

Abstract
We recently developed a method to measure mitochondrial proteome dynamics with heavy water (2H2O)-based metabolic labeling and high resolution mass spectrometry. We reported the half-lives and synthesis rates of several proteins in the two cardiac mitochondrial subpopulations, subsarcolemmal and interfibrillar (SSM and IFM), in Sprague Dawley rats. In the present study, we tested the hypothesis that the mitochondrial protein synthesis rate is reduced in heart failure, with possible differential changes in SSM versus IFM. Six to seven week old male Sprague Dawley rats underwent transverse aortic constriction (TAC) and developed moderate heart failure after 22 weeks. Heart failure and sham rats of the same age received heavy water (5% in drinking water) for up to 80 days. Cardiac SSM and IFM were isolated from both groups and the proteins were separated by 1D gel electrophoresis. Heart failure reduced protein content and increased the turnover rate of several proteins involved in fatty acid oxidation, electron transport chain and ATP synthesis, while it decreased the turnover of other proteins, including pyruvate dehydrogenase subunit in IFM, but not in SSM. Because of these bidirectional changes, the average overall half-life of proteins was not altered by heart failure in both SSM and IFM. The kinetic measurements of individual mitochondrial proteins presented in this study may contribute to a better understanding of the mechanisms responsible for mitochondrial alterations in the failing heart.
Keywords: Deuterium, cardiac failure, proteome dynamics, mitochondria
1. Introduction
Decreased mitochondrial ATP generating capacity in myocardium is a hallmark of heart failure, as evidenced by a decrease in maximal ADP-stimulated oxidative phosphorylation in intact cardiomyocytes or in isolated mitochondria [1–3]. The activity of mitochondrial oxidative enzymes in whole tissue homogenates and the yield of isolated mitochondria are decreased in models of advanced heart failure induced by chronic arterial pressure overload [2, 4]. This suggests loss of mitochondrial protein content in failing myocardium, which could be due to lower rates of protein synthesis.
A new method to measure the synthesis rate of individual proteins in vivo was recently developed using heavy water (2H2O) to label the precursor amino acids and then to assess the time course of 2H incorporation into newly synthesized proteins using advanced tandem mass spectrometry [5–8]. We recently refined this method to measure the rate of synthesis of proteins in the two spatially distinct subpopulations of cardiac mitochondria: interfibrillar (IFM, located between the myofibrils) and subsarcolemmal (SSM, found along the perimeter of the cell) [9]. In healthy rats, we identified multiple tryptic peptides from 28 proteins in both SSM and IFM, and found time-dependent increases in heavy mass isotopomers that were consistent within a given protein [9]. The rate of protein turnover was relatively slow (average half-life of 30 days, 2.4% per day), and was correlated between IFM and SSM, although it was ~15% lower in SSM than in IFM. This may have particular relevance to mitochondrial dysfunction in heart failure, as previous studies found differential effects of heart failure in SSM and IFM, which could be due to different rates of protein turnover. Several groups found that IFM are more susceptible than SSM to aging, diabetes and heart failure-induced damage [10–15]. However, the differences between the two populations in heart failure are not entirely clear. For instance, some authors described respiratory defects in IFM but not SSM in cardiomyopathic hamsters [16], although this is not a consistent finding [17, 18]. Rats with advanced pressure overload- induced heart failure have greater dysfunction in IFM than SSM [4], suggesting a more impaired protein synthesis in IFM than in SSM.
The goal of the present investigation was to test the hypothesis that heart failure decreases the rate of protein synthesis and therefore oxidative capacity in mitochondria, with a more pronounced effect in IFM than in SSM. We used a well-established rat model of heart failure induced by chronic aortic constriction, which results in left ventricular remodeling and dysfunction and loss of mitochondrial yield and respiratory function [1, 2, 4, 19]. Protein synthesis was assessed using our previously established method with a heavy water tracer and advanced mass spectrometry, analysis of multiple peptide fragments within proteins of interest, and rigorous exclusion criteria applied across experimental groups and mitochondrial subpopulations.
2. MATERIALS AND METHODS
2.1. Experimental design and surgery
All experiments were performed according to the guidelines provided for the Care and Use of Laboratory Animals by the National Institutes of Health (Publication 85–23) and were approved by the Institutional Animal Care and Use Committee at the University of Maryland Baltimore School of Medicine. Male Sprague Dawley rats (6–8 weeks old, Harlan Laboratories, IN) underwent either sham surgery or transverse aorta constriction (TAC) to induce cardiac hypertrophy and heart failure (n=14/group), as previously described in detail [19]. Briefly, rats were anesthetized with isoflurane, intubated, and a partial median sternotomy was performed to expose the transverse aorta. A tantalum clip (0.5mm internal diameter) was positioned around the transverse aorta between the brachiocephalic trunk and the left common carotid artery [20]. Sham animals underwent similar surgical procedure without the insertion of the clip. All rats were fed a custom manufactured standard diet (Research Diets, New Brunswick, NJ, USA) and maintained for 22 weeks post-surgery and then euthanized to obtain cardiac mitochondria. Drinking water was supplemented with 5% heavy water (2H2O; Sigma-Aldrich, St. Louis, MO, USA) for either 0, 3, 10, 20, 40, 60 and 80 days before euthanasia (n=2 rats per time point). Food and water were provided ad libitum. Pooled mitochondria from two sham and heart failure rats at each time points were used for isolation of SSM and IFM sub-populations and proteomics studies. Thus, n=7 samples per group were analyzed by mass spectrometry. Since only negligible 2H-labeling was detected after 3 days of 2H2O exposure, this time point was excluded from the kinetic analysis.
2.2. Echocardiographic measurements
Left ventricular chamber size and function were assessed 21 weeks after surgery by echocardiography using a high-resolution imaging system (Vevo 2100 High-Resolution Imaging System, MS250 transducer, VisualSonics Inc., Toronto, Canada). Animals were anesthetized with isoflurane (1.5%), and images were acquired with rats in the supine position on a warming platform and analyzed as previously described [21].
2.3. Tissue harvest and mitochondrial isolation
Cardiac tissue was harvested at 22 weeks post-surgery. Fed animals were anaesthetized with 5.0% isoflurane between 3 and 6 h after initiation of the light phase. The thorax was opened and blood was collected from the left ventricle, immediately placed on ice, and centrifuged to obtain plasma. The heart was removed, and a section of the left ventricle free wall was immediately frozen in liquid nitrogen and stored at −80°C for later assessment of mitochondrial enzyme activities in whole tissue. The remainder of the left ventricle was used for mitochondrial isolation. The two subpopulations of mitochondria, SSM and IFM, were isolated as described previously in detail [22] with a minor modification [23]. Briefly, SSM was released by the treatment of the heart with Polytron homogenizer. To release IFM, trypsin (5 mg/g wet heart weight) was added to the remaining tissue. After rapid homogenization and centrifugation the supernatant containing the IFM fraction was isolated. To prevent protease digestion of IFM proteins, trypsin activity was immediately stopped by the soebean trypsin inhibitor (2.5 mg/g wet weight). IFM and SSM samples were stored at −80 °C for later analysis.
Mitochondrial protein content was determined by the Lowry method using bovine serum albumin as a standard [24].
2.4. Assessment of mitochondrial function
Mitochondrial respiration was measured as previously described in detail [25, 26]. Briefly, 0.2 mg/ml of mitochondrial proteins were suspended in a respiration buffer containing 100 mM KCl, 50 mM MOPS, 5 mM KH2PO4, 1mg/mL BSA/Fraction V and 1 mM EGTA. State 3 (ADP-stimulated) and 4 respiration (ADP-limited) were measured at 37°C with glutamate + malate (10mM and 5mM respectively), and palmitoylcarnitine (40 μM). Succinate with Rotenone (20mM and 7.5μM respectively) was used to assess respiration through Complex II of the ETC exclusively. State 3 respiration was measured in the presence of 200 μM ADP. State 4 respiration was assessed after ADP consumption. Respiratory Control Ratio, the ratio of State 3 to State 4 was calculated to assess the control of oxygen consumption by phosphorylation. The ratio of ADP added in the chamber to the total amount of oxygen consumed in state 3 (ADP:O ratio) were calculated as an index of the efficiency of oxidative phosphorylation.
The maximal activities of the mitochondrial citric acid cycle enzyme citrate synthase and the fatty acid β-oxidation enzyme medium chain acyl-CoA dehydrogenase (MCAD), markers of mitochondrial content, were measured in whole tissue homogenates as previously described [27]. Activities were normalized to the wet mass of tissue. In addition, citrate synthase activity was measured in isolated IFM and SSM so that the extraction of mitochondria from the myocardium could be measured.
2.5. Protein separation and sample preparation
Deuterium labeled plasma water was assessed by 2H-acetone exchange method previously described [9]. Cardiac mitochondrial proteins from two rats at each time point pooled together and were separated using one-dimensional gel electrophoresis using gradient gels (Biorad, Hercules, CA, USA) with 4–12% bis-tris gels. Initial protein concentration was measured using Lowry’s method of protein estimation with bovine serum albumin (BSA) as a standard. Each gel lane was loaded with 20μg of SSM or IFM mitochondrial protein. 1D gel electrophoresis using NuPAGE running buffer (Invitrogen, Grand Island, NY, USA) was carried out for 90 minutes at 130–150V and 20A and further overnight gel staining was performed by incubating the gel with Coomassie brilliant blue dye at 4°C.
2.6. Proteome dynamic analysis
For the analysis of protein synthesis rates and half-lives, we followed protocol similar to the one in our previous study [9], with minor modifications. Mitochondrial SSM and IFM proteins separated by 1D gel electrophoresis were reduced and alkylated prior to digestion with trypsin (Promega, Madison, WI, USA) at room temperature overnight. To extract the peptides from the digested proteins in gel pieces, 30 μl of 50% acetonitrile with 5% formic acid were combined with polyacrylamide gel and resuspended in 1% acetic acid. Further analysis of samples was carried out by nano-flow LC-MS/MS with a Thermo Acclaim PepMap RSLC C18 nano-column (150mm × 75 μm, 2μm, 100Å) using mobile phase A and B (1% formic acid in water and 0.1% formic acid in acetonitrile respectively). Tandem mass spectrometry was performed on an LTQ Oribtrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) in positive ion mode. Each full scan MS at a resolution of 60,000 (at 400 m/z) was followed by 15 Collisionally Induced Dissociation (CID) MS/MS (or MS2) scans. Dynamic exclusion was triggered by 3 consecutive selections within 30 seconds and last for 90 seconds.
For protein identification, all the CID spectra obtained from the mass spectrometer were transformed into a Mascot Generic Format (MGF) peak list file using Thermo Proteome Discoverer software (Version 1.3), and the MGF file was searched using Mascot 2.3 (Matrix Science, London, UK) software against National Center for Biotechnology Information (NCBI) rattus norvegicus reference sequence database (Release 50, 11/09/2011, 29,369 sequences, 15,267,967 residues). All the searches were performed as previously described [9]. The mass tolerance of the parent and product ions were set at 10 ppm and 1.2 Th, respectively. Trypsin was used as protease with 2 missed cleavage allowed. Peptide scores > 35 with p<0.05 were considered valid peptide identifications, and minimum of 2 peptides were required for a protein identification.
In order to obtain a list of proteins and peptides we used custom-made software (https://ispace.utmb.edu/users/rgsadygo/Proteomics/HeavyWater) that works with the data obtained from the mass spectrometer to list all the proteins and integrates corresponding extracted ion chromatograms of peptides [9]. The isotopic distribution was computed based on high resolution full scan spectra using the Mass Isotopomer Distribution Analysis and the percent distribution of the isotopomeric peaks were calculated. Data were modeled using a one-compartmental model by fitting a time course of total labeling of a peptide [Epeptide (t)] to an exponential growth curve equation: Epeptide (t) = Ess*(1−e−kt), where Ess is a asymptotical steady-state labeling, and k is the rate constant. The data from multiple peptides were aggregated to calculate the averaged rate constant and half-life (t1/2=ln2/k) of a protein. All the peptides used for quantification and analysis were unique to their respective proteins. To minimize errors, only the peptides with a regression coefficient (R2) value greater than 0.95 were included in the data analysis. Peptides with coefficients of variation (CV) greater than 27 % in their rate constants were eliminated as outliers.
2.7. Statistical analysis
Results are presented as mean ± standard deviation (SD). Differences in cardiac mass and echocardiographic parameters were assessed using a 2-tailed t-test. Although each time point represents one pooled sample from two rats, all six time points in each group were used to model the rate constants using non-linear regression analysis. The calculated 95% confidence intervals for the rate constants were used as the criteria for determining the differences when we compared the 2H enrichment profiles. Differences in protein half-life between SSM and IFM within and between the sham and heart failure groups were assessed using a one-way ANOVA with a Bonferroni test for multiple comparisons. Differences between proteins based on function (citric acid cycle, electron transport chain and fatty acid oxidation) or location (inner membrane, outer membrane, and matrix) were evaluated by 2-way repeated measures ANOVA for mitochondrial subpopulation and surgery (sham vs heart failure) with a Bonferroni post hoc test for multiple comparisons. A P <0.05 was considered significant.
3. RESULTS
3.1. Development of Heart Failure in Response to Pressure Overload
Heart failure became manifest at 22 weeks following aortic constriction, as indicated by a 45% increase in LV mass, hypertrophy of the atria and right ventricle, greater LV wall thickness, significant increases in LV end systolic and diastolic volumes and a decreased ejection fraction (Table 1).
Table 1.
Body and tissue mass, and left ventricular (LV) functional parameters including LV wall thickness, end diastolic volume and ejection fraction assessed by echocardiography (n=14/group).
| Sham | Heart Failure | P value | |
|---|---|---|---|
| Body Mass (g) | 440 ± 9 | 431 ± 12 | NS |
| Atria Mass (mg) | 82 ± 5 | 286 ± 27 | 0.000001 |
| RV Mass (mg) | 207 ± 7 | 367 ± 30 | 0.00002 |
| LV Mass (mg) | 976 ± 27 | 1419 ± 72 | 0.000004 |
| Tibia Length (mm) | 43.1 ± 0.4 | 41.8 ± 0.5 | NS |
| LV mass/tibia length (mg/mm) | 24.0 ± 0.5 | 34.6 ± 4.3 | <0.000005 |
| LV wall thickness (anterior + posterior (mm) | 5.0 ± 0.1 | 5.9 ± 0.2 | 0.0001 |
| LV End Diastolic Diameter (mm) | 6.1±0.3 | 7.2 ± 0.4 | 0.046 |
| End Diastolic Volume (mL) | 0.27 ± 0.04 | 0.42 ± 0.05 | 0.0005 |
| Ejection Fraction (%) | 83 ± 2 | 61 ± 3 | <0.000005 |
| LV End Systolic Diameter (mm) | 3.1 ± 0.3 | 5.1 ± 0.4 | 0.0003 |
| Fractional Shortening (%) | 50.3 ± 3.1 | 29.2 ± 2.3 | 0.00001 |
3.2. Mitochondrial yield and respiratory function
The heart failure group had a significant 35% decrease in IFM yield and a 24% decrease in total mitochondrial yield (mg of mitochondrial protein extracted per gram of myocardium) (Figure 1). Further, the activities of citrate synthase and MCAD per gram of tissue were both decreased by 30% (Figure 1). Thus the heart failure group had a lower total mitochondrial content in myocardium. To confirm that low mitochondrial yield in the heart failure group was not related to poor extraction, we assessed the extraction efficiency in both groups. The extraction of mitochondria from the myocardium (calculated from the sum of the yield of each subpopulation times its citrate synthase activity divided by the activity in whole tissue) was not different between sham and heart failure groups (Table 2).
Figure 1.
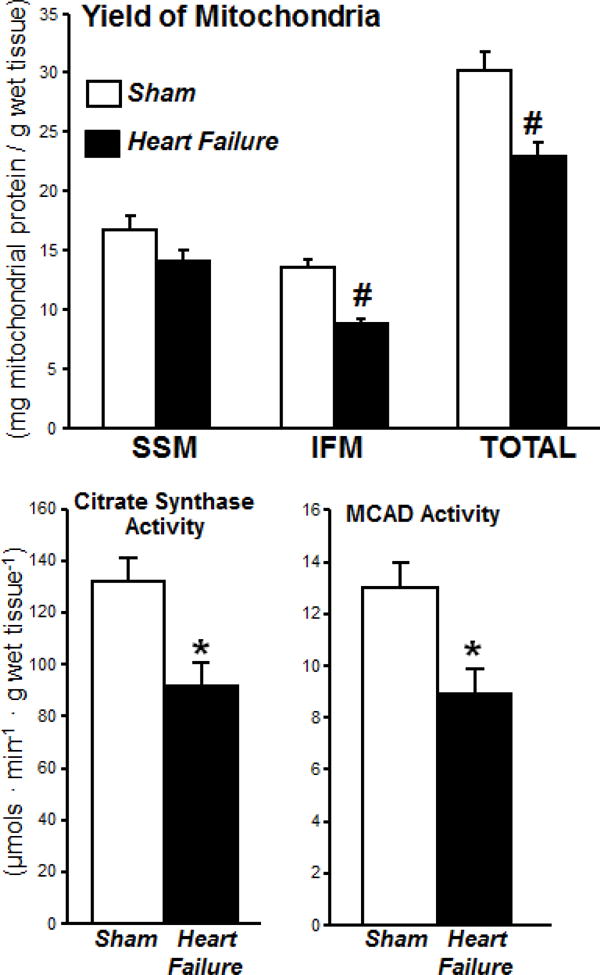
Mitochondrial yield for isolated subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM) (upper panel) and activity of the citric acid cycle enzyme citrate synthase and the β-oxidation enzyme medium chain acyl-CoA dehydrogenase (MCAD) measured in whole tissue homogenates (n=14/group, * P<0.01, # P<0.001)
Table 2.
Mitochondrial yield, activity of citrate synthase, and respiration. All respiration rates are expressed in Atoms O · mg mitochondrial protein−1 · min−1, the respiratory control ratio (state 3/state 4), and the ratio of ADP:O in isolated subsarcolemmal and interfibrillar mitochondria (n=14/group, *P<0.05 compared to the sham group).
| Sham | Heart Failure | |||
|---|---|---|---|---|
| Extraction(% of total tissue citrate synthase activity in tissue recovered in IFM + SSM) | 67 ± 8% | 64 ± 10% | ||
|
| ||||
| SSM | IFM | |||
| SHAM | Heart Failure | SHAM | Heart Failure | |
|
| ||||
| Mitochondrial Citrate | ||||
| Synthase Activity (pmols ■ mg protein-1-min-1) | 2.59 ± 0.21 | 2.25 ± 0.09 | 2.95 ± 0.27 | 2.59 ± 0.20 |
| SHAM | Heart Failure | SHAM | Heart Failure | |
| Gluatamate + Malate | ||||
| State 3 | 199 ± 9 | 219 ± 15 | 243 ± 13 | 245 ± 13 |
| State 4 | 34.1 ± 2.7 | 39.7 ± 3 | 37.9 ± 3.7 | 46.2 ± 3.4 |
| Respiratory Control Ratio | 6.16 ± 0.4 | 5.61 ± 0.3 | 6.95 ± 0.6 | 5.45 ± 0.3* |
| ADP: O | 2.02 ± 0.1 | 1.81 ± 0.1 | 2.27 ± 0.1 | 2.09 ± 0.1 |
| Succinate+Rotenone | ||||
| State 3 | 357 ± 22 | 371 ± 22 | 467 ± 16 | 498 ± 31 |
| State 4 | 131 ± 7 | 131 ± 9 | 173 ± 8 | 188 ± 12 |
| Respiratory Control Ratio | 2.75 ± 0.1 | 2.90 ± 0.1 | 2.85 ± 0.1 | 2.71 ± 0.2 |
| ADP: O | 1.38 ± 0.1 | 1.42 ± 0.1 | 1.44 ± 0.1 | 1.25 ± 0.1 |
| Palmitoylcarnitine | ||||
| State 3 | 242 ± 16 | 218 ± 13 | 333 ± 28 | 258 ± 19* |
| State 4 | 55.7 ± 3.1 | 59.7 ± 2.3 | 66.3 ± 4.8 | 68.4 ± 3.6 |
| Respiratory Control Ratio | 4.35 ± 0.23 | 3.67 ± 0.21* | 5.14 ± 0.45 | 3.81 ± 0.25* |
| ADP: O | 2.20 ± 0.08 | 2.29 ± 0.11 | 2.35 ± 0.11 | 2.32 ± 0.11 |
To study the effect of heart failure on different functional components of IFM and SSM, we measured mitochondrial respiration with different substrates (palmitoylcarnitine, glutamate+malate, and succinate) that use distinct mitochondrial transport, oxidative pathways and provide specific substrates to Complex I and Complex II. Palmitoylcarnitine oxidation reflects mitochondrial palmitoylcarnitine transport, palmitate oxidation, the activity of entire ETC and the phosphorylation process. The combination of glutamate and malate as substrate produces NADH, source of electron for Complex I, thus allowing the study of the aspartate shuttle. Succinate is oxidized in Complex II (bypassing Complex I) and the addition of rotenone, an inhibitor of Complex I, allows assessing the activity of complex II. The respiratory control ratio was decreased in IFM, but not SSM, with glutamate+malate as substrates. The maximal rate of mitochondrial respiration (state 3) in isolated mitochondria expressed per mg of mitochondrial protein was decreased by 25% in IFM from heart failure animals with palmitoylcarnitine as the substrate, with no other significant differences in state 4 between groups (Table 2). The respiratory control ratio (state 3/state 4), an index of the ability of mitochondria to increase ATP generation above basal values in response to maximally stimulating conditions, was decreased in both SSM and IFM in heart failure when palmitoylcarnitine was used as substrate. When state 3 respiration was expressed relative to whole myocardial tissue (calculated as the product of state 3 rate in isolated IFM and yield of IFM per gram of tissue) there was a decrease in respiration with all substrates (Figure 2). In SSM there was a decrease in respiration with palmitoylcarnitine and succinate+rotenone, but not with glutamate+malate (Figure 2). Despite these differences in the respiratory control ratios, ADP/O ratios were similar in SSM and IFM isolated from sham or heart failure rats. Taken together, heart failure resulted in loss of mitochondrial content and tissue respiratory capacity in IFM, but had a modest effect on the respiratory function of SSM.
Figure 2.
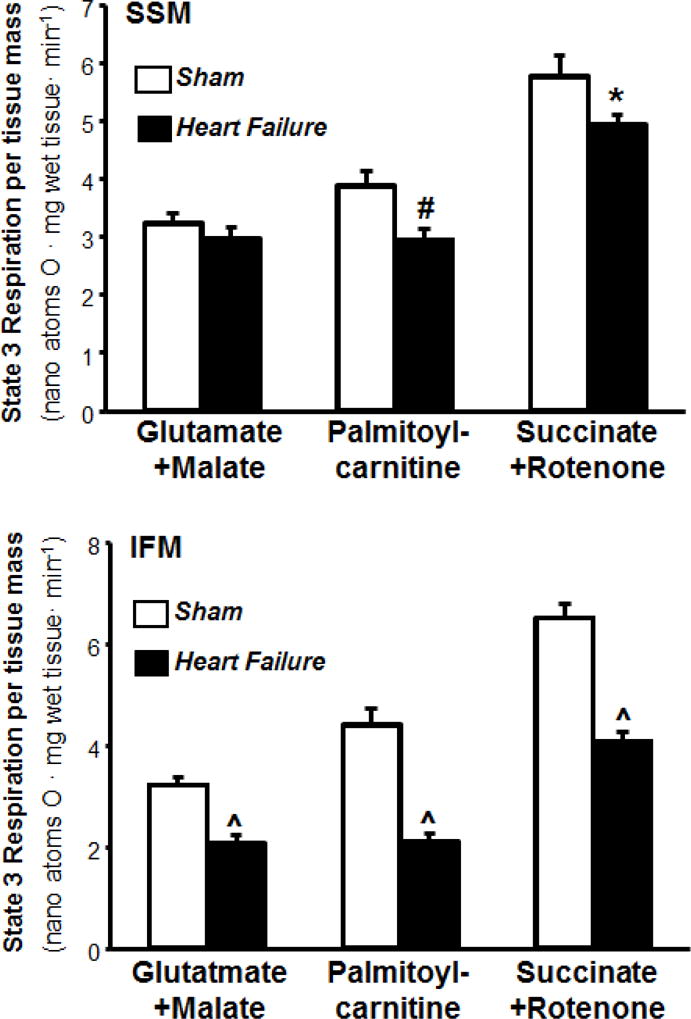
State 3 respiration rates in isolated subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM) expressed per mg of wet tissue. Values were calculated as the product of the state 3 rate in isolated mitochondria and the mitochondrial yield (n=14/group, * P<0.05, # P<0.005, ^ P<0.000005).
3.3. Mitochondrial protein synthesis rates
3.3.1. Half-life of proteins in sham and heart failure rats
In this study we used a simplified dosing method in which we introduced the 2H2O tracer only in the drinking water without priming bolus injection. The time course labeling of 2H2O enrichment of plasma water reached ~3.5% after 20 days and stayed constant until 80 days of labeling period (Table S1). To account for the slow increase in body water labeling, we fit the measured body water enrichment into the first order exponential curve that yields the body water turnover curve with the 2H-enrichment rate constant of 0.063–0.086 day−1 (t1/2 = 6.3–8.6 days). Then, the modeled continuous body water curve was used for the estimation of kinetically relevant body water enrichment required for accurate calculation of protein synthesis rates. A similar approach has previously been used when the 2H2O tracer was administered in the drinking water without a priming bolus injection [28]. Selected high protein abundant SDS gel bands (Supplementary Figure 1S) from cardiac mitochondria protein separation were analyzed. All analyzed bands were below 100 kDa protein marker. However, SSM, but not IFM fractions contained two intense bands above the molecular weight of 100 kDa marker. Our proteomics analysis revealed that these bands corresponded to heavy-chain myosin contaminants from myofibrils (Table 3S), therefore they were excluded from kinetic analysis. In the remaining six analyzed bands, a total of 90 proteins were identified in both sham and heart failure groups in both SSM and IFM (Table S2), and 48 of these 90 proteins met the previously described criteria for quantification [9]. The proteins were subdivided based on their location in the mitochondria (Table 3). The average half-life of protein synthesis was not significantly different in sham SSM vs IFM (Figure 3). We previously found that in normal rats the half-life of mitochondrial proteins was 15% longer in SSM than IFM (P<0.05) with an evaluation of 28 proteins [9]. In the present study the differences between average half-lives of sham SSM and sham IFM did not reach statistical significance threshold (P=0.08 by 2-tailed t-test).
Table 3.
Half-lives of proteins in cardiac SSM and IFM of sham and heart failure groups. Rats were exposed to 5% 2H2O in the drinking water up to 80 days. Pooled cardiac mitochondria from two sham and heart failure rats at six time points were used for the analysis of SSM and IFM proteins. Only proteins that were identified in both treatment groups, mitochondrial subpopulations, and meet our analytic constraints are listed. The gradual increase in total labeling of the tryptic peptides was modeled to calculate the turnover rate constant and half-life. Proteins are grouped by their primary location within the mitochondrion. Data are presented as mean ± SD, where mean and SD (standard deviation) were calculated from all analyzed peptides for a given protein. (n=6, *P<0.05 vs IFM SHAM, #P<0.05 vs IFM TAC, $p<0.05 vs SSM SHAM)
| Sham | Heart Failure | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| IFM | SSM | IFM | SSM | ||||||
| Accession | protein | # of analyzed peptides | t 1/2 (days) Mean ± SD | # of analyzed peptides | t 1/2 (days) Mean ± SD | # of analyzed peptides | t 1/2 (days) Mean ± SD | # of analyzed peptides | t 1/2 (days) Mean ± SD |
| OUTER MEMBRANE | |||||||||
| gi|13786200 | VDAC-1 | 3 | 17.02 ± 4.59 | 8 | 16.26 ± 2.49 | 5 | 18.22 ± 1.89 | 7 | 16.87 ± 1.37 |
| gi|32189355 | ADP/ATP translocase 1 | 5 | 22.81 ± 2.70 | 8 | 17.81 ± 2.58 | 3 | 26.18 ± 0.87 | 4 | 23.61 ± 1.83 |
| gi|6978703 | Carnitine Palmitoyltransferase-1 | 2 | 20.33 ± 0.41 | 9 | 13.04 ± 2.38* | 2 | 11.62 ± 2.21* | 11 | 13.85 ± 2.18# |
| gi|25742739 | Long chain - coA Ligase | 7 | 1 3.57 ± 1.76 | 16 | 14.53 ± 2.64 | 6 | 13.38 ± 1.33 | 20 | 15.59 ± 2.96 |
| INNER MEMBRANE | |||||||||
| gi|54792127 | ATP Synthase beta subunit | 5 | 22.76 ± 1.87 | 8 | 23.97 ± 2.67 | 6 | 16.96 ± 2.58 | 14 | 18.58 ± 2.43 |
| gi|8393180 | Cytochrome C Oxidase 4 isoform 1 | 9 | 22.91 ± 3.33 | 6 | 19.89 ± 2.57 | 10 | 18.57 ± 2.32 | 11 | 22.44 ± 2.54 |
| gi|6981260 | NADH dehydrogenase | 5 | 12.76 ± 1.93 | 3 | 9.32 ± 0.48 | 5 | 15.50 ± 2.53 | 4 | 15.88 ± 1.81 |
| gi|40538742 | ATP Synthase alpha subunit | 8 | 24.49 ± 4.40 | 9 | 23.24 ± 4.34 | 8 | 20.55 ± 2.88 | 9 | 21.30 ± 3.52 |
| gi|189011657 | Cytochrome bc subunit 7 | 6 | 20.56 ± 3.93 | 3 | 23.70 ± 2.93 | 6 | 16.05 ± 3.35 | 5 | 14.26 ± 2.74$ |
| gi|32189350 | ADP/ATP translocase 2 | 6 | 27.58 ± 3.65 | 3 | 23.38 ± 5.37 | 4 | 20.96 ± 2.40* | 5 | 23.40 ± 3.56 |
| gi|39930503 | ATP Synthase subunit gamma | 5 | 28.83 ± 3.09 | 3 | 21.28 ± 6.08 | 4 | 32.66 ± 1.41 | 4 | 31.44 ± 7.32 |
| gi|18426858 | Succinate dehydrogenase | 4 | 20.32 ± 3.31 | 6 | 17.93 ± 2.25 | 3 | 12.26 ± 2.21* | 7 | 17.54 ± 3.67 |
| gi|61557127 | NAD(P) transhydrogenase | 4 | 21.92 ± 1.13 | 4 | 19.00 ± 3.97 | 4 | 29.48 ± 3.67 | 4 | 25.35 ± 4.77 |
| gi|55741544 | Cytochrome bc subuit 2 | 7 | 18.90 ± 2.80 | 8 | 22.81 ± 4.20 | 8 | 23.47 ± 2.69* | 11 | 25.43 ± 4.01 |
| gi|58865384 | NADH dehydrogenase iron sulfur protein 2 | 8 | 18.77 ± 3.35 | 6 | 18.91 ± 2.61 | 8 | 20.24 ± 3.23 | 5 | 17.88 ± 3.75 |
| gi|53850628 | NADH Ubiquinone Oxidoreductase 75KD | 15 | 16.36 ± 2.57 | 24 | 14.26 ± 2.14 | 17 | 13.61 ± 2.16 | 24 | 14.99 ± 2.24 |
| gi|51948476 | Cytochrome bc subunit 1 | 4 | 10.81 ± 2.55 | 8 | 25.23 ± 2.90 | 3 | 11.42 ± 1.47 | 8 | 25.11 ± 3.54 |
| gi|158749584 | Succinyl CoA ligase beta | 2 | 26.57 ± 6.21 | 7 | 21.08 ± 4.64 | 5 | 21.44 ± 3.74 | 9 | 21.35 ± 4.91 |
| gi|6341995 | NADH dehydrogenase iron sulfur protein 4 | 7 | 20.87 ± 4.23 | 6 | 15.86 ± 1.60 | 6 | 17.93 ± 4.84 | 5 | 18.04 ± 3.34 |
| gi|8394328 | Superoxide dismutase | 5 | 21.09 ± 6.59 | 2 | 16.01 ± 4.40 | 3 | 26.64 ± 6.34 | 3 | 28.76 ± 4.20 |
| gi|164565371 | NADH dehydrogenase 1 alpha subcomplex 12 | 5 | 14.57 ± 2.31 | 6 | 13.91 ± 1.68 | 5 | 12.56 ± 3.87 | 5 | 11.30 ± 1.76 |
| gi|24233541 | cytochrome c oxidase subunit 5A | 4 | 23.00 ± 4.47 | 3 | 30.63 ± 7.11 | 2 | 28.75 ± 1.92 | 4 | 27.53 ± 5.33 |
| gi|20806141 | Phosphate carrier protein | 5 | 19.84 ± 2.57 | 5 | 22.22 ± 4.85 | 3 | 21.73 ± 0.77 | 3 | 19.52 ± 3.11 |
| gi|189083744 | Creatinine Kinase S type | 9 | 19.26 ± 4.16 | 16 | 17.45 ± 2.56 | 14 | 14.71 ± 2.81 | 15 | 15.71 ± 3.96* |
| gi|6980956 | glutamate dehydrogenase 1 mitochondrial precursor | 4 | 21.77 ± 3.49 | 4 | 16.75 ± 2.02 | 5 | 20.97 ± 4.58 | 11 | 21.05 ± 3.21 |
| gi|55741424 | NADH dehydrogenase [ubiquinone] flavoprotein 1 mitochondrial precursor | 10 | 15.30 ± 2.16 | 9 | 12.37 ± 1.62 | 11 | 11.78 ± 3.01 | 12 | 7.19 ± 2.20 |
| MATRIX | |||||||||
| gi|40538860 | Aconitase | 5 | 21.35 ± 2.77 | 3 | 17.02 ± 3.46 | 8 | 20.43 ± 1.58 | 5 | 18.34 ± 4.19 |
| gi|62079055 | Isocitrate dehydrogenase | 8 | 23.00 ± 3.41 | 8 | 15.04 ± 2.15 | 8 | 27.55 ± 3.90 | 4 | 17.84 ± 3.16 |
| gi|18426866 | 3- Ketoacyl CoA thiolase | 3 | 27.42 ± 3.25 | 7 | 25.87 ± 5.62 | 8 | 19.80 ± 3.00 | 8 | 20.57 ± 2.87 |
| gi|148747393 | Trifunctional protein alpha subunit | 4 | 20.24 ± 3.03 | 6 | 15.44 ± 2.39 | 4 | 14.74 ± 4.09 | 7 | 12.19 ± 1.40 |
| gi|42476181 | Malate Dehydrogenase | 2 | 24.22 ± 3.08 | 4 | 13.88 ± 2.23 | 3 | 21.70 ± 5.65 | 2 | 23.88 ± 0.37 |
| gi|6980972 | Aspartate Aminotransferase | 10 | 22.99 ± 2.55 | 17 | 22.43 ± 3.38 | 15 | 26.12 ± 3.29 | 13 | 23.62 ± 3.25 |
| gi|6978435 | VLC Acyl CoA dehydrogenase | 4 | 12.18 ± 1.17 | 3 | 17.49 ± 3.15 | 3 | 10.75 ± 2.50 | 3 | 16.87 ± 3.74 |
| gi|14192933 | Aldehyde dehydrogenase | 4 | 11.12 ± 1.59 | 11 | 10.80 ± 1.39 | 6 | 10.80 ± 1.62 | 21 | 9.77 ± 2.01 |
| gi|16758404 | Peroxiredoxin-5 | 5 | 23.54 ± 3.99 | 3 | 20.58 ± 4.69 | 2 | 23.09 ± 2.45 | 5 | 25.19 ± 4.51 |
| gi|56090293 | pyruvate dehydrogenase E1 component subunit beta | 2 | 28.68 ± 0.43 | 11 | 22.85 ± 4.84 | 2 | 27.64 ± 5.59 | 4 | 31.22 ± 3.92 |
| gi|145651820 | methylmalonate-semialdehyde dehydrogenase | 3 | 23.19 ± 0.92 | 13 | 20.74 ± 3.87 | 2 | 21.00 ± 4.12 | 9 | 21.15 ± 3.39 |
| gi|17105336 | Hydroxy acylCoA deH2 | 2 | 29.01 ± 4.49 | 14 | 23.87 ± 2.36 | 3 | 35.39 ± 1.72 | 8 | 28.84 ± 4.81 |
| gi|78365255 | Diydrolipoyllysine acetyltransferase of PD | 3 | 23.47 ± 2.50 | 2 | 26.61 ± 4.41 | 2 | 29.42 ± 1.11 | 2 | 24.83 ± 1.10 |
| gi|6981112 | Isovaleryl CoA deH2 | 10 | 14.78 ± 3.20 | 10 | 16.67 ± 2.04 | 9 | 15.28 ± 3.70 | 11 | 14.09 ± 2.53 |
| gi|48675862 | Acyl CoA thioesterase 2 | 5 | 20.70 ± 4.63 | 8 | 22.51 ± 2.78 | 3 | 24.18 ± 3.21 | 12 | 21.15 ± 4.34 |
| gi|11693174 | Branched chain aa aminotransferase | 2 | 23.99 ± 2.08 | 3 | 25.76 ± 7.47 | 3 | 22.37 ± 4.37 | 3 | 28.43 ± 3.01 |
| gi|158303308 | Propionyl coA carboxylase alpha | 3 | 21.57 ± 6.16 | 8 | 18.63 ± 0.79 | 8 | 19.05 ± 4.76 | 17 | 19.00 ± 4.26 |
| gi|40786469 | dihydrolipoyl dehydrogenase mitochondrial precursor | 8 | 23.16 ± 4.17 | 13 | 23.20 ± 3.97 | 10 | 24.29 ± 3.90 | 12 | 35.99 ± 3.46$# |
| gi|6980956 | glutamate dehydrogenase 1 mitochondrial precursor | 4 | 21.77 ± 3.49 | 4 | 16.75 ± 2.02 | 5 | 20.97 ± 4.58 | 11 | 21.05 ± 3.21 |
| gi|62945278 | 2-oxoglutarate dehydrogenase mitochondrial precursor | 5 | 16.03 ± 2.79 | 8 | 13.74 ± 2.83 | 5 | 14.39 ± 3.09 | 10 | 16.03 ± 2.79 |
| gi|12018236 | thioredoxin reductase 2 mitochondrial precursor | 2 | 25.35 ± 6.34 | 4 | 35.70 ± 4.71 | 3 | 25.30 ± 5.37 | 4 | 22.43 ± 3.77 |
| gi|157818027 | acetyl-coenzyme A synthetase 2-like mitochondrial | 5 | 13.40 ± 1.70 | 3 | 15.70 ± 3.70 | 6 | 12.93 ± 1.54 | 3 | 13.76 ± 3.10 |
Figure 3.
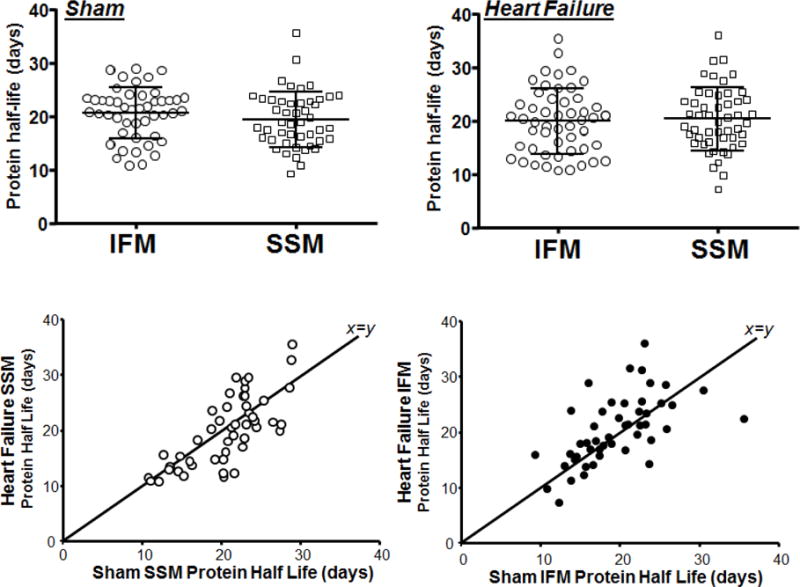
Half-life of proteins in sham and heart failure rats. Each data point represents the half-life of distinct protein calculated using non-linear regression analysis of 2H-labeling of peptides at six time points. The top left panel shows that the protein synthesis rate was slightly, but not significantly slower in IFM than in SSM sham (P=0.08). This trend was not present in animals with heart failure. The bottom panels plot the half-life of protein synthesis for heart failure rats as a function of values in sham animals for SSM (left) and IFM (right).
Surprisingly, in spite of the changes in the mitochondrial protein content, the average rate of protein synthesis was similar between sham and heart failure groups. This was due to bidirectional changes in the synthesis rate of different mitochondrial proteins. Although the half-lives of several proteins differed largely between SSM and IFM, when the half-lives of all analyzed proteins were plotted against each other, they were strongly correlated (r2=0.90, p<0.0001 for sham and r2=0.82, p<0.005 for heart failure groups, Figure 3, bottom panel).
Protein stability is regulated by physico-chemical characteristics including molecular weight, charge, protein order, sequence motif and the N-terminal amino acid of mature protein. Previously, based on the radiolabeling approach it has been proposed that there is a relationship between the degradation rates and both molecular weights and isoelectric points (pI) of proteins. However, consistent with a recent stable isotope-based study assessing proteome turnover [29], we found no correlation between the turnover rate constants and molecular weight or pI values of mitochondrial proteins (data not shown).
Next, we analyzed protein stability based on the N-end rule. According to this rule N-terminal amino acids of mature proteins are classified as either stabilizing (M, I, P, G, V) or destabilizing (Q, N, D, E, C, R, K, H, F, L, W, A, S, T) [30]. Thus, based on the N-end rule, mitochondrial proteins in our study were classified as with stabilizing (n=7) and destabilizing (n=40) N-terminal end. We found significant differences between the half-lives of proteins with stabilizing and destabilizing N-terminal residues in both IFM (p<0.05) and SSM (p<0.005) fractions (Figure 4), confirming that proteins with destabilizing amino acid residues are less stable.
Figure 4.
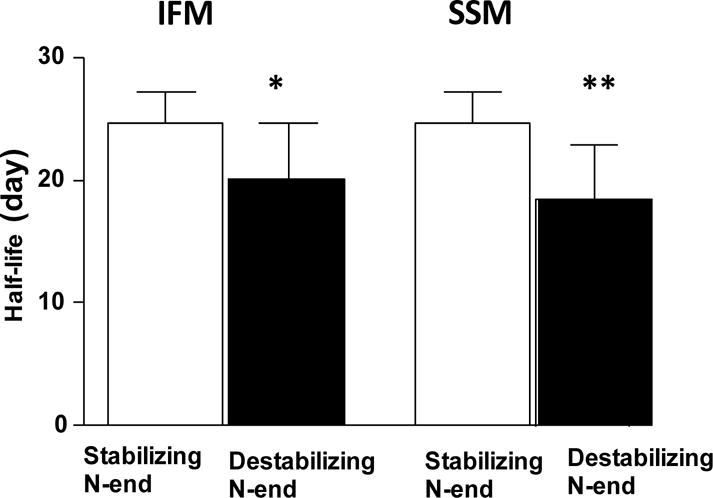
Effect of the N-terminal end on protein stability. Proteins were grouped based on the N-terminal amino acid of a mature protein. Proteins with the stabilizing amino acids (n=7) have significantly longer half-lives than proteins and destabilizing amino acids (n=40). *P=0.02, **P=0.001.
Finally, we assessed protein stability based on PEST motifs calculated by the online software (http://emboss.bioinformatics.nl/cgi-bin/emboss/epestfind). It has been proposed that the proteins containing segments with 10–50 amino acids sequences enriched in proline (P), glutamate (E), serine (S) and threonine (T) have faster turnover rates [31]. The software identified only 3 proteins (out of 48) with PEST motif. Comparison of the half-lives of proteins with PEST versus proteins with poor or no PEST indicated no association of protein turnover with the PEST motifs. Although we analyzed only a limited number of proteins, similar conclusion was reached in a recent study with the large number (>550) of proteins [29].
3.3.2. Comparison of half-lives of mitochondrial proteins based on intra-organelle location and their metabolic functions
Analysis of protein synthesis based on protein location within the mitochondrion revealed a shorter half-life for outer membrane proteins than inner matrix proteins for both sham and heart failure groups in both SSM and IFM (Figure 5), consistent with our previous observations in normal rats [9].
Figure 5.
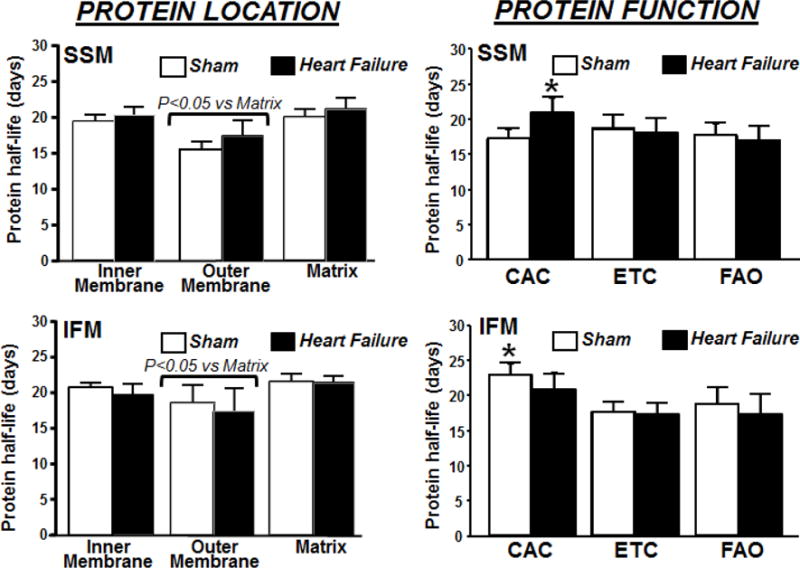
Comparison of the half-lives of cardiac mitochondrial proteins based on intra-organelle location (left panels) and their metabolic function (right panel) in SSM and IFM from sham and heart failure animals. The data are derived using non-linear regression analysis of peptides 2H-labeling to generate the best-fit curves describing protein turnover (n=6/group), * P<0.05 vs. SSM sham. CAC, citric acid cycle; ETC, electron transfer chain; FAO, fatty acid oxidation. See Table 3 for the list of specific proteins.
Mitochondrial proteins were further sorted by function (citric acid cycle, electron transport chain or fatty acid oxidation (see Supplemental Table 3). SSM of heart failure hearts had a 20% longer half-life for proteins involved in the citric acid cycle compared to sham (Figure 5). This slowing of protein turnover by heart failure was not observed in citric acid cycle proteins of IFM, in contrast there was a trend toward increased degradation. On the other hand, the turnover rate was faster for enzymes involved in the citric acid cycle in SSM compared to IFM (Figure 5), suggesting a better preserved turnover of proteins in IFM, a mitochondria population that is critical for energy supply to the contractile apparatus.
The kinetic analysis of individual mitochondrial proteins reveals that heart failure had a differential effect on the turnover rates of mitochondrial proteins. For instance, several proteins from IFM in rats with heart failure had faster turnover rates compared to that in sham animals: the clearance rates of carnitinepalmitoyl transferase 1 and trifunctional protein, involved in mitochondrial fatty acid transport and oxidation, were increased in failing hearts by ~42% (P<0.05) and ~27% (P<0.05), respectively (Figure 6). Similarly, heart failure caused a ~40% (P<0.05) increase in the turnover rate constant of succinate dehydrogenase, a protein involved in the citric acid cycle and Complex II. In contrast, heart failure increased ~25% (P=0.05) the degradation rate of dihydrolipoyllysine acetyltranferase which is part of the pyruvate dehydrogenase complex, in SSM, but not in IFM. In addition, the kinetics of 2 out of 11 analyzed ETC proteins was affected by heart failure. Cytochrome bc subunit 7, a protein of Complex III, displayed increased degradation rates in SSM (~40%, P<0.05) and a similar trend was observed in IFM (~22%, P=0.06) fractions from failing heart. A similar 25% (but not statistically significant) increase in the turnover rate of cytochrome c oxidase subunit 5A caused by heart failure was observed in the IFM. These changes in ETC protein turnover were also associated with alterations in the kinetics of ADP/ATP translocase from IFM (but not SSM) which showed a ~25% increased degradation rate (P<0.05). Taken together, these results suggest that heart failure increased the clearance of several IFM proteins involved in fatty acid transport, oxidation, ETC and ATP synthesis.
Figure 6.
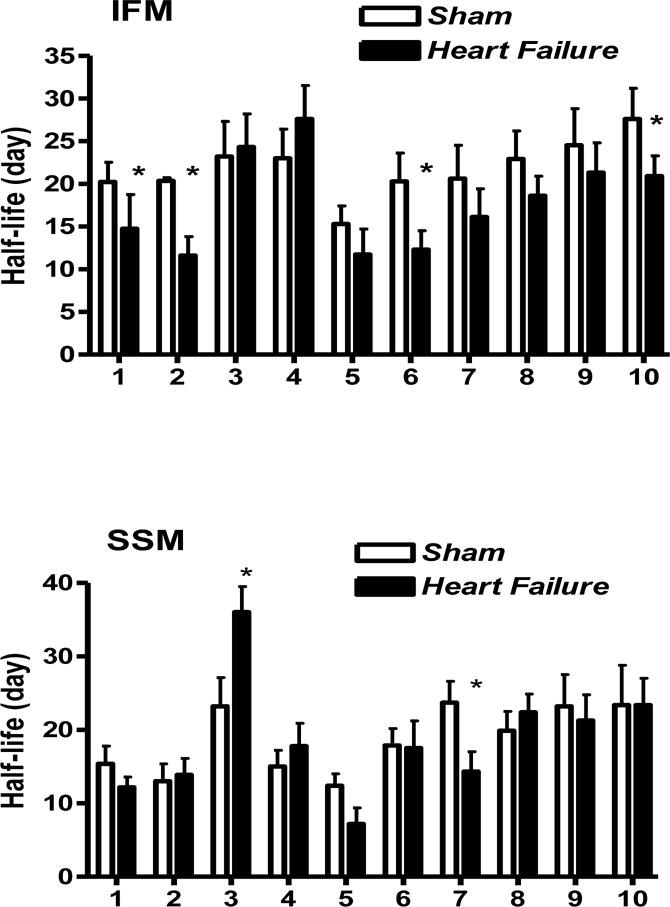
Comparison of the half-lives of selected individual cardiac mitochondrial proteins with different metabolic functions in IFM (top panel) and SSM (bottom panel) from sham and heart failure rats. The data are derived from Table 3. 1: Trifunctional protein, 2: carnitinepalmitoyl transferase 1, 3: Dihydropollysine acetyltranferase (a component of pyruvate dehydrogenase complex), 4: Isocitrate Dehydrogenase, 5: NADH Dehydrogenase Flavoprotein, 6: Succinate Dehydrogenase, 7: Citochrome bc, subunit 7, 8: Cytochrome C Oxidase subunit 4 isoform 1, 9: ATP Synthase α, 10: ADP/ATP translocase 2 (n=6/group, *P<0.05 vs. sham).
4. DISCUSSION
This study was designed to assess the effect of pressure overload-induced heart failure on protein synthesis rate in two diverse mitochondrial populations, i.e. SSM and IFM, characterized with distinct biochemical properties and functions. The main findings are that pressure overload-induced heart failure in rats reduces total mitochondrial protein content, with more pronounced changes in IFM. These alterations are associated with the impairment of basal and stimulated mitochondrial respiration in IFM, while the effects on SSM were only modest. Our proteome dynamics measurements suggest that the increase in degradation of IFM proteins involved in fatty acid oxidation, ETC and ATP synthesis may account, at least in part, for mitochondrial dysfunction in the failing heart. However, despite the mitochondrial defects, the integrated rates of fractional protein synthesis/degradation were similar between sham and heart failure groups due to bidirectional changes in multiple proteins kinetics.
The reduction of the maximal rate of respiration only in IFM from heart failure rats, when palmitoylcarnitine was used as a substrate, indicates that the decreased fatty acid oxidation and ATP generation capacity is more specific for IFM rather than for SSM. Our results point to the down regulation of major fatty acid oxidation pathways as the cause of an energy deficit in the failing heart. This is consistent with the notion that the healthy heart mainly relies on fatty acids oxidation for its energetic needs [32]. The oxidative defect observed in the IFM population is also consistent with the known susceptibility of this mitochondrial subpopulation to oxidative damage [33]. Reductions in mitochondrial protein content and respiration highlight that similar to aging, heart failure-induced mitochondrial dysfunction selectively involves IFM. These results are consistent with the initial studies by Hoppel’s group demonstrating that a decrease in mitochondrial respiration with Complex I and Complex III substrates was present only in IFM (but not in SSM) isolated from hamster hearts with dystrophic cardiomyopathy [16].
Reduced mitochondrial protein content and impaired respiratory function observed in this and previous studies suggest that defects in IFM may contribute to contractile dysfunction in heart failure. Although several mechanisms have been proposed, the underlying mechanisms contributing to mitochondrial dysfunction in heart failure are not yet fully understood. Recent proteomics- based discovery studies showed proteome remodeling in pressure overload-induced heart failure and highlighted the role of mitochondrial oxidative stress [2, 34]. Those reports demonstrated that the changes in mitochondrial proteins are bidirectional, with down regulation of proteins involved in fatty acid metabolism and increased expression of proteins involved in glycolysis, apoptosis, mitochondrial unfolded protein response and proteolysis. In the present study we tested the hypothesis that the decreased mitochondrial proteins turnover may contribute to the reduced mitochondrial protein content and respiratory dysfunction in heart failure. The 2H-metabolic labeling approach we used allows determination of the turnover rate constant (a fraction of pool that is turned over per unit of time). At the steady state, the fractional synthesis rate (FSR) is equal to the fractional catabolic rate (FCR) and therefore the rate constant is often referred to as the FCR. The production rate of a protein can be calculated by multiplying the FCR by the pool size (i.e. absolute quantity of an individual mitochondrial protein) [35]. We have measured only FCR (k), but not the levels of individual mitochondrial proteins resulting from the balance between synthesis and degradation. Previous studies demonstrated that proteins involved in fatty acid oxidation and electron transport chain are mostly down regulated in failing hearts [2, 36], although this is not a consistent finding [34]. In this study, we demonstrated that heart failure caused an increased clearance rate of proteins involved in mitochondrial fatty acid oxidation (palmitoylcarnitine transferase I and trifunctional protein). Importantly, our results show that these changes take place predominantly in IFM. Taken together with our data on respiration of mitochondria fed with palmitoylcarnitine, these findings suggest that heart failure-induced impairment in fatty acid oxidation in IFM could be related to the increased degradation of key proteins involved in mitochondrial fatty acid metabolism. Of note, in a normal heart IFM has higher oxidative capacity than SSM and it is more susceptible to oxidative damage [37].
In addition, our results indicate that heart failure also increases the rate of degradation of key ETC proteins of Complex I (NADH Dehydrogenase ubiquinone flavoprotein), Complex II (Succinate Dehydrogenase), and Complex III (Cytochrome bc subunit7). Although not significant, but a trend towards increased degradation was observed for a protein of Complex IV (cytochrome c oxidase subunit 5A). It has been recently proposed that defects in the supercomplex consisting of Complex I/Complex III dimer/Complex IV (the major respirasome essential for the oxidative phosphorylation) is responsible in mitochondrial dysfunction in heart failure [38] and minor changes in proteins levels may affect supercomplex formation. Thus, it is possible that the increased degradation of ETC proteins may contribute to their decreased levels and impaired respirosome integrity. Increased turnover of these ETC proteins could be related to heart failure-induced oxidative stress.
Although heart failure increased clearance of several mitochondrial proteins, we observed a decrease turnover rates for several mitochondrial proteins. Particularly, increased half-life was found for dihydrolipoyl transacetylase (a component of pyruvate dehydrogenase complex pyruvate oxidation). A similar but not significant trend was observed for isocitrate dehydrogenase, enzymes involved in citric acid cycle. Previous studies demonstrated increased expression levels of several glycolytic proteins in heart failure [2, 34]. However, it was unknown whether these changes are due to increased production or decreased degradation of these proteins. A decreased turnover rate constant (increased half-life) of pyruvate dehydrogenase subunit due to heart failure observed in our study suggests that decreased clearance of this protein could be responsible for its increased expression. Combined with our results on increased degradation of fatty acid metabolizing enzymes, these changes may reflect compensatory fuel switching from fatty acid to glucose utilization in failing heart [32]. Since the decrease in fatty acid oxidation precedes the decline of contractility, altered turnover of mitochondrial proteins observed in this study could contribute, but not be a key factor in impairing myocardial functional integrity.
The protein turnover assessed in our study represents the net result of protein synthesis and degradation. Mitochondrial protein synthesis is regulated by multiple signaling pathways including peroxisome proliferator-activated receptors (PPARα, PPARβ and PPARγ). PPARγ co-activators PGC1α and PGC1β contribute to both nuclear and mitochondrial protein synthesis. While PGC1α and PGCβ play overlapping roles in mitochondrial biogenesis, PGC1β mainly maintains mitochondrial function through the regulation of OXPHOS [39] and PGC1α directly stimulates the transcription of mytofusin1 (Mfn1) involved in mitochondrial fusion in non-stressed heart [40]. In contrast to PGC1β, PGC1α also controls the expression of fatty acid oxidation enzymes and responsible for metabolic shift from fatty acid to glucose oxidation [41–43]. Although during compensated cardiac hypertrophy the adaptive increase in PGC1α and PGC1β signaling stimulates mitochondrial biogenesis and oxidative metabolism, in the decompensated heart failure a reduction of both PPARγ co-activators leads to compromised oxidative defense and reduced expression of oxidative phosphorylation, and fatty acid oxidation genes [39, 44]. Thus, observed alterations in the turnover of fatty acid oxidizing and ETC proteins in our study could be related to the decrease in PGC signaling in heart failure.
Decreased half-lives of multiple IFM proteins in our study indicate that pressure overload-induced heart failure stimulates mitochondrial protein degradation in rats. However, from our in vivo turnover study it is difficult to conclude which protein degradation pathway, i.e. mitochondrial proteolysis, autophagy or cytosolic proteolysis, is involved in heart failure-induced alterations. In contrast to the autophagy pathway that is responsible for the turnover of intact organelles [45], protein degradation through ubiquitin proteasome system (UPS) is the major pathway of degradation of individual proteins [46]. The wide range differences in mitochondrial proteins half-lives (from ~10 to ~35 days) observed in this study suggest that UPS, but not autophagy, is the most likely pathway of mitochondrial protein degradation. The signal for UPS degradation is determined by the presence of variety of structural features of a target protein, including charge state, N-terminal residue, specific motifs such as the PEST sequences, and post-translational modifications [29, 47, 48]. In this study, we found that only N-terminal residue was strongly associated with the half-lives of analyzed mitochondrial proteins indicating that the specificity of both ubiquinating enzymes (E1, E2 and E3) and protein sequences determines the distinct turnover rates of individual mitochondrial proteins.
Recent in vitro proteome stability studies suggest that, in addition to mitochondrial proteases, cytosolic proteasomes may also contribute to mitochondrial protein turnover in stressed mitochondria [49]. It has been demonstrated that oxidatively damaged proteins are not efficient substrates for mitochondrial degradation due to diminished proteolytic capacity of mitochondrial 26S proteasome [49]. In contrast, extra-mitochondrial 20S proteasome (20S) is more resistant to oxidative stress and if accessible, mitochondrial proteins could be degraded by 20S. Based on IFM’s susceptibility to oxidative stress [33] it is conceivable that in heart failure IFM proteins are preferentially degraded by extra-mitochondrial proteasomes.
Consistent with our previous finding [9], this study demonstrated that the protein half-lives are associated with their intra-organelle location. In particular, outer-membrane proteins had shorter half-lives than matrix proteins in both sham and heart failure groups, suggesting that the degradation of outer-membrane proteins occurs through the extra-mitochondrial proteasomal pathway due to their cytosolic proximity.
From our study, quantitative (mitochondrial content) and qualitative (mitochondrial function) differences in SSM and IFM do not seem to affect the overall average rate of mitochondrial proteins synthesis in response to pressure overload. This was probably the result of differential regulation of mitochondrial proteins due to heart failure. Comprehensive studies of mitochondrial proteome dynamics with simultaneous quantification of both protein abundance and kinetics in the same experiment would substantially enhance the understanding of the effect of heart failure on mitochondrial proteome remodeling.
5. CONCLUSION
In conclusion, pressure overload-induced heart failure causes a decrease of mitochondrial proteins and respiratory capacity in IFM, but not in SSM. Our proteome dynamics study suggests that these changes might be in part related to increased proteasomal degradation of several proteins involved in fatty acid oxidation and ETC. However, due to bidirectional changes in the kinetics of multiple proteins, the overall rate of protein turnover was not affected in the failing heart. Our results indicates that measuring the kinetics of individual proteins provides a valuable tool to uncover changes in mitochondrial proteome due to heart disease that cannot be obtained by simply measuring their static expression at any given time point.
Supplementary Material
Highlights.
Heart failure (HF) significantly decreased cardiac mitochondrial content in rat.
HF also decreased the maximal rate of interfibrillar mitochondrial respiration.
HF had only modest effect on the respiratory function of subsarcolemmal mitochondria.
HF increased degradation of proteins involved in mitochondrial energy metabolism.
The average half-life of total mitochondrial proteins was not altered due to HF.
Acknowledgments
We are grateful to Dongmei Zhang for processing mitochondrial protein samples for proteomics analysis. We thank Dr. Stephen Previs, who pioneered the heavy water-based metabolic labeling approach, for his insight and efforts on our global proteome dynamic studies. We also are grateful to Drs. Charles Hoppel and Janos Kerner for the fruitful discussion.
SOURCES OF FUNDING
This work was supported by National Institutes of Health Grants HL-074237 (WS), HL-114407 (WS, TK), P01 HL-74237 and R01 HL108213 (FAR), NLBIHHSN268201000037C (RGS) and American Heart Grant 131RG14700011 (TK). The Thermo Orbitrap Elite mass spectrometer used in this study was purchased using NIH shared instrument grant 1S10RR031537-01 (BW).
Abbreviations
- 2H2O
heavy water
- IFM
interfibrillar mitochondria
- SSM
subsarcolemmal mitochondria
- TAC
transverse aorta constriction
- ETC
electron transport chain
- LV
left ventricular
- LTQ
linear trap quadrupole
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- MCAD
medium chain acyl-CoA dehydrogenase
- FCR
fractional catabolic rate
- FSR
fractional synthesis rate
- CID
collisionally induced dissociation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
The authors declare no conflicts of interest, financial or otherwise.
References
- 1.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. The Journal of physiology. 2003;551:491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovascular research. 2010;85:376–84. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 3.Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. Journal of molecular and cellular cardiology. 1992;24:1333–47. doi: 10.1016/0022-2828(92)93098-5. [DOI] [PubMed] [Google Scholar]
- 4.Schwarzer M, Schrepper A, Amorim PA, Osterholt M, Doenst T. Pressure overload differentially affects respiratory capacity in interfibrillar and subsarcolemmal mitochondria. American journal of physiology Heart and circulatory physiology. 2013;304:H529–37. doi: 10.1152/ajpheart.00699.2012. [DOI] [PubMed] [Google Scholar]
- 5.Rachdaoui N, Austin L, Kramer E, Previs MJ, Anderson VE, Kasumov T, et al. Measuring proteome dynamics in vivo: as easy as adding water? Molecular & cellular proteomics : MCP. 2009;8:2653–63. doi: 10.1074/mcp.M900026-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Willard B, Rachdaoui N, Kirwan JP, Sadygov RG, Stanley WC, et al. Plasma proteome dynamics: analysis of lipoproteins and acute phase response proteins with 2H2O metabolic labeling. Molecular & cellular proteomics : MCP. 2012;11:M111 014209. doi: 10.1074/mcp.M111.014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price JC, Holmes WE, Li KW, Floreani NA, Neese RA, Turner SM, et al. Measurement of human plasma proteome dynamics with (2)H(2)O and liquid chromatography tandem mass spectrometry. Analytical biochemistry. 2012;420:73–83. doi: 10.1016/j.ab.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 8.Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, et al. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Molecular & cellular proteomics : MCP. 2012;11:1586–94. doi: 10.1074/mcp.M112.021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasumov T, Dabkowski ER, Shekar KC, Li L, Ribeiro RF, Jr, Walsh K, et al. Assessment of cardiac proteome dynamics with heavy water: slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. American journal of physiology Heart and circulatory physiology. 2013;304:H1201–14. doi: 10.1152/ajpheart.00933.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. Journal of molecular and cellular cardiology. 2001;33:1065–89. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 11.Hofer T, Servais S, Seo AY, Marzetti E, Hiona A, Upadhyay SJ, et al. Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mechanisms of ageing and development. 2009;130:297–307. doi: 10.1016/j.mad.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williamson CL, Dabkowski ER, Baseler WA, Croston TL, Alway SE, Hollander JM. Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. American journal of physiology Heart and circulatory physiology. 2010;298:H633–42. doi: 10.1152/ajpheart.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:419–21. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 14.Asemu G, O’Connell KA, Cox JW, Dabkowski ER, Xu W, Ribeiro RF, Jr, et al. Enhanced resistance to permeability transition in interfibrillar cardiac mitochondria in dogs: effects of aging and long-term aldosterone infusion. American journal of physiology Heart and circulatory physiology. 2013;304:H514–28. doi: 10.1152/ajpheart.00674.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, et al. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. American journal of physiology Heart and circulatory physiology. 2010;299:H529–40. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoppel CL, Tandler B, Parland W, Turkaly JS, Albers LD. Hamster cardiomyopathy. A defect in oxidative phosphorylation in the cardiac interfibrillar mitochondria. The Journal of biological chemistry. 1982;257:1540