basic drugs in medicinal chemistry: a perspective. J. Med. Chem. 2014, 57, 9701–9717. 10.1021/jm501000a. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe D. A. Variability in Caco-2 and MDCK cell-based intestinal permeability assays. J. Pharm. Sci. 2008, 97, 712–725. 10.1002/jps.21010. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Keseru G. M. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discovery 2012, 11, 355–365. 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- Johnson T. W.; Dress K. R.; Edwards M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. 10.1016/j.bmcl.2009.08.045. [DOI] [PubMed] [Google Scholar]
- Somanathan R.; Rivero I. A.; Nunez G. I.; Hellberg L. H. Convenient synthesis of 1-oxa-3,8-diazaspiro-4,5-decan-2-ones. Synth. Commun. 1994, 24, 1483–1487. 10.1080/00397919408011753. [DOI] [Google Scholar]
- Yamada H.; Hayashi T.; Usuki T. Bull. Chem. Soc. Jpn. 2015, 88, 673–683. 10.1246/bcsj.20140394. [DOI] [Google Scholar]
- Kay A. J.; Woolhouse A. D.; Gainsford G. J.; Haskell T. G.; Wyss C. P.; Giffin S. M.; McKinnie I. T.; Barnes T. H. Simple zwitterionic merocyanines as potential NLO chromophores. J. Mater. Chem. 2001, 11, 2271–2281. 10.1039/b103148c. [DOI] [Google Scholar]
- Bavetsias V.; Large J. M.; Sun C.; Bouloc N.; Kosmopoulou M.; Matteucci M.; Wilsher N. E.; Martins V.; Reynisson J.; Atrash B.; Faisal A.; Urban F.; Valenti M.; de Haven Brandon A.; Box G.; Raynaud F. I.; Workman P.; Eccles S. A.; Bayliss R.; Blagg J.; Linardopoulos S.; McDonald E. Imidazo[4,5-b]pyridine derivatives as inhibitors of Aurora kinases: lead optimization studies toward the identification of an orally bioavailable preclinical development candidate. J. Med. Chem. 2010, 53, 5213–5228. 10.1021/jm100262j. [DOI] [PubMed] [Google Scholar]
- Li C.; Ai J.; Zhang D.; Peng X.; Chen X.; Gao Z.; Su Y.; Zhu W.; Ji Y.; Chen X.; Geng M.; Liu H. Design, Synthesis, and Biological Evaluation of Novel Imidazo[1,2-a]pyridine Derivatives as Potent c-Met Inhibitors. ACS Med. Chem. Lett. 2015, 6, 507–512. 10.1021/ml5004876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Zhou W.; Weisberg E.; Wang J.; Zhang J.; Sasaki T.; Nelson E.; Griffin J. D.; Janne P. A.; Gray N. S. An amino-indazole scaffold with spectrum selective kinase inhibition of FLT3, PDGFRalpha and kit. Bioorg. Med. Chem. Lett. 2012, 22, 4579–4584. 10.1016/j.bmcl.2012.05.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler R. C.; Baxter E.; Campbell I. B.; Macdonald S. J. F. A General, One-Step Synthesis of Substituted Indazoles using a Flow Reactor. Org. Process Res. Dev. 2011, 15, 565–569. 10.1021/op100288t. [DOI] [Google Scholar]
- Neumann L.; von Konig K.; Ullmann D. HTS reporter displacement assay for fragment screening and fragment evolution toward leads with optimized binding kinetics, binding selectivity, and thermodynamic signature. Methods Enzymol. 2011, 493, 299–320. 10.1016/B978-0-12-381274-2.00012-1. [DOI] [PubMed] [Google Scholar]
- Workman P.; Aboagye E. O.; Balkwill F.; Balmain A.; Bruder G.; Chaplin D. J.; Double J. A.; Everitt J.; Farningham D. A.; Glennie M. J.; Kelland L. R.; Robinson V.; Stratford I. J.; Tozer G. M.; Watson S.; Wedge S. R.; Eccles S. A. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.