

ABSTRACT
Plasmodial species are protozoan parasites that infect erythrocytes. As such, they are in close contact with microvascular endothelium for most of the life cycle in the mammalian host. The host-parasite interactions of this stage of the infection are responsible for the clinical manifestations of the disease that range from a mild febrile illness to severe and frequently fatal syndromes such as cerebral malaria and multi-organ failure. Plasmodium falciparum, the causative agent of the most severe form of malaria, is particularly predisposed to modulating endothelial function through either direct adhesion to endothelial receptor molecules, or by releasing potent host and parasite products that can stimulate endothelial activation and/or disrupt barrier function. In this review, we provide a critical analysis of the current clinical and laboratory evidence for endothelial dysfunction during severe P. falciparum malaria. Future investigations using state-of-the-art technologies such as mass cytometry and organs-on-chips to further delineate parasite-endothelial cell interactions are also discussed.
KEYWORDS: barrier function, cytoadherence, endothelial cell death, endothelial junctional proteins, endothelium, inflammatory mediators, malaria, P. falciparum
Introduction
Plasmodium species are intracellular protozoan parasites of erythrocytes. The life cycle of malaria parasites is complex, with asexual reproduction occurring in the mammalian host and sexual reproduction in the anopheline mosquito vectors.1 The parasites are transmitted to humans in the form of sporozoites through the bite of a female anopheline mosquito. After circulating briefly in the bloodstream, sporozoites invade hepatocytes in which they undergo asexual reproduction to form a large intracellular schizont that contains thousands of merozoites when they are mature within 5–15 d of sporozoite inoculation. The enlarged hepatocyte eventually bursts, releasing merozoites into the bloodstream where they rapidly invade erythrocytes via specific surface receptors. Approximately 50% of released merozoites effectively reinvade erythrocytes to initiate the erythrocytic cycle. Inside the erythrocyte, the parasite develops within a membrane-bound parasitophorous vacuole first as a trophozoite and then as a schizont. When the schizont matures, the infected erythrocyte ruptures, liberating merozoites that rapidly invade fresh erythrocytes in the general circulation, thus continuing the erythrocytic life cycle. Some merozoites develop into sexual forms (gametocytes), which are taken into the mosquito mid-gut with a blood meal. These may fuse to form a zygote and then undergo meiosis to form first an ookinete and later an oocyst in the gut wall. The oocysts burst, liberating large numbers of sporozoites that migrate to the salivary glands where they are injected into the human host during the next blood meal to continue the exoerythrocytic life cycle.
Because of the intravascular localization, infected red blood cells (IRBC) are in intimate contact with vascular endothelium throughout the erythrocytic cycle. The host-parasite interactions of this stage of the infection are responsible for the clinical manifestations of the disease that range from a mild febrile illness to severe and frequently fatal syndromes such as cerebral malaria and multi-organ failure. However, despite intense investigations, many questions remain regarding the role of endothelial cells in the pathogenesis of severe malaria. In particular, the part played by endothelial barrier function is controversial. Is barrier function compromised in severe malaria and to what degree? Is barrier dysfunction a direct effect of parasite adhesion or is it secondary to endothelial activation by pro-inflammatory mediators? Is there significant endothelial cell death? In this review, we will focus on the endothelial response to Plasmodium falciparum, the major cause of mortality and morbidity among the 5 known plasmodial species that infect humans. Endothelial pathology by P. vivax that has been associated with severe disease will also be discussed.
Endothelial cell barriers
Endothelial cells that line the vasculature separate the vascular and parenchymal compartments of all organs and thereby regulate gas exchange, metabolism, trafficking of immune cells and dissemination of blood borne infections. The homotypic and heterotypic cell-cell interactions formed on the lateral membrane that maintain paracellular barrier integrity include adherens and tight junctions, with a lesser contribution by gap junctions.2 Adherens junctions (AJ) are composed of VE-cadherin and nectins while tight junctions (TJ) are composed of junctional adhesion molecules (JAM), claudins, and occludin all of which are anchored to the endothelial cytoskeleton via various plaque proteins such as p120, afadin, catenins, and zonula occludens (ZO) (Fig. 1). Plaque proteins also serve to localize regulators of junction assembly and disassembly, including Src-family kinases (SFK) and vascular endothelial cell–specific phosphotyrosine phosphatase (VE-PTP). In general, endothelial barrier function is maintained by preserving transmembrane AJ and TJ proteins VE-cadherin, claudin-5 and occludin in an unphosphorylated state.2,3 AJ serve as the primary regulators of endothelial barrier function in most organs, with the notable exception of the blood brain barrier (BBB) where the thin microvascular endothelial cells are bound together to a large extent by TJ, creating a high resistance paracellular barrier. Permeability results when junctional proteins are relocated or their expression downregulated, as occurs in many inflammatory conditions.
Figure 1.
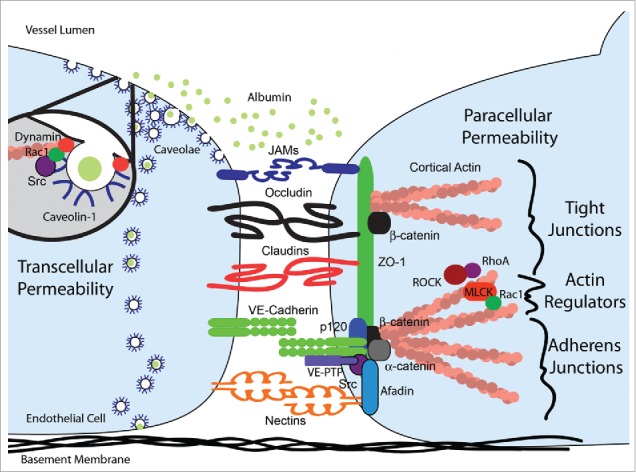
Schematic representation of the transcellular and paracellular endothelial permeability pathways. Transcellular permeability is mediated by the transcytosis of albumin through endothelial cells by membrane invaginations called caveolae. The process is regulated by dynamin which pinches off the neck and thereby internalizes caveolae in response to c-Src and Rac1 signaling. Paracellular permeability is regulated by tight (TJ) and adherens (AJ) junctions. Within each endothelial cell, TJ and AJ transmembrane proteins are anchored to the actin cytoskeleton by a multitude of plaque proteins including α-catenin and p120 plakoglobin for AJ and β-catenin and ZO-1 for both TJ and AJ. Src family kinases critically regulate paracellular permeability by regulating the homotypic or heterotypic association of transmembrane and plaque proteins and the cytoskeleton. The maintenance of cortical actin by myosin light chain kinase (MLCK) is regulated positively by Rac1 signaling and negatively by RhoA and Rho-associated protein kinase (ROCK) signaling.
In addition to the paracellular barrier maintained by junctional proteins, there has been increasing appreciation of the role of transcytosis that serves to transport albumin across resting endothelium under physiological conditions, but is increased as a result of injury and disease. Transcytosis of albumin, the major oncotic protein of serum, involves the intracellular trafficking of caveolae carrying albumin that has been taken up via multiple receptors.4 Invagination of caveolae leads to scission from the plasmalemma by the GTPase dynamin 2 and c-src.5,6 The vesicles move into the endothelial cell and fuse with the basolateral membrane and release albumin into the interstitial space. The process of albumin transcytosis in resting and thrombin-stimulated human lung endothelial cells is regulated by acid sphingomyelinase production of ceramide which results in increases in caveolin-1, the major structural protein of caveolae.7 Besides SFK, pathologic increases in transcytosis of albumin (transcellular leak) and paracellular leak are co-regulated by dynamin and Rac1.8 In addition to albumin, transcytosis transports immunoglobulins, transferrin, aminopeptidase P and numerous other molecules. Microvascular endothelial cells can regulate permeability by both transcellular and paracellular routes except for brain microvascular endothelial cells that have few caveolae under basal conditions.
Endothelial barrier function also depends on surrounding cell types which in turn reflect the primary function of a given organ system. For example, in the brain where maintaining strict metabolic homeostasis while preserving a high integrity barrier is paramount, the microvascular endothelial cells are surrounded by pericytes, a basement membrane, and astrocytes with foot processes that are in contact with neurons.9 The relative importance of stromal cells in maintaining the BBB phenotype is supported by recent evidence that pericyte-deficient mice have increased permeability to water and different solutes through an increase in transcytosis, a process that can be inhibited by the tyrosine kinase inhibitor imatinib.10 Interestingly, the distribution of junctional proteins in the mutant cells remained unaltered, although irregular stretches of endothelial overlap were commonly seen. An absence of pericytes also affected the normal polarization of the astrocyte end-feet, while the deposition of basement membrane proteins did not change. These results confirm that the fine regulation of endothelial barrier function is organ-specific and involves multiple cell types.
In vascular beds such as the lung, the thick cortical actin layer in microvascular endothelial cells contributes to determining cell shape and stabilizing endothelial AJ, TJ and focal adhesions through protein bridges to the actin cytoskeleton.11 Under basal conditions, cortical actin is preserved by Rac1 signaling which inhibits myosin light chain kinase (MLCK) and actin stress fiber formation. However, under pathologic conditions, the inflammatory mediators thrombin and TNF-α can activate intracellular SFK, calcium release, protein kinase C and RhoA signaling which together result in Rho-associated protein kinase (ROCK)-mediated phosphorylation of MLCK. MLCK activation generates actinomyosin-mediated contractile units with subsequent changes in cell shape, disassembly of the cortical actin layer and the formation of cytoplasmic actin stress fibers. Concurrently, pathways upstream of ROCK/MLCK also induce tyrosine phosphorylation of claudin-5, occludin, VE-cadherin and plaque proteins. Phosphorylation is driven by SFK and RhoA activation and the inhibition of phosphatases such as VE-PTP. The result is disassembly of junctional protein complexes and internalization of transmembrane receptors that, coupled with stress fiber induced cell retraction, lead to increased paracellular permeability.
In addition to well established endogenous barrier protective signaling pathways such as the ligand/receptor pairs sphingosine-1-phosphate (S1P)/S1P receptor 1(S1P1R), Angiopoietin-1 (Ang-1)/TEK receptor tyrosine kinase2 (Tie2), activated protein C (APC)/endothelial protein C receptor (EPCR) and Slit guidance ligand 2 (Slit2)/Roundabout guidance receptor 4 (Robo4),12,13 new barrier protective pathways have been described. These include Frizzled7 which localizes to endothelial cell-cell junctions and prevents β-catenin activation and VE-cadherin disruption through Wnt signaling.11 Adrenomedulin, and the related protein intermedin or adrenomedulin 2, are also known to enhance barrier function through the GPCR calcitonin receptor-like receptor (CRLR).14 In response to adrenomedulins, CRLR partners with receptor activity-modifying proteins or RAMPS to increase cAMP and Rac1 signaling while decreasing p38MAPK signaling to reduce endothelial actin stress fiber formation, permeability and pro-inflammatory molecule expression. These complex pathways display significant crosstalk where, for example, EPCR signaling regulates S1P1R activation through PAR-115 and Tie2 transactivation through PAR-3 cleavage16 to maintain barrier function. The maintenance of endothelial integrity ultimately depends on the balance of disruptive and protective pathways.
Clinical evidence of barrier dysfunction in severe malaria
By far the most studied and most controversial endothelial barrier in patients with falciparum malaria is the BBB that is linked to the clinical syndrome called cerebral malaria (CM). The complication is characterized by coma, seizures and focal neurological deficits. This complication carries a mortality rate of 15–20% despite optimal therapy. CM also results in long-term cognitive defects in about 20% of survivors. CM is more common in children than adults. Both pediatric and adult BBB function has been studied clinically using fluorescein angiography of the retina,13 cerebrospinal fluid (CSF) protein analysis,17-19 and more recently magnetic resonance imaging (MRI) studies of the brain.20,21 In children with cerebral malaria, there is clinical evidence of increased intracranial pressure leading to brainstem herniation.22,23 In a recent ante-mortem MRI study of 168 children with CM in Malawi, brain swelling emerges as the most reliable predictor of severity.20 The cause of brain swelling is postulated to be due to cytotoxic edema from impaired perfusion, metabolic injury and cell death. Endothelial cell activation leading to barrier disruption may also occur. Increase in blood volume due to vascular congestion and increased cerebral blood flow are other possibilities. The absence of classical postmortem findings of cerebral edema is attributed to the rapidity with which death occurs in children with CM.
In adults, cerebral edema has not been seen in several imaging studies.21,24 Mild diffuse brain swelling is found in a variety of anatomical sites. This swelling is partly attributed to venous congestion by the sequestered IRBC mass causing increased cerebral blood volume.25 Brain herniation is not seen, nor is there evidence of toxigenic or vasogenic edema.21 This would be in keeping with a lack of detection of albumin or IgG leakage into the CSF of adult patients with CM, in contrast to the significant leakage seen during bacterial meningitis.19 More importantly, none of the MRI findings are different between patients with cerebral and non-cerebral malaria, and there are no differences between fatal and nonfatal infections. However, more marked changes in BBB have been observed in postmortem samples, including endothelial vacuolar degeneration and junctional protein disruption.26-28 Whether these findings reflect the disease process or postmortem changes in the BBB is unclear. To date, the only adequately powered randomized trial on modulating BBB function during cerebral malaria was on the use of mannitol. Unfortunately, mannitol infusion failed to show therapeutic benefit but instead was associated with prolongation of coma.24
The contribution of leukocytes to endothelial barrier function in CM is also controversial. Histological sections of the adult brain in CM are notable for the almost complete absence of leukocyte infiltrates.26 In fact, the intriguing question has been why leukocytes are not recruited when the pro-inflammatory milieu for recruitment to occur is present. More recent studies in pediatric patients have revealed the presence of some monocytes and platelets in the cerebral microvascular.17,27,29 There is also a suggestion that there may be systemic neutrophil activation which may contribute to cerebral pathology through soluble mediators such as myeloperoxidase and elastase that are released.30
In contrast to the findings in the brain, features consistent with acute lung injury are frequently found in the pulmonary microvasculature at autopsy of both adult and pediatric patients with severe falciparum malaria.31-33 The findings include edema, microthrombi and leukocyte infiltration. The microvessels are densely packed with IRBC. Leukocytes, particularly malaria pigment laden monocytes and less so neutrophils, are also frequently found. Clinical studies on lung function in adult and pediatric patients support the presence of capillary dysfunction manifested as impaired gas exchange and reduced peripheral reactive hyperemic index.34,35 However, despite evidence of acute lung injury, the presence of overt pulmonary edema in severe malaria varies from 5 to 25% in adults and less than 10% in children.32 In the latter group, respiratory distress is believed to be mainly central nervous system (CNS)-driven as a response to systemic metabolic acidosis.36
Pulmonary manifestations are also associated with infection with the parasite P. vivax, an infection that has often been considered mild and chronic rather than fulminant. There is evidence of alveolar-capillary membrane permeability37,38 and systemic endothelial activation39 despite a lower parasite burden and the relative absence of significant IRBC sequestration.
In addition to the brain and lung as major sites of pathology, acute kidney injury leading to renal failure approaches 40% in adults and 10% in children under 5 years of age.40,41 Renal pathology has been largely attributed to acute tubular necrosis secondary to sequestration of IRBC and accumulation of mononuclear cells within both glomerular and peritubular capillaries.42 Indirect measures of parasite sequestration and monocyte activation levels have been found to correlate with renal failure requiring replacement therapy.43 Mild to moderate glomerulonephritis has also been observed as seen by glomerular hypercellularity and collapse.40 Histological assessment of renal tissue from severe malaria patients for ZO-1 distribution reveals marked and diffuse reductions in glomerular ZO-1 staining including within glomerular endothelial cells suggestive of extensive paracellular junction disruption.44
Mechanisms of endothelial dysfunction in severe malaria
Endothelial permeability in response to IRBC has been studied using different sources of primary endothelial cells or endothelial cell lines in vitro.45-49 Permeability is assessed either as a change in transendothelial resistance, or the flux of fluorescein-labeled solutes such as dextran or albumin. While there is general agreement that IRBC do induce permeability, the mechanisms appear to differ depending on the experimental design and cell types used. The unresolved issues include i) whether direct contact between IRBC and endothelium is necessary; ii) what inflammatory mediators are involved; and iii) whether there is associated endothelial apoptosis or cell death. Each of these issues will be discussed in the following sections, with a summary schematic representation in Fig. 3.
Figure 3.
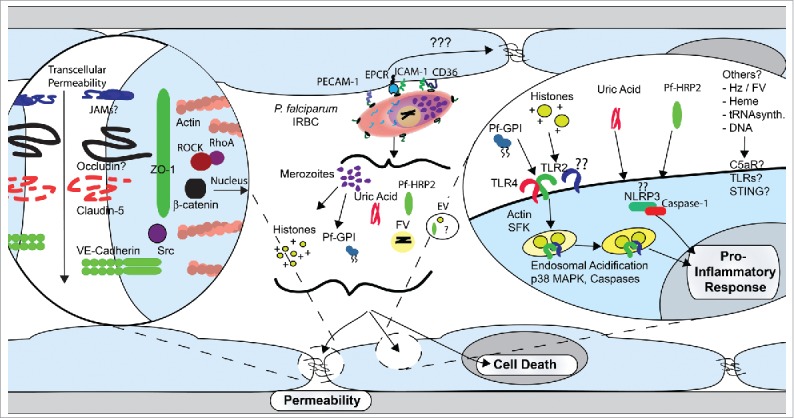
Proposed model of the induction of endothelial proinflammatory response and barrier dysfunction by P. falciparum. In the second half of the erythrocytic life cycle, P. falciparum-infected erythrocytes (IRBC) either adhere to endothelial receptors or are in close contact with microvascular endothelium. Adhesion through receptors such as CD36, ICAM-1 or α5β1 integrin may lead to intracellular signaling events with resultant actin cytoskeletal changes and disruption of junctional protein expression. In addition, numerous host and parasite products are released in the proximity of endothelial cells at IRBC rupture, where they may induce a pro-inflammatory response or exert a disruptive effect on endothelial barrier function. Transcytosis of albumin and other host molecules may further add to vascular leakage.
Cytoadherence
Over a century ago, Marchiafava and Bignami made the seminal observation in postmortem examination that IRBC are sequestered in the capillaries and postcapillary venules of the brain, resulting in the reduction of the vascular lumen to create a mechanical obstruction to blood flow (Fig. 2).50 Detailed histopathological studies of human postmortem tissues performed in the past 3 decades show that sequestration is in fact widespread in infected patients, and the degree and organ specificity of sequestration correlated with clinical manifestations.50-53 More recently, obstruction of the microcirculation that is reversed after anti-malarial therapy has also been documented in vivo in acutely infected patients.25
Figure 2.
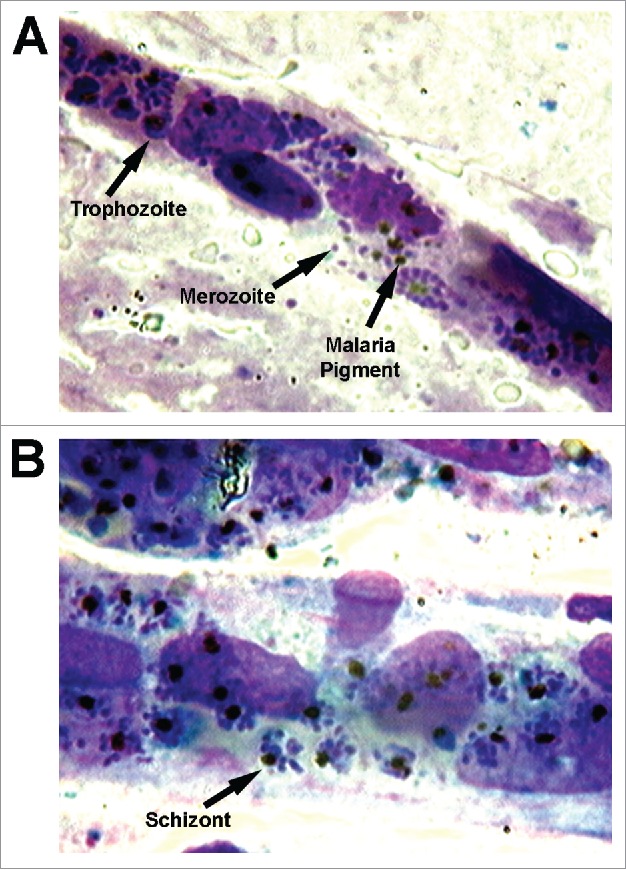
Sequestration of P. falciparum-infected erythrocytes in human brain microvessels. Post-mortem brain smears taken from an adult Vietnamese patient who died from cerebral malaria showing intense packing of IRBC in the microvasculature. The brain smears were made by placing brain tissue obtained within 6 hours of death between 2 glass microscope slides, pressing them together, and then sliding them apart to obtain thin smears.51 This method preserves long fragments of capillaries and venules. Slides were fixed in absolute methanol and stained by the reverse Field's method. The blood vessels were examined at 1000x magnification under oil immersion using an Olympus BH2 microscope. The arrows in (A) indicate an IRBC with a mature trophozoite, and a free merozoite and malaria pigment after schizont rupture. The arrow in (B) indicates an intact schizont with a cluster of merozoites within an IRBC. Note the absence of any inflammatory infiltrate. The micrographs, courtesy of Dr. K. Silamut, Mahidol Oxford Research Unit, Bangkok, Thailand, were originally published in Blood. Ho M. EPCR: Holy Grail of Malaria Cytoadhesion? 2014;123:157–159 © the American Society of Hematology.
Sequestration results from the adhesion, or cytoadherence, of IRBC to vascular endothelial cells, and the process is mediated by the variant parasite ligand Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) expressed on the surface of IRBC, and endothelial receptors, of which a number have been implicated.54,55 Cytoadherence occurs to any significant extent only with P. falciparum that infects human erythrocytes, which explains why there is no appropriate animal model to study this phenomenon. The majority of PfEMP1 variants bind to CD36, an 88-kD class B scavenger protein expressed on a number of cell types, including macrophages and endothelial cells.54 Adhesion of IRBC to CD36 has been shown to recruit CD36 and actin to the site of attachment in human dermal microvascular endothelial cells through the activation of p130 CAS and Src family kinases.56 Although there is no clear evidence of stress fiber formation, the actin cytoskeletal rearrangement may lead to a change in cell shape and hence affect the integrity of adherens junctions. Moreover, adhesion of IRBC to CD36 recruits α5β1 integrin to the same site, which strengthens the adhesive force.57 α5β1 has been shown to induce breakdown of endothelial barriers through binding to cANGPTL4 which can directly activate Rho-GTPase Rac1/PAK signaling to cause downstream disruption of VE-cadherin and claudin 5.58 Paradoxically, α5β1 also appears to have a critical role in barrier maintenance through localization of the integrin to cell-cell contacts and adhesion to extracellular matrix such as fibronectin.59 These divergent results suggest that the subcellular location of integrin engagement (junctional/basolateral versus apical) may differentially regulate endothelial permeability. Whether engagement of α5β1 by PfEMP1 affects endothelial permeability has not been explored.
IRBC adhesion to ICAM-1 can also lead to similar cytoskeletal changes. Induction of ICAM-1 clustering by antibody-coated beads on human umbilical vein endothelial cells (HUVEC) that do not express CD36 results in Src- and Pyk2-dependent phosphorylation of VE-cadherin that is implicated in the promotion of neutrophil transmigration via the paracellular pathway.60 Binding of ICAM-1 on human lung endothelial cells by neutrophils also leads to increased transcellular permeability through activation of caveolin-dependent albumin transport.61 Interestingly, IRBC adhesion to ICAM-1 on a brain endothelial cell line (hCMEC/D3) has been linked to the formation of endothelial cup-like structures and trogocytosis of membrane fragments of IRBC, and the processes are associated with disruption of barrier integrity.62 As endothelial docking structures or transmigrating cups are well described during leukocyte transmigration,63 their formation around adherent IRBC that do not transmigrate is somewhat surprising. Moreover, transmigrating cups around neutrophils has been shown to be a mechanism for the maintenance rather than disruption of barrier function.64 These discrepancies will need to be resolved in future studies.
IRBC adhesion may also affect permeability indirectly by inducing tissue factor production on endothelial cells and subsequent activation of the coagulation cascade,65 as evidenced by elevated levels of circulating thrombin-antithrombin III complex,66,67 the presence of activated thrombin68 and fibrin deposition69 on brain microvascular endothelium in postmortem brain tissues. Thrombin is known to induce permeability through PAR-1 which heterodimerizes with PAR-3 leading to Gα13-dependent calcium signaling, actin stress fiber formation and transient increases in endothelial permeability.70 Moreover, a subset of parasites from patients with CM has been shown to bind to EPCR, and may compete with APC for the binding to this receptor.55,71,72 As APC exerts a protective effect on thrombin induced barrier dysfunction, IRBC adhesion may exaggerate the barrier dysfunction elicited by thrombin.71,73,74
It should be emphasized that the in vitro evidence for the disruption of barrier function as a direct effect of cytoadherence is not robust, as each process has been demonstrated by only a few IRBC, or by using surrogates, such as antibody or recombinant PfEMP1-coated beads. It is conceivable that the number of interactions could be much higher in vivo, in view of the fact that microvessels are densely packed with IRBC (Fig. 2) compared with the relatively sparse IRBC adhesion to endothelial monolayers in vitro. It has also been demonstrated that IRBC adhesion at a given time may be mediated by multiple, some as yet unidentified, endothelial receptors,75 so that the downstream effect is likely the summation of the activation of multiple pathways.
Host inflammatory mediators
A growing number of host-derived pro-inflammatory cytokines, chemokines, metabolic and lipid mediators known to increase endothelial permeability has been shown to be elevated in severe malaria.76,77 These inflammatory mediators are identified mostly by measuring circulating levels using various immunoassays, or more recently by transcriptomics. Very few of the mediators have been studied functionally in relation to the infection, and as a result, the elevated levels do not distinguish between whether a particular inflammatory mediator actually contributes to disease pathology or merely serves as a biomarker of severity.
The most studied pro-inflammatory cytokine in relation to severe falciparum malaria is tumor necrosis factor-α (TNF-α) following reports of elevated levels of this cytokine in African children that correlated with severity and mortality.78 Cytokine levels have also been measured in P. vivax malaria. TNF-α levels during paroxysms of fever in vivax malaria are either much higher than in severe and fatal falciparum malaria,79 or levels are similar in the 2 infections.39 Among its protean effects, TNF-α could induce adhesion molecule expression and endothelial barrier dysfunction. However, although animal studies with the murine parasite P. berghei suggested the efficacy of anti-TNF-α therapy,80 results of anti-TNF-α in human clinical trials in both adult and pediatric patient populations were disappointing.81,82 The lack of efficacy of the monotherapy, as in the case of sepsis,83 was likely due to the fact that by the time patients present for treatment, other inflammatory or inhibitory pathways would have been activated. In at least the adult patients, the anti-inflammatory cytokine IL-10 was also found to be markedly elevated,84 which suggests that perhaps the ratio and time course of TNF-α to IL-10 production may be more predictive of outcome.
Another mediator that has been studied extensively in malaria is nitric oxide (NO). NO could derive from different nitric oxide synthase (NOS) isoenzymes in neuronal tissue (nNOS), or vascular endothelium (eNOS), or from an inducible form found in phagocytic cells (iNOS). The isoenzyme eNOS is most concerned with regulation of microvascular hyperpermeability. eNOS mediates increased endothelial permeability in response to VEGF through tyrosine phosphorylation of VE-cadherin and Rho GTPase-dependent actin stress fiber formation.85
Nitric oxide has been postulated to have a protective role in severe falciparum malaria. Similar to its anti-inflammatory role on leukocyte recruitment,86 it inhibits adhesion of IRBC on HDMEC in vitro.87 In patients with both P. falciparum and P. vivax malaria, an inverse relationship between the severity of the infection and nitric oxide production/nitric oxide synthase expression has been demonstrated by several studies.35,88 The decreased NO production and its precursor L-arginine were attributed to a deficiency of arginine and increased arginase activity. These derangements are compounded by the increased cell-free hemoglobin circulating in malaria patients secondary to hemolysis, leading to increased NO scavenging and plasma L-arginine catabolism, and an overall reduction in NO bioavailability as seen in sickle cell disease.89 Increased arginase activity has also been demonstrated in monocytes of the M2 phenotype in children with falciparum malaria.90 However, in all the above studies concerning the role of NO in severe malaria, microvascular permeability was not directly measured. The readouts were reactive hyperemia-peripheral arterial tonometry, which is more a measure of vascular tone, and gaseous exchange that could be affected by perfusion. Clinical trials of L-arginine in patients with severe falciparum malaria have led to some improvement in vascular tone,35 but larger randomized trials will be needed to assess effects on disease severity or mortality. A clinical trial in pediatric malaria showed that inhaled nitric oxide did not affect biomarkers of severe disease.91
More recently, the role of NO in severe malaria has also implicated the angiogenic factor Ang-2 which is stored in Weibel-Palade bodies (WPB) in endothelial cells and secreted in conjunction with von Willebrand Factor.92 The decreased vascular NO bioavailability found in severe malaria is proposed to increase exocytosis of WPB with a resultant increase in Ang-2 release. Independently of NO, elevations in Ang-2 in severe malaria are strongly predictive of disease severity, particularly when measured as a ratio with its barrier protective homolog Ang-1.93 Ang-2 is known to disrupt AJ and can lead to lung injury in mice by mediating the dissociation of Ang-1 from its receptor Tie2 leading to VE-cadherin disruption.94,95 While murine models of cerebral malaria suggest the Ang-1/-2 axis plays a role in severe disease,96 analysis of altered Ang-1, Ang-2 and Tie2 protein levels in human brain specimens from adults patients with CM did not discriminate CM from non-CM cases.97 At present, Ang-2/Ang-1 ratios remain an excellent marker for severe disease, but whether this pathway contributes critically to human cerebral malaria or acute lung injury as seen in sepsis remains unclear.98
Host-derived products released at the time of red blood cell rupture such as free heme and its parent compound, hemoglobin, have also been implicated in pediatric99 and adult100 severe malaria as well as in endothelial dysfunction in malaria.101 Heme is known to disrupt endothelial barriers directly through induction of free radical generation and mitochondrial reactive oxygen species signaling in endothelial cells.102 Heme can also indirectly induce endothelial permeability through induction of histone- and DNA-rich neutrophil extracellular traps103 or by complement activation which induces the formation of interendothelial gaps in HUVEC.104 In addition, the barrier disruptive effects of heme could be mediated by NLRP3,105 TLR4106 or other MyD88 signaling pathways.107 Heme induced inflammatory effects can be inhibited by hemoglobin- and heme-binding proteins such as haptoglobin108 and hemopexin,109 or detoxification of hemoglobin breakdown products by nitric oxide,110 heme oxygenase-1 or carbon monoxide which converts toxic methemoglobin to carboxyhemoglobin.111
Parasite products
In addition to host pro-inflammatory molecules, parasite products may be released as soluble factors during the intra-erythrocytic growth phase, or released as insoluble factors when the infected red cell and mature parasite ruptures, releasing merozoites, food vacuoles and hemozoin. Plasmodium falciparum histidine rich protein 2 (PfHRP2) is an example of a parasite product that is transported from the parasitophorous vacuole to the cytoplasm and is released throughout the life cycle and at the time of red cell rupture.112 PfHRP2 has been shown to activate the NLRP3 inflammasome activation in the brain endothelial cell line hCMEC/D3, leading to the production of IL-1β- and MyD88-dependent induction of vascular permeability.113
More potent parasite products known to mediate endothelial leakage are released on IRBC rupture. In P. falciparum infection, merozoites are released every 48 hours and contain glycophosphatidyl inositols (GPI) that anchor parasite proteins to the plasma membrane. GPI are true pathogen associated molecular patterns that are unique to protozoan parasites, and are recognized by TLR2/TLR1 and to a lesser extent TLR4 on human and mouse macrophages.114 Malarial GPI have been shown to stimulate NO production and upregulate the expression of the adhesion molecules ICAM-1, VCAM-1 and E-selectin on HUVEC, leading to increased leukocyte and parasite adhesion.115 The effect of GPI can be inhibited by the Src-family kinase antagonist herbimycin A. Merozoite proteins, independently of TLR2 stimulation, can also increase endothelial permeability in a Src family kinase-dependent manner by inducing the redistribution of ZO-1, VE-cadherin and claudin-5 away from cell-cell junctions in HDMEC.49
As part of hemoglobin degradation, P. falciparum generates a relatively insoluble crystal of heme monomers called hemozoin or malaria pigment in parasitophorous food vacuoles. Similar to merozoites, food vacuoles are released every 48 hours at the time of IRBC rupture. Purified food vacuoles have been found to induce endothelial cell death and barrier dysfunction in HDMEC.49 Food vacuoles may also activate complement and coagulation pathways,116 both of which could increase endothelial permeability through the production of C5a117 and thrombin, respectively. Although hemozoin itself does not induce endothelial barrier dysfunction,47,49 it has been shown to activate NF-κB signaling pathways as shown by increased IL-8 and MCP-1 (CCL2) production in HUVEC.118
Other parasite derived products such as DNA,119,120 histones,121 tRNA synthetase,120 uric acid122 and heat shock proteins (HSP)123 have been shown to activate the host immune system. In the case of histones, they can disrupt endothelia into renal arteries l barrier function. P. falciparum histones H3 and H4, which share >95% homology with human histones, were shown to increase endothelial permeability in primary human lung cells based on their strongly cationic charge.121 Histone-induced disruption of VE-cadherin, claudin-5 and ZO-1 expression was partially Src family kinase-dependent, and may also involve caspase-independent cell death. In vivo, the direct injection of bovine histones, which share high homology to P. falciparum histones, into murine renal arteries induced TLR2/4-dependent endothelial cell death and edema culminating in acute kidney injury.124 Other soluble parasite proteins, such as the DNA-binding proteins high mobility group box (HMGB) 1 and 2, may induce permeability through activation of RAGE as does its human counterpart.125 Plasmodia also produce HSP, and PfHSP70 is thought to have adjuvant and pro-inflammatory activities through TLR2/4 and TLR4/MyD88 activation respectively.123
The importance of parasite products in inducing endothelial permeability is underscored in a recent study which demonstrated that as yet unidentified product(s) released on IRBC rupture activated β-catenin signaling and subsequent disruption of claudin-5 expression in primary human brain endothelial cells.126 Inhibition of β-catenin–induced T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factor activity in the nucleus prevented the disruption of endothelial junctions. Nuclear translocation of β-catenin and TCF/LEF is known to mediate crosstalk between AJ and TJ following VE-cadherin disruption.127 Nuclear β-catenin stabilizes TCF/LEF transcription factor binding to the claudin-5 promoter and suppresses claudin-5 production. These studies are consistent with previous results showing late stage (schizont) parasite extracts and merozoite proteins decreased endothelial claudin-5 production in conjunction with disruption in VE-cadherin localization to cell-cell junctions.49 Interestingly, the blockade of the angiotensin II type 1 receptor (AT1) or stimulation of the type 2 receptor (AT2) abrogated IRBC–induced activation of β-catenin and prevented the disruption of brain endothelial monolayers.
Most recently, IRBC-derived extracellular vesicles (EV) have been shown to transfer host microRNA451a (miR451a) that silences cav-1 and atf2 mRNA.128 Endothelial permeability in bone marrow endothelial cells was augmented by the transfer of EV containing RNA-induced silencing complex (RISC) proteins, including the Argonaut protein Ago2. The findings are consistent with the reported occurrence of trogocytosis,62 and the contact-dependent but cytoadherence-independent activation of endothelial cells by intact IRBC. Parasite-derived proteins are also contained within EV,129,130 so that there may be intracellular delivery of parasite products to endothelial cells. Depending on the route of delivery, parasite products may elicit different signaling pathways with potentially different outcomes. A difference in the outcome of intracellular vs. extracellular activation has been shown for LPS that activates via TLR4 on the cell surface, but induces non-canonical inflammasome activation involving caspase 4/11 when presented intracellularly.131 These findings suggest that P. falciparum products may target multiple pathways in the maintenance of endothelial integrity.
Cell death
Closely linked to permeability is whether endothelial activation is associated with cell death. Endothelial cell apoptosis has been implicated as a mechanism of increased endothelial permeability,46,132-134 and different clinical isolates of P. falciparum appeared to induce apoptosis to varying degrees in primary endothelial cells derived from human lung and brain.133 The induction of endothelial apoptosis was sensitive to the environmental pH and required direct contact between the parasite and the endothelial cell, although adhesion to specific receptors was not essential. Moreover, the extent of induced apoptosis in the 2 endothelial cell types was inhibited by pan-caspase or caspase-8 inhibitors and varied with the isolate. Analysis of parasite gene transcripts suggest that the activation of different parasite pathways, such as plasmodium apoptosis–linked pathogenicity factors, could play a part in the observed susceptibility to cell death. The increase in endothelial permeability and apoptosis induced by IRBC has also been linked to Rho A activation and was partially inhibited by the ROCK inhibitor fasudil but the parasite agonists, host receptors and downstream effectors, such as actin stress fiber formation, were not determined.134
In other studies, cell death is either not observed47,49,135 or could not be attributed to the activation of caspases. Treatment of primary dermal and lung endothelial cells with P. falciparum histones has been shown to induce cell death which was insensitive to a pan-caspase inhibitor.121 In addition, PfHRP2 has been shown to induce redistribution of the tight junction protein claudin-5 leading to compromised integrity of the brain endothelial cell line hCMEC/D3.113 Although the parasite protein activated caspase 1, apoptosis and pyroptosis were excluded by the absence of DNA fragmentation and the lack of an inhibitory effect of a pan-caspase inhibitor at 6 hours when a change in permeability was first detectable. Nicking of DNA was however evident at 24 hours.
The extent of endothelial cytotoxicity in vivo also remains to be determined. Based on histopathological53 and transmission electron microscopy26 studies of postmortem tissues, generalized endothelial cell death is not a prominent feature of acute falciparum malaria, consistent with the largely reversible nature of neurologic dysfunction in those that survive the infection. However, in a small autopsy series of pediatric and adult with CM, cleaved caspase-3, the terminal effector of apoptosis pathways, was seen in >70% of brain endothelial cells examined.136 The presence of ring hemorrhages around cerebral microvessels at autopsy,53 or as part of the retinopathy observed in patients137 would also be compatible with the occurrence of endothelial cell death. Indeed, the high levels of circulating nucleosomes of both parasite and human origins detected in the plasma of patients with severe falciparum malaria, compared with the much lower levels detected in septic patients,138 would suggest that cell death in this infection may be more extensive than previously appreciated. The host cell types that are affected and cell death pathways involved remain to be defined.
Future directions
Imaging studies
Current understanding of the pathophysiology of severe malaria is largely based on autopsy studies. New imaging techniques are now available to perform some of the studies in the clinical setting. Novel ultrasound techniques that allow high resolution imaging of microvessels (10µm) could be performed non-invasively to quantify microvascular perfusion within the CNS in severe malaria patients in conjunction with MRI studies to better correlate the relationship between brain swelling and sequestration.139 In fatal cases, ante-mortem ultrasound imaging would also be useful to correlate microvascular perfusion deficits with abnormalities found at autopsy.
Basic questions of whether changes in endothelial permeability in severe malaria are caused by trans- vs. paracellular permeability and in what organs also remain to be addressed. Significant increases in transcellular permeability can occur in conjunction with paracellular permeability as shown in murine models of endotoxin-induced acute respiratory distress syndrome,61 or transcellular permeability can occur independently as seen during dengue virus infection140 or HMGB1 stimulation of mouse lung microvascular endothelial cells in vitro.141 Detailed analysis of clinical tissue samples for the activation of key signaling pathways associated with dissociation of junctions and transcytosis may provide additional insight into potential therapeutic targets. In this regard, mass cytometry has become an extremely powerful tool for the detection of over 100 molcules per cell using metal-labeled antibodies that could allow for the simultaneous characterization of multiple cell types, inflammatory mediators, junctional proteins, and activation of signaling molecules.142 Moreover, localizing the presence and relative abundance of parasite products such as histones, GPI, and PfHRP2 at sites of apparent barrier dysfunction might also provide insight into which agonists could be targeted for blockade. Mass cytometry based techniques could be supplemented by CLARITY protocols which entail embedding fixed tissues, primarily brain, into a synthetic hydrogel matrix followed by electrophoresis to remove lipids.143 The resulting optically transparent tissue allows high resolution 3D imaging of large (cm) sections of tissue and could provide a more comprehensive overview of endothelial junctional protein distribution in severe malaria. Beyond the brain and lung, applying these methods to the analysis of renal tissue from patients with severe malaria could provide additional insight into acute kidney injury in both children144 and adults43 which is associated with renal endothelial cell hypertrophy and cytoplasmic vacuolation as well as monocyte infiltration.42
Organs-on-chips
The lack of an appropriate animal model has hampered the study of cytoadherence in severe falciparum malaria, including its effect on barrier function. Recent advances in 3D co-culture techniques145 that can recapitulate the complex interactions of human cells in physiological matrices such as collagen I or fibronectin constitute a promising approach for determining the relative contribution of specific cells to organ function in states of health and disease.146 Coculture models of the human blood brain barrier including pericytes and astrocytes along with brain endothelial cells under continuous flow (1dyne/cm2) have confirmed a long suspected role for both stromal populations in regulating BBB function.147 Cocultures in this model showed a 2-fold increase in barrier integrity as measured by 3kDa dextran flux and 2- to 5-fold increase in responsiveness to TNF-α as measured by IL-6, IL-8 and G-CSF production compared with endothelial cells alone. Neurons have also been integrated into microfluidic chips containing endothelial cells, astrocytes and pericytes, which results in a 5-fold decrease in 10kDa dextran flux in comparison to endothelial cells alone.148 Moreover, these coculture systems allow for the examination of neuronal calcium signaling.149 Similarly, 3D cultures of lung epithelial cells, fibroblasts and endothelial cells to recapitulate the features of the small airways including barrier function and ciliary movement on epithelial cells have been used to assess the effects of drugs on leukocyte recruitment in inflammatory lung diseases and their mechanism of action.150 Microfluidic models have also allowed for analysis of cellular interactions within microvascular networks (10–100μm) using either de novo generated vascular networks in 3D matrices151 or in patterned devices devoid of matrices.152
The microfluidic models would be ideal for studying endothelial-IRBC interactions in capillaries (5–15µm) and post-capillary venules (30–70µm) where host-pathogen interactions occur in vivo. The hemodynamics of IRBC-endothelial interaction in microfluidic microvessels are likely to vary significantly from the more traditional 2D cultures, including changes in shear stress through high variability in hematocrit, viscosity and parasitemia within microvessels which can lead to vessel obstruction, changes in mechanoreceptor signaling and in turn endothelial function.137 Although not in direct contact with IRBC, stromal cells such as pericytes, astrocytes, neurons, fibroblasts are likely to be exposed to inflammatory mediators that in turn would modulate endothelial function. There is suggestive clinical evidence for abnormalities in some of these cell types in falciparum malaria, as shown by pericyte and axonal degeneration along with microglial and astrocyte end foot retraction,26,27 but the full extent of the involvement of these cells in endothelial barrier function remains to be systematically examined.
Metabolomics
A relatively unexplored area of study is the effect of metabolites on endothelial barrier function. Imbalances in common metabolites have been shown to affect immune functions.153 In P. falciparum malaria, an increase in permeability of an immortalized brain endothelial cell line (hCMEC/D3) was shown to occur in an acidic environment attributed to parasite metabolic products.45 As metabolic acidosis secondary to lactate production by both host and parasites is one of the main indicators of a poor outcome of severe malaria36 lactic acid too could contribute to barrier dysfunction. Moreover, late stage P. falciparum in culture releases high levels of amino acids such as histidine and glutamate.154 Histidine can be converted by histidine-L-decarboxylase to histamine which is elevated in severe malaria155 and is a well described barrier disrupting agent acting through the G-protein coupled receptor H1 leading to subsequent RhoA/ROCK-dependent actin contraction.156 Extracellular glutamate can also increase BBB permeability in vitro148 and in vivo through the GPCR mGluR pathways.157 In a mouse model of CM with the murine parasite P. berghei, CNS levels of glutamate are found to be increased while glutamine is decreased at the onset of neurologic dysfunction.158 Blockade of glutamine breakdown to glutamate using chemical inhibitors administered at the time of neurologic deterioration increased glutamine levels, reversed BBB permeability and brain swelling and rescued mice from CM. The source of excess glutamate in vivo remains unclear although both host cells and parasites have been implicated.
In future studies, inhibition of parasite metabolic pathways by genetic manipulation using CRISPR-Cas9 technology may shed further light on the relative importance of specific parasite derived metabolites.159 As well, confirming the timing, location and severity of metabolic dysfunction in human CM by analyzing tissue and fluid samples (e.g. blood and CSF) with metabolomics techniques including mass spectrometry and nuclear magnetic resonance analysis will be important. Ultimately, advances in metabolomics analysis in vivo and in vitro may provide insight into metabolic pathways with ideal characteristics for intervention, such as the ability to be rapidly turned off or switched on, as has been attempted with L-arginine administration to boost NO production.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Support for the research by the authors was provided by an operating grant from the Government of Canada/Canadian Institutes of Health Research (CIHR) to MH (MT14104) and a studentship to MRG.
References
- [1].Cowman AF, Healer J, Marapana D, Marsh K. Malaria: biology and disease. Cell 2016; 167:610-24; PMID:27768886; http://dx.doi.org/ 10.1016/j.cell.2016.07.055 [DOI] [PubMed] [Google Scholar]
- [2].Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci 2013; 126:2545-9; PMID:23781019; http://dx.doi.org/ 10.1242/jcs.124529 [DOI] [PubMed] [Google Scholar]
- [3].Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, Persidsky Y, Ikezu T. Phosphorylation of claudin-5 and occludin by Rho kinase in brain endothelial cells. Am J Pathol 2010; 172:521-33; http://dx.doi.org/ 10.2353/ajpath.2008.070076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Merlot AM, Kalinowski DS, Richardson DR. Unraveling the mysteries of serum albumin-more than just a serum protein. Front Physiol 2014; 5:1-7; PMID:24478714; http://dx.doi.org/ 10.3389/fphys.2014.00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tiruppathi C, Finnegan A, Malik AB. Isolation and characterization of a cell surface albumin-binding protein from vascular endothelial cells. Proc Natl Acad Sci U S A 1996; 93:250-4; PMID:8552615; http://dx.doi.org/ 10.1073/pnas.93.1.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. Endothelial cell-surface gp60 activates vesicle formation and trafficking via Gi-coupled Src kinase signaling pathway. J Cell Biol 2000; 150:1057-69; PMID:10973995; http://dx.doi.org/ 10.1083/jcb.150.5.1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kuebler WM, Wittenberg C, Lee WL, Reppien E, Goldenberg NM, Lindner K, Gao Y, Winoto-Morbach S, Drab M, Mühlfeld C, et al.. Thrombin stimulates albumin transcytosis in lung microvascular endothelial cells via activation of acid sphingomyelinase. Am J Physiol Lung Cell Mol Physiol 2016; 310:720-32. [DOI] [PubMed] [Google Scholar]
- [8].Armstrong SM, Khajoee V, Wang C, Wang T, Tigdi J, Yin J, Kuebler WM, Gillrie M, Davis SP, Ho M, et al.. Co-regulation of transcellular and paracellular leak across microvascular endothelium by dynamin and Rac. Am J Pathol 2012; 180:1308-23; PMID:22203054; http://dx.doi.org/ 10.1016/j.ajpath.2011.12.002 [DOI] [PubMed] [Google Scholar]
- [9].Daneman R, Prat A. The blood – brain barrier. Cold Spring Harb Perspect Biol 2015; 7:1-24; http://dx.doi.org/ 10.1101/cshperspect.a020412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al.. Pericytes regulate the blood-brain barrier. Nature 2010; 468:557-61; PMID:20944627; http://dx.doi.org/ 10.1038/nature09522 [DOI] [PubMed] [Google Scholar]
- [11].García-Ponce A, Citalán-Madrid AF, Velázquez-Avila M, Vargas-Robles H, Schnoor M. The role of actin-binding proteins in the control of endothelial barrier integrity. Thromb Haemost 2014; 113:20-36; http://dx.doi.org/ 10.1160/TH14-04-0298 [DOI] [PubMed] [Google Scholar]
- [12].Hawkes M, Elphinstone RE, Conroy AL, Kain KC. Contrasting pediatric and adult cerebral malaria: the role of the endothelial barrier. Virulence 2013; 4:543-55; PMID:23924893; http://dx.doi.org/ 10.4161/viru.25949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mosnier LO, Lavstsen T. The role of EPCR in the pathogenesis of severe malaria. Thromb Res 2016; 141:S46-9; PMID:27207424; http://dx.doi.org/ 10.1016/S0049-3848(16)30364-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].García-ponce A, Paredes SC, Castro KF, Schnoor M, Ch S. Regulation of endothelial and epithelial barrier functions by peptide hormones of the adrenomedullin family the adrenomedullin family. Tissue Barriers 2016; 4:1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bouwens EAM, Stavenuiter F, Mosnier LO. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J Thromb Haemost 2013; 11:242-53; PMID:23809128; http://dx.doi.org/ 10.1111/jth.12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stavenuiter F, Mosnier LO. Noncanonical PAR3 activation by factor Xa identifies a novel pathway for Tie2 activation and stabilization of vascular integrity. Blood 2015; 124:3480-90; http://dx.doi.org/ 10.1182/blood-2014-06-582775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Brown H, Rogerson S, Taylor T, Tembo M, Mwenechanya J, Molyneux M, Turner G. Blood-brain barrier function in cerebral malaria in Malawian children. Am J Trop Med Hyg 2001; 64:207-13; PMID:11442219 [DOI] [PubMed] [Google Scholar]
- [18].Warrell DA, Looareesuwan S, Phillips RE, White NJ, Warrell MJ, Chapel HM, Areekul S, Tharavanij S. Function of the blood-cerebrospinal fluid barrier in human cerebral malaria: rejection of the permeability hypothesis. Am J Trop Med Hyg 1986; 35:882-9; PMID:2429567 [DOI] [PubMed] [Google Scholar]
- [19].Brown HC, Chau TTH, Mai NTH, Day NPJ, Sinh DX. Blood – brain barrier function in cerebral malaria and CNS infections in Vietnam. Neurology 2000; 55:104-11; PMID:10891914; http://dx.doi.org/ 10.1212/WNL.55.1.104 [DOI] [PubMed] [Google Scholar]
- [20].Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner D a, Muwalo FW, Birbeck GL, Bradley WG, Fox LL, Glover SJ, et al.. Brain swelling and death in children with cerebral malaria. N Engl J Med 2015; 372:1126-37; PMID:25785970; http://dx.doi.org/ 10.1056/NEJMoa1400116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Maude RJ, Barkhof F, Hassan MU, Ghose A, Hossain A, Abul Faiz M, Choudhury E, Rashid R, Sayeed AA, Charunwatthana P, et al.. Magnetic resonance imaging of the brain in adults with severe falciparum malaria. Malar J 2014; 13:1-8; PMID:24383426; http://dx.doi.org/ 10.1186/1475-2875-13-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Newton CR, Peshu N, Kendall B, Kirkham FJ, Sowunmi A, Waruiru C, Mwangi I, Murphy SA, Marsh K. Brain swelling and ischaemia in Kenyans with cerebral malaria. Arch Dis Child 1994; 70:281-7; PMID:8185359; http://dx.doi.org/ 10.1136/adc.70.4.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Newton CRJC, Marsh K, Peshu N, Kirkham FJ, Crjc N, Marsh K, Peshu N, Perturba- KFJ. Perturbations of cerebral hemodynamics in Kenyans with cerebral malaria. Pediatr Neurol 1996; 15:41-9; PMID:8858700; http://dx.doi.org/ 10.1016/0887-8994(96)00115-4 [DOI] [PubMed] [Google Scholar]
- [24].Mohanty S, Mishra SK, Patnaik R, Dutt AK, Pradhan S, Das B, Patnaik J, Mohanty AK, Lee SJ, Dondorp AM. Brain swelling and mannitol therapy in adult cerebral malaria: A randomized trial. Clin Infect Dis 2011; 53:349-55; PMID:21810747; http://dx.doi.org/ 10.1093/cid/cir405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dondorp AM, Ince C, Charunwatthana P, Hanson J, Van Kuijen A, Faiz MA, Rahman MR, Hasan M, Bin Yunus E, Ghose A, et al.. Direct in vivo assessment of microcirculatory dysfunction in severe falciparum malaria. J Infect Dis 2008; 197:79-84; PMID:18171289; http://dx.doi.org/ 10.1086/523762 [DOI] [PubMed] [Google Scholar]
- [26].MacPherson G, Warrell M, White N, Looareesuwan S, Warrell D. Human cerebral malaria: a quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol 1985; 119:385-401; PMID:3893148 [PMC free article] [PubMed] [Google Scholar]
- [27].Pongponratn E, Turner GDH, Day NPJ, Phu NH, Simpson J a, Stepniewska K, Mai NTH, Viriyavejakul P, Looareesuwan S, Hien TT, et al.. An ultrastructural study of the brain in fatal Plasmodium falciparum malaria. Am J Trop Med Hyg 2003; 69:345-59; PMID:14640492 [PubMed] [Google Scholar]
- [28].Brown Hien, Day Mai, Chuong Chau, Loc Phu, Bethell Farrar, et al.. Evidence of blood–brain barrier dysfunction in human cerebral malaria. Neuropathol Appl Neurobiol 1999; 25:331-40; PMID:10476050; http://dx.doi.org/ 10.1046/j.1365-2990.1999.00188.x [DOI] [PubMed] [Google Scholar]
- [29].Hochman S, Madaline T, Wassmer S, Mbale E, Namjong C, Seydel K, Whitten RO, Varughese J, Grau GER, Kamiza S, et al.. Fatal pediatric cerebral malaria is associated with intravascular monocytes and platelets that are increased with HIV coinfection. MBio 2015; 6:e01390-15; PMID:26396242; http://dx.doi.org/ 10.1128/mBio.01390-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Feintuch C, Saidi A, Seydel K, Chen G. Activated neutrophils are associated with pediatric cerebral malaria vasculopathy in Malawian children. MBio 2016; 7:e01300-15; PMID:26884431; http://dx.doi.org/ 10.1128/mBio.01300-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brooks MH, Kiel FW, Sheehy TW, Barry KG. Acute pulmonary edema in falciparum malaria. N Engl J Med 1968; 279:732-7; http://dx.doi.org/ 10.1056/NEJM196810032791402 [DOI] [Google Scholar]
- [32].Taylor WRJ, Hanson J, Turner GDH, White NJ, Dondorp AM. Respiratory manifestations of malaria. Chest 2012; 142:492-505; PMID:22871759; http://dx.doi.org/ 10.1378/chest.11-2655 [DOI] [PubMed] [Google Scholar]
- [33].Milner D, Factor R, Whitten R, Carr RA, Kamiza S, Pinkus G, Molyneux M, Taylor T. Pulmonary pathology in pediatric cerebral malaria. Hum Pathol 2013; 44:2719-26; PMID:24074535; http://dx.doi.org/ 10.1016/j.humpath.2013.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Maguire GP, Handojo T, Pain MCF, Kenangalem E, Ric N, Tjitra E, Anstey NM. Lung injury in uncomplicated and severe falciparum malaria: a longitudinal study in Papua, Indonesia. J Infect Dis 2008; 192:1966-74; http://dx.doi.org/ 10.1086/497697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, McNeil YR, Darcy CJ, Granger DL, Weinberg JB, Lopansri BK, et al.. Impaired nitric oxide bioavailability and L-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med 2007; 204:2693-704; PMID:17954570; http://dx.doi.org/ 10.1084/jem.20070819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Maitland K, Marsh K. Pathophysiology of severe malaria in children. Acta Trop 2004; 90:131-40; PMID:15177139; http://dx.doi.org/ 10.1016/j.actatropica.2003.11.010 [DOI] [PubMed] [Google Scholar]
- [37].Anstey NM, Jacups SP, Cain T, Pearson T, Ziesing PJ, Fisher D a, Currie BJ, Marks PJ, Maguire GP. Pulmonary manifestations of uncomplicated falciparum and vivax malaria: cough, small airways obstruction, impaired gas transfer, and increased pulmonary phagocytic activity. J Infect Dis 2002; 185:1326-34; PMID:12001051; http://dx.doi.org/ 10.1086/339885 [DOI] [PubMed] [Google Scholar]
- [38].Anstey NM, Handojo T, Pain MCF, Kenangalem E, Tjitra E, Price RN, Maguire GP. Lung injury in vivax malaria: pathophysiological evidence for pulmonary vascular sequestration and posttreatment alveolar-capillary inflammation. J Infect Dis 2007; 195:589-96; PMID:17230420; http://dx.doi.org/ 10.1086/510756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Barber BE, William T, Grigg MJ, Parameswaran U, Piera KA, Price RN, Yeo TW, Anstey NM. Parasite biomass-related inflammation, endothelial activation, microvascular dysfunction and disease severity in vivax malaria. PLoS Pathog 2015; 11:e1004558; PMID:25569250; http://dx.doi.org/ 10.1371/journal.ppat.1004558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barsoum RS. Malarial acute renal failure. J Am Soc Nephrol 2000; 11:2147-54; PMID:11053494 [DOI] [PubMed] [Google Scholar]
- [41].White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet 2014; 383:723-35; PMID:23953767; http://dx.doi.org/ 10.1016/S0140-6736(13)60024-0 [DOI] [PubMed] [Google Scholar]
- [42].Nguansangiam S, Day NPJ, Hien TT, Mai NTH, Chaisri U, Riganti M, Dondorp AM, Lee SJ, Phu NH, Turner GDH, et al.. A quantitative ultrastructural study of renal pathology in fatal Plasmodium falciparum malaria. Trop Med Int Health 2007; 12:1037-50; PMID:17875015; http://dx.doi.org/ 10.1111/j.1365-3156.2007.01881.x [DOI] [PubMed] [Google Scholar]
- [43].Plewes K, Royakkers A a, Hanson J, Hasan MMU, Alam S, Ghose A, Maude RJ, Stassen PM, Charunwatthana P, Lee SJ, et al.. Correlation of biomarkers for parasite burden and immune activation with acute kidney injury in severe falciparum malaria. Malar J 2014; 13:91; PMID:24618154; http://dx.doi.org/ 10.1186/1475-2875-13-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wichapoon B, Punsawad C, Chaisri U, Viriyavejakul P. Glomerular changes and alterations of zonula occludens-1 in the kidneys of Plasmodium falciparum malaria patients. Malar J 2014; 13:176; PMID:24884882; http://dx.doi.org/ 10.1186/1475-2875-13-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zougbédé S, Miller F, Ravassard P, Rebollo A, Cicéron L, Couraud P, Mazier D, Moreno A. Metabolic acidosis induced by Plasmodium falciparum intraerythrocytic stages alters blood–brain barrier integrity. J Cereb Blood Flow & Metab 2011; 31:514-26; http://dx.doi.org/ 10.1038/jcbfm.2010.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pino P, Vouldoukis I, Kolb JP, Mahmoudi N, Desportes-Livage I, Bricaire F, Danis M, Dugas B, Mazier D. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis 2003; 187:1283-90; PMID:12696008; http://dx.doi.org/ 10.1086/373992 [DOI] [PubMed] [Google Scholar]
- [47].Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J Infect Dis 2007; 195:942-50; PMID:17330783; http://dx.doi.org/ 10.1086/512083 [DOI] [PubMed] [Google Scholar]
- [48].Wassmer SC, Candal FJ, Grau GE. Platelets potentiate brain endothelial alterations induced by Plasmodium falciparum. Infect Immun 2006; 74:645-53; PMID:16369021; http://dx.doi.org/ 10.1128/IAI.74.1.645-653.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gillrie MR, Krishnegowda G, Lee K, Buret AG, Robbins SM, Looareesuwan S, Gowda DC, Ho M. Src-family kinase dependent disruption of endothelial barrier function by Plasmodium falciparum merozoite proteins. Blood 2007; 110:3426-35; PMID:17693580; http://dx.doi.org/ 10.1182/blood-2007-04-084582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].White NJ, Turner GDH, Day NPJ, Dondorp AM. Lethal malaria: Marchiafava and Bignami were right. J Infect Dis 2013; 208:192-8; PMID:23585685; http://dx.doi.org/ 10.1093/infdis/jit116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Silamut K, Phu NH, Whitty C, Turner GD, Louwrier K, Mai NT, Simpson J a, Hien TT, White NJ. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am J Pathol 1999; 155:395-410; PMID:10433933; http://dx.doi.org/ 10.1016/S0002-9440(10)65136-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ponsford MJ, Medana IM, Prapansilp P, Hien TT, Lee SJ, Dondorp AM, Esiri MM, Day NPJ, White NJ, Turner GDH. Sequestration and microvascular congestion are associated with coma in human cerebral malaria. J Infect Dis 2012; 205:663-71; PMID:22207648; http://dx.doi.org/ 10.1093/infdis/jir812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Milner DA, Whitten RO, Kamiza S, Carr R, Liomba G, Dzamalala C, Seydel KB, Molyneux ME, Taylor TE. The systemic pathology of cerebral malaria in African children. Front Cell Infect Microbiol 2014; 4:1-13; PMID:24478989; http://dx.doi.org/ 10.3389/fcimb.2014.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Smith JD, Rowe JA, Higgins MK, Lavstsen T. Malaria's deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cell Microbiol 2013; 15:1976-83; PMID:23957661; http://dx.doi.org/ 10.1111/cmi.12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE V, Avril M, Brazier AJ, Freeth J, Jespersen JS, Nielsen M a, et al.. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature 2013; 498:502-5; PMID:23739325; http://dx.doi.org/ 10.1038/nature12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Davis SP, Amrein M, Gillrie MR, Lee K, Muruve D a, Ho M. Plasmodium falciparum-induced CD36 clustering rapidly strengthens cytoadherence via p130CAS-mediated actin cytoskeletal rearrangement. FASEB J 2012; 26:1119-30; PMID:22106368; http://dx.doi.org/ 10.1096/fj.11-196923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Davis SP, Lee K, Gillrie MR, Roa L, Amrein M, Ho M. CD36 recruits α5β1 integrin to promote cytoadherence of P. falciparum-infected erythrocytes. PLoS Pathog 2013; 9:e1003590; PMID:24009511; http://dx.doi.org/ 10.1371/journal.ppat.1003590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Huang RL, Teo Z, Chong HC, Zhu P, Tan MJ, Tan CK, Lam CRI, Sng MK, Leong DTW, Tan SM, et al.. ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood 2011; 118:3990-4002; PMID:21841165; http://dx.doi.org/ 10.1182/blood-2011-01-328716 [DOI] [PubMed] [Google Scholar]
- [59].Lampugnani MG, Resnati M, Dejana E, Marchisio PC. The role of integrins in the maintenance of endothelial monolayer integrity. J Cell Biol 1991; 112:479-90; PMID:1899416; http://dx.doi.org/ 10.1083/jcb.112.3.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol 2007; 179:4053-64; PMID:17785844; http://dx.doi.org/ 10.4049/jimmunol.179.6.4053 [DOI] [PubMed] [Google Scholar]
- [61].Hu G, Vogel SM, Schwartz DE, Malik AB, Minshall RD. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ Res 2008; 102:e120-31; PMID:18511851; http://dx.doi.org/ 10.1161/CIRCRESAHA.107.167486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jambou R, Combes V, Jambou M-J, Weksler BB, Couraud P-O, Grau GE. Plasmodium falciparum adhesion on human brain microvascular endothelial cells involves transmigration-like cup formation and induces opening of intercellular junctions. PLoS Pathog 2010; 6:e1001021; PMID:20686652; http://dx.doi.org/ 10.1371/journal.ppat.1001021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Carman C V, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 2004; 167:377-88; PMID:15504916; http://dx.doi.org/ 10.1083/jcb.200404129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Petri B, Kaur J, Long EM, Li H, Parsons SA, Butz S, Phillipson M, Vestweber D, Patel KD, Robbins SM, et al.. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood 2011; 117:942-52; PMID:21030556; http://dx.doi.org/ 10.1182/blood-2010-02-270561 [DOI] [PubMed] [Google Scholar]
- [65].Francischetti I, Seydel K, Monteiro R, Whitten R, Erexson C, Noronha A, Ostera G, Kamiza S, Molyneux M, Ward J, et al.. Plasmodium falciparum-infected erythrocytes induce tissue factor expression in endothelial cells and support the assembly of multimolecular coagulation complexes. J Thromb Haemost 2007; 5:155-65; PMID:17002660; http://dx.doi.org/ 10.1111/j.1538-7836.2006.02232.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Moxon CA, Chisala N V, Mzikamanda R, Maccormick I, Harding S, Downey C, Molyneux M, Seydel KB, Taylor TE, Heyderman RS, et al.. Laboratory evidence of disseminated intravascular coagulation is associated with a fatal outcome in children with cerebral malaria despite an absence of clinically evident thrombosis or bleeding. J Thromb Haemost 2015; 13:1653-64; PMID:26186686; http://dx.doi.org/ 10.1111/jth.13060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Clemens R, Pramoolsinsap C, Lorenz R, Pukrittayakamee S, Bock HL, White NJ. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br J Haematol 1994; 87:100-5; PMID:7947233; http://dx.doi.org/ 10.1111/j.1365-2141.1994.tb04877.x [DOI] [PubMed] [Google Scholar]
- [68].Gillrie MR, Renaux B, Russell-Goldman E, Avril M, Brazier AJ, Mihara K, Di Cera E, Milner DA, Hollenberg MD, Smith JD, et al.. Thrombin cleavage of Plasmodium falciparum erythrocyte membrane protein 1 inhibits cytoadherence. MBio 2016; 7:e01120-16; PMID:27624125; http://dx.doi.org/ 10.1128/mBio.01120-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Moxon CA, Wassmer SC, Milner DA, Chisala N V, Taylor TE, Seydel KB, Molyneux ME, Faragher B, Esmon CT, Downey C, et al.. Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood 2013; 122:842-51; PMID:23741007; http://dx.doi.org/ 10.1182/blood-2013-03-490219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A 2007; 104:5662-7; PMID:17376866; http://dx.doi.org/ 10.1073/pnas.0700763104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bernabeu M, Danziger SA, Avril M, Vaz M, Babar PH, Brazier AJ, Herricks T, Maki JN, Pereira L, Mascarenhas A, et al.. Severe adult malaria is associated with specific PfEMP1 adhesion types and high parasite biomass. Proc Natl Acad Sci 2016; :201524294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lau CKY, Turner L, Jespersen JS, Lowe ED, Petersen B, Wang CW, Petersen JEV, Lusingu J, Theander TG, Lavstsen T, et al.. Structural conservation despite huge sequence diversity allows EPCR Binding by the PfEMP1 family implicated in severe childhood malaria. Cell Host Microbe 2015; 17:118-29; PMID:25482433; http://dx.doi.org/ 10.1016/j.chom.2014.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gillrie MR, Avril M, Brazier AJ, Davis SP, Stins MF, Smith JD, Ho M. Diverse functional outcomes of Plasmodium falciparum ligation of EPCR: Potential implications for malarial pathogenesis. Cell Microbiol 2015; 17:1883-99; PMID:26119044; http://dx.doi.org/ 10.1111/cmi.12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Petersen JE V, Bouwens EAM, Tamayo I, Turner L, Wang CW, Stins M, Theander TG, Hermida J, Mosnier LO, Lavstsen T. Protein C system defects inflicted by the malaria parasite protein PfEMP1 can be overcome by a soluble EPCR variant. Thromb Haemost 2015; 114:1038-48; PMID:26155776; http://dx.doi.org/ 10.1160/TH15-01-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Avril M, Brazier AJ, Melcher M, Sampath S, Smith JD. DC8 and DC13 var genes associated with severe malaria bind avidly to diverse endothelial cells. PLoS Pathog 2013; 9:e1003430; PMID:23825944; http://dx.doi.org/ 10.1371/journal.ppat.1003430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sahu PK, Satpathi S, Behera PK, Mishra SK, Stumhofer JS. Pathogenesis of cerebral malaria: new diagnostic tools, biomarkers, and therapeutic approaches. Front Cell Infect Microbiol 2015; 5:1-13; PMID:25674541; http://dx.doi.org/ 10.3389/fcimb.2015.00075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Storm J, Craig AG. Pathogenesis of cerebral malaria-inflammation and cytoadherence. Front Cell Infect Microbiol 2014; 4:100; PMID:25120958; http://dx.doi.org/ 10.3389/fcimb.2014.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Grau GE, Taylor TE, Molyneux ME, Wirima JJ, Vassalli P, Hommel M, Lambert PH. Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med 1989; 320:1586-91; PMID:2657427; http://dx.doi.org/ 10.1056/NEJM198906153202404 [DOI] [PubMed] [Google Scholar]
- [79].Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci U S A 1992; 89:3200-3; PMID:1565611; http://dx.doi.org/ 10.1073/pnas.89.8.3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 1987; 237:1210-1212; PMID:3306918; http://dx.doi.org/ 10.1126/science.3306918 [DOI] [PubMed] [Google Scholar]
- [81].Looareesuwan S, Sjostrom L, Krudsood S, Wilairatana P, Porter RS, Hills F, Warrell DA. Polyclonal anti-tumor necrosis factor-alpha Fab used as an ancillary treatment for severe malaria. Am J Trop Med Hyg 1999; 61:26-33; PMID:10432050 [DOI] [PubMed] [Google Scholar]
- [82].van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, Memming H, Frenkel J, Enwere G, Bennett S, et al.. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis 1996; 174:1091-7; PMID:8896514; http://dx.doi.org/ 10.1093/infdis/174.5.1091 [DOI] [PubMed] [Google Scholar]
- [83].Alejandria MM, Lansang MAD, Dans LF, Mantaring JB III. Intravenous immunoglobulin for treating sepsis, severe sepsis and septic shock. Cochrane Database Syst Rev 2013; CD001090; PMID:24043371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Day NPJ, Hien TT, Schollaardt T, Loc PP, Van Chuong L, Chau TTH, Mai NTH, Phu NH, Sinh DX, White NJ, et al.. The prognostic and pathophysiologic role of pro- and antiinflammatory cytokines in severe malaria. J Infect Dis 1999; 180:1288-97; PMID:10479160; http://dx.doi.org/ 10.1086/315016 [DOI] [PubMed] [Google Scholar]
- [85].Di Lorenzo A, Lin MI, Murata T, Landskroner-Eiger S, Schleicher M, Kothiya M, Iwakiri Y, Yu J, Huang PL, Sessa WC. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J Cell Sci 2013; 126:5541-52; PMID:24046447; http://dx.doi.org/ 10.1242/jcs.115972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A 1991; 88:4651-5; PMID:1675786; http://dx.doi.org/ 10.1073/pnas.88.11.4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Serirom S, Raharjo WH, Chotivanich K, Loareesuwan S, Kubes P, Ho M. Anti-adhesive effect of nitric oxide on Plasmodium falciparum cytoadherence under flow. Am J Pathol 2003; 162:1651-60; http://dx.doi.org/ 10.1016/S0002-9440(10)64299-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Barber BE, William T, Grigg MJ, Piera KA, Chen Y, Wang H, Weinberg JB, Yeo TW, Anstey NM. Nitric oxide dependent endothelial dysfunction and reduced arginine bioavailability in vivax malaria but no greater increase in intravascular haemolysis in severe disease. J Infect Dis 2016; 214:1557-64; PMID:27630198; http://dx.doi.org/ 10.1093/infdis/jiw427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 2002; 8:1383-9; PMID:12426562; http://dx.doi.org/ 10.1038/nm1202-799 [DOI] [PubMed] [Google Scholar]
- [90].Weinberg JB, Volkheimer AD, Rubach MP, Florence SM, Mukemba JP, Kalingonji AR, Langelier C, Chen Y, Bush M, Yeo TW, et al.. Monocyte polarization in children with falciparum malaria: relationship to nitric oxide insufficiency and disease severity. Sci Rep 2016; 6:29151; PMID:27385484; http://dx.doi.org/ 10.1038/srep29151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Hawkes MT, Conroy AL, Opoka RO, Hermann L, Thorpe KE, McDonald C, Kim H, Higgins S, Namasopo S, John C, et al.. Inhaled nitric oxide as adjunctive therapy for severe malaria: a randomized controlled trial. Malar J 2015; 14:421; PMID:26510464; http://dx.doi.org/ 10.1186/s12936-015-0946-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, Piera K, Price RN, Duffull SB, Celermajer DS, Anstey NM. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc Natl Acad Sci U S A 2008; 105:17097-102; PMID:18957536; http://dx.doi.org/ 10.1073/pnas.0805782105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lovegrove FE, Tangpukdee N, Opoka RO, Lafferty EI, Rajwans N, Hawkes M, Krudsood S, Looareesuwan S, John CC, Liles WC, et al.. Serum angiopoietin-1 and −2 levels discriminate cerebral malaria from uncomplicated malaria and predict clinical outcome in African children. PLoS One 2009; 4:1-8; http://dx.doi.org/ 10.1371/journal.pone.0004912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Milam KE, Parikh SM. The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation. Tissue barriers 2015; 3:e957508; PMID:25838975; http://dx.doi.org/ 10.4161/21688362.2014.957508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, Sukhatme VP. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med 2006; 3:356-70; http://dx.doi.org/ 10.1371/journal.pmed.0030046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Higgins S, Purcell LA, Silver KL, Tran V, Crowley V, Hawkes M, Conroy AL, Opoka RO, Hay JG, Quaggin SE, et al.. Dysregulation of angiopoietin-1 plays a critical mechanistic role in the pathogenesis of cerebral malaria. Sci Transl Med 2016; 8:1-27; http://dx.doi.org/ 10.1126/scitranslmed.aaf6812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Prapansilp P, Medana I, Mai NTH, Day NPJ, Phu NH, Yeo TW, Hien TT, White NJ, Anstey NM, Turner GDH. A clinicopathological correlation of the expression of the angiopoietin-Tie-2 receptor pathway in the brain of adults with Plasmodium falciparum malaria. Malar J 2013; 12:50; PMID:23383853; http://dx.doi.org/ 10.1186/1475-2875-12-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Ghosh CC, David S, Zhang R, Berghelli A, Milam K, Higgins SJ, Hunter J, Mukherjee A, Wei Y, Tran M, et al.. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc Natl Acad Sci U S A [Internet] 2016; 113:2472-7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26884170; http://dx.doi.org/ 10.1073/pnas.1519467113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Elphinstone RE, Conroy AL, Hawkes M, Hermann L, Namasopo S, Warren HS, John CC, Liles WC, Kain KC. Alterations in systemic extracellular heme and hemopexin are associated with adverse clinical outcomes in Ugandan children with severe malaria. J Infect Dis 2016; 214:1268-75; PMID:27515862; http://dx.doi.org/ 10.1093/infdis/jiw357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Dalko E, Das B, Herbert F, Fesel C, Pathak S, Tripathy R, Cazenave P-A, Ravindran B, Sharma S, Pied S. Multifaceted roles of heme during severe Plasmodium falciparum infections in India. Infect Immun 2015; 83:3793-9; PMID:26169278; http://dx.doi.org/ 10.1128/IAI.00531-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ferreira A, Balla J, Jeney V, Balla G, Soares MP. A central role for free heme in the pathogenesis of severe malaria: The missing link? J Mol Med 2008; 86:1097-111; PMID:18641963; http://dx.doi.org/ 10.1007/s00109-008-0368-5 [DOI] [PubMed] [Google Scholar]
- [102].Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol 2014; 5:1-20; PMID:24478702; http://dx.doi.org/ 10.3389/fphar.2014.00115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014; 123:3818-27; PMID:24620350; http://dx.doi.org/ 10.1182/blood-2013-10-529982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013; 122:282-92; PMID:23692858; http://dx.doi.org/ 10.1182/blood-2013-03-489245 [DOI] [PubMed] [Google Scholar]
- [105].Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, Zamboni DS, Bozza MT. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A 2014; 111:4110-8; http://dx.doi.org/ 10.1073/pnas.1405023111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014; 123:377-90; PMID:24277079; http://dx.doi.org/ 10.1182/blood-2013-04-495887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lisk C, Kominsky D, Ehrentraut S, Bonaventura J, Nuss R, Hassell K, Nozik-Grayck E, Irwin DC. Hemoglobin-induced endothelial cell permeability is controlled, in part, via a myeloid differentiation primary response gene-88-dependent signaling mechanism. Am J Respir Cell Mol Biol 2013; 49:619-26; PMID:23713977; http://dx.doi.org/ 10.1165/rcmb.2012-0440OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Schaer C, Deuel J, Bittermann A, Rubio I, Schoedon G, Spahn D, Wepf R, Vallelian F, Schaer D. Mechanisms of haptoglobin protection against hemoglobin peroxidation triggered endothelial damage. Cell Death Differ 2013; 20:1569-79; PMID:23995229; http://dx.doi.org/ 10.1038/cdd.2013.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Vinchi F, Gastaldi S, Silengo L, Altruda F, Tolosano E. Hemopexin prevents endothelial damage and liver congestion in a mouse model of heme overload. Am J Pathol 2008; 173:289-99; PMID:18556779; http://dx.doi.org/ 10.2353/ajpath.2008.071130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos J a, Intaglietta M, van der Heyde HC. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med 2006; 12:1417-22; PMID:17099710; http://dx.doi.org/ 10.1038/nm1499 [DOI] [PubMed] [Google Scholar]
- [111].Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, et al.. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 2007; 13:703-10; PMID:17496899; http://dx.doi.org/ 10.1038/nm1586 [DOI] [PubMed] [Google Scholar]
- [112].Desakorn V, Chotivanichl K, Angus B, Sahassananda D, Chotivanich K, Suntharasamai P, Simposon J, White N. Semi-quantitative blood and plasma measurement of Plasmodium falciparum antigen PfHRP2 in blood and plasma. Trans R Soc Trop Med Hyg 1997; 91:479-83; PMID:9373661; http://dx.doi.org/ 10.1016/S0035-9203(97)90292-3 [DOI] [PubMed] [Google Scholar]
- [113].Pal P, Daniels BP, Oskman A, Diamond MS, Klein RS, Goldberg DE. Plasmodium falciparum histidine-rich protein II compromises brain endothelial barriers and may promote cerebral malaria pathogenesis. MBio 2016; 7:e00617-16; PMID:27273825; http://dx.doi.org/ 10.1128/mBio.00617-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Gowda DC. TLR-mediated cell signaling by malaria GPIs. Trends Parasitol 2007; 23:596-604; PMID:17980663; http://dx.doi.org/ 10.1016/j.pt.2007.09.003 [DOI] [PubMed] [Google Scholar]
- [115].Schofield L, Novakovic S, Gerold P, Schwarz RT, McConville MJ, Tachado SD. Glycosylphosphatidylinositol toxin of Plasmodium up-regulates intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin expression in vascular endothelial cells and increases leukocyte and parasite cytoadherence via tyrosine kin. J Immunol 1996; 156:1886-96; PMID:8596041 [PubMed] [Google Scholar]
- [116].Dasari P, Heber SD, Beisele M, Torzewski M, Reifenberg K, Orning C, Fries A, Zapf A-L, Baumeister S, Lingelbach K, et al.. Digestive vacuole of Plasmodium falciparum released during erythrocyte rupture dually activates complement and coagulation. Blood 2012; 119:4301-10; PMID:22403252; http://dx.doi.org/ 10.1182/blood-2011-11-392134 [DOI] [PubMed] [Google Scholar]
- [117].Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol 2002; 169:2102-10; PMID:12165538; http://dx.doi.org/ 10.4049/jimmunol.169.4.2102 [DOI] [PubMed] [Google Scholar]
- [118].Griffith JW, Sun T, McIntosh MT, Bucala R. Pure hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol 2009; 183:5208-20; PMID:19783673; http://dx.doi.org/ 10.4049/jimmunol.0713552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wu X, Gowda NM, Kumar S, Gowda DC. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol 2010; 184:4338-48; PMID:20231693; http://dx.doi.org/ 10.4049/jimmunol.0903824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Bhatt TK, Khan S, Dwivedi VP, Banday MM, Sharma A, Chandele A, Camacho N, Ribas de Pouplana L, Wu Y, Craig AG, et al.. Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nat Commun 2011; 2:530; PMID:22068597; http://dx.doi.org/ 10.1038/ncomms1522 [DOI] [PubMed] [Google Scholar]
- [121].Gillrie MR, Lee K, Gowda DC, Davis SP, Monestier M, Cui L, Hien TT, Day NPJ, Ho M. Plasmodium falciparum histones induce endothelial proinflammatory response and barrier dysfunction. Am J Pathol 2012; 180:1028-39; PMID:22260922; http://dx.doi.org/ 10.1016/j.ajpath.2011.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Orengo JM, Evans JE, Bettiol E, Leliwa-Sytek A, Day K, Rodriguez A. Plasmodium-induced inflammation by uric acid. PLoS Pathog 2008; 4:e1000013; PMID:18369465; http://dx.doi.org/ 10.1371/journal.ppat.1000013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Qazi KR, Oehlmann W, Singh M, López MC, Fernández C. Microbial heat shock protein 70 stimulatory properties have different TLR requirements. Vaccine 2007; 25:1096-103; PMID:17049413; http://dx.doi.org/ 10.1016/j.vaccine.2006.09.058 [DOI] [PubMed] [Google Scholar]
- [124].Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, Hagemann JH, Rupanagudi K V, Ryu M, Schwarzenberger C, et al.. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 2012; 23:1375-88; PMID:22677551; http://dx.doi.org/ 10.1681/ASN.2011111077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Wolfson RK, Chiang ET, Garcia JGN. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res 2011; 81:189-97; PMID:21146549; http://dx.doi.org/ 10.1016/j.mvr.2010.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Gallego-Delgado J, Basu-Roy U, Ty M, Alique M, Fernandez-Arias C, Movila A, Gomes P, Weinstock A, Xu W, Edagha I, et al.. Angiotensin receptors and β-catenin regulate brain endothelial integrity in malaria. J Clin Invest 2016; 119:4016-4029; http://dx.doi.org/10.117:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 2008; 10:923-34; PMID:18604199; http://dx.doi.org/ 10.1038/ncb1752 [DOI] [PubMed] [Google Scholar]
- [128].Mantel P-Y, Hjelmqvist D, Walch M, Kharoubi-Hess S, Nilsson S, Ravel D, Ribeiro M, Grüring C, Ma S, Padmanabhan P, et al.. Infected erythrocyte-derived extracellular vesicles alter vascular function via regulatory Ago2-miRNA complexes in malaria. Nat Commun 2016; 7:12727; PMID:27721445; http://dx.doi.org/ 10.1038/ncomms12727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Mantel PY, Hoang AN, Goldowitz I, Potashnikova D, Hamza B, Vorobjev I, Ghiran I, Toner M, Irimia D, Ivanov AR, et al.. Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe 2013; 13:521-34; PMID:23684304; http://dx.doi.org/ 10.1016/j.chom.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Regev-Rudzki N, Wilson DW, Carvalho TG, Sisquella X, Coleman BM, Rug M, Bursac D, Angrisano F, Gee M, Hill AF, et al.. Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 2013; 153:1120-33; PMID:23683579; http://dx.doi.org/ 10.1016/j.cell.2013.04.029 [DOI] [PubMed] [Google Scholar]
- [131].Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187-92; PMID:25119034; http://dx.doi.org/ 10.1038/nature13820 [DOI] [PubMed] [Google Scholar]
- [132].Wilson NO, Huang M-B, Anderson W, Bond V, Powell M, Thompson WE, Armah HB, a Adjei A, Gyasi R, Tettey Y, et al.. Soluble factors from Plasmodium falciparum-infected erythrocytes induce apoptosis in human brain vascular endothelial and neuroglia cells. Mol Biochem Parasitol 2008; 162:172-6; PMID:18848585; http://dx.doi.org/ 10.1016/j.molbiopara.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].N'Dilimabaka N, Taoufiq Z, Zougbédé S, Bonnefoy S, Lorthiois A, Couraud PO, Rebollo A, Snounou G, Mazier D, Sabater AM. P. falciparum isolate-specific distinct patterns of induced apoptosis in pulmonary and brain endothelial cells. PLoS One 2014; 9:e90692; http://dx.doi.org/ 10.1371/journal.pone.0090692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Taoufiq Z, Gay F, Balvanyos J, Ciceron L, Tefit M, Lechat P, Mazier D. Rho kinase inhibition in severe malaria: thwarting parasite-induced collateral damage to endothelia. J Infect Dis 2008; 197:1062-73; PMID:18419473; http://dx.doi.org/ 10.1086/528988 [DOI] [PubMed] [Google Scholar]
- [135].Chakravorty SJ, Carret C, Nash GB, Ivens A, Szestak T, Craig AG. Altered phenotype and gene transcription in endothelial cells, induced by Plasmodium falciparum-infected red blood cells: Pathogenic or protective? Int J Parasitol 2007; 37:975-87; PMID:17383656; http://dx.doi.org/ 10.1016/j.ijpara.2007.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Punsawad C, Maneerat Y, Chaisri U, Nantavisai K, Viriyavejakul P. Nuclear factor kappa B modulates apoptosis in the brain endothelial cells and intravascular leukocytes of fatal cerebral malaria. Malar J 2013; 12:260; PMID:23890318; http://dx.doi.org/ 10.1186/1475-2875-12-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Maccormick IJC, Beare NA V, Taylor TE, Barrera V, White VA, Hiscott P, Molyneux ME, Dhillon B, Harding SP. Cerebral malaria in children: Using the retina to study the brain. Brain 2014; 137:2119-42; PMID:24578549; http://dx.doi.org/ 10.1093/brain/awu001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Zeerleder S, Zwart B, Wuillemin WA, Aarden LA, Groeneveld ABJ, Caliezi C, van Nieuwenhuijze AEM, van Mierlo GJ, Eerenberg AJM, Lammle B, et al.. Elevated nucleosome levels in systemic inflammation and sepsis. Crit Care Med 2003; 31:1947-51; PMID:12847387; http://dx.doi.org/ 10.1097/01.CCM.0000074719.40109.95 [DOI] [PubMed] [Google Scholar]
- [139].Errico C, Pierre J, Pezet S, Desailly Y, Lenkei Z, Couture O, Tanter M. Ultrafast ultrasound localization microscopy for deep super-resolution vascular imaging. Nature 2015; 527:499-502; PMID:26607546; http://dx.doi.org/ 10.1038/nature16066 [DOI] [PubMed] [Google Scholar]
- [140].Chanthick C, Kanlaya R, Kiatbumrung R, Pattanakitsakul S, Thongboonkerd V. Caveolae-mediated albumin transcytosis is enhanced in dengue-infected human endothelial cells: A model of vascular leakage in dengue hemorrhagic fever. Sci Rep 2016; 6:31855; PMID:27920426; http://dx.doi.org/ 10.1038/srep31855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Shang D, Peng T, Gou S, Li Y, Wu H, Wang C, Yang Z. High mobility group box protein 1 boosts endothelial albumin transcytosis through the RAGE/Src/Caveolin-1 pathway. Sci Rep 2016; 6:32180; PMID:27572515; http://dx.doi.org/ 10.1038/srep32180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schuffler PJ, Grolimund D, Buhmann JM, Brandt S, et al.. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Meth 2014; 11:417-22; http://dx.doi.org/ 10.1038/nmeth.2869 [DOI] [PubMed] [Google Scholar]
- [143].Chung K, Wallace J, Kim S-Y, Kalyanasundaram S, Andalman AS, Davidson TJ, Mirzabekov JJ, Zalocusky KA, Mattis J, Denisin AK, et al.. Structural and molecular interrogation of intact biological systems. Nature 2013; 497:332-7; PMID:23575631; http://dx.doi.org/ 10.1038/nature12107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Conroy AL, Hawkes M, Elphinstone RE, Morgan C, Hermann L, Barker KR, Namasopo S, Opoka RO, John CC, Liles WC, et al.. Acute kidney injury is common in pediatric severe malaria and is associated with increased mortality. Open Forum Infect Dis 2016; 3:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].van der Helm MW, van der Meer AD, Eijkel JCT, van den Berg A, Segerink LI. Microfluidic organ-on-chip technology for blood-brain barrier research. Tissue Barriers 2016; 4:e1142493; http://dx.doi.org/ 10.1080/21688370.2016.1142493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol 2014; 32:760-72; PMID:25093883; http://dx.doi.org/ 10.1038/nbt.2989 [DOI] [PubMed] [Google Scholar]
- [147].Herland A, Van Der Meer AD, FitzGerald EA, Park TE, Sleeboom JJF, Ingber DE. Distinct contributions of astrocytes and pericytes to neuroinflammation identified in a 3D human blood-brain barrier on a chip. PLoS One 2016; 11:1-21; http://dx.doi.org/ 10.1371/journal.pone.0150360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Brown JA, Pensabene V, Markov DA, Allwardt V, Neely MD, Shi M, Britt CM, Hoilett OS, Yang Q, Brewer BM, et al.. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015; 9:1-15; http://dx.doi.org/ 10.1063/1.4934713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Adriani G, Ma D, Pavesi A, Goh ELK, Kamm RD. Modeling the blood-brain barrier in a 3D triple co-culture microfluidic system. Conf Proc IEEE Eng Med Biol Soc 2015; 2015:338-41; PMID:26736268 [DOI] [PubMed] [Google Scholar]
- [150].Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee H, Alves SE, Salmon M, Ferrante TC, Weaver JC, et al.. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods 2016; 13:151-7; PMID:26689262; http://dx.doi.org/ 10.1038/nmeth.3697 [DOI] [PubMed] [Google Scholar]
- [151].Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc Natl Acad Sci 2012; 109:13515-20; PMID:22869695; http://dx.doi.org/ 10.1073/pnas.1210182109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Tsai M, Kita A, Leach J, Rounsevell R, Huang JN, Moake J, Ware RE, Fletcher DA, Lam WA. In vitro modeling of the microvascular occlusion and thrombosis that occur in hematologic diseases using microfluidic technology. J Clin Invest 2012; 122:408-18; PMID:22156199; http://dx.doi.org/ 10.1172/JCI58753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].O'Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol 2016; 16:553-65; http://dx.doi.org/ 10.1038/nri.2016.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Wallqvist A, Fang X, Tewari SG, Ye P, Reifman J. Metabolic host responses to malarial infection during the intraerythrocytic developmental cycle. BMC Syst Biol 2016; 10:58; PMID:27502771; http://dx.doi.org/ 10.1186/s12918-016-0291-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Srichaikul T, Archararit N, Siriasawakul T, Viriyapanich T. Histamine changes in Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg 1976; 70:36-8; PMID:772896; http://dx.doi.org/ 10.1016/0035-9203(76)90004-3 [DOI] [PubMed] [Google Scholar]
- [156].Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, Masedunskas A, Weigert R, Chavakis T, Adams RH, et al.. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat Commun 2015; 6:6725; PMID:25857352; http://dx.doi.org/ 10.1038/ncomms7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Collard CD, Park KA, Montalto MC, Alapati S, Buras JA, Stahl GL, Colgan SP. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem 2002; 277:14801-11; PMID:11847215; http://dx.doi.org/ 10.1074/jbc.M110557200 [DOI] [PubMed] [Google Scholar]
- [158].Gordon EB, Hart GT, Tran TM, Waisberg M, Akkaya M, Kim AS, Hamilton SE, Pena M, Yazew T, Qi C-F, et al.. Targeting glutamine metabolism rescues mice from late-stage cerebral malaria. Proc Natl Acad Sci U S A 2015; 112:13075-80; PMID:26438846; http://dx.doi.org/ 10.1073/pnas.1516544112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Wagner JC, Platt RJ, Goldfless SJ, Zhang F, Niles JC. Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nat Meth 2014; 11:915-8; http://dx.doi.org/ 10.1038/nmeth.3063 [DOI] [PMC free article] [PubMed] [Google Scholar]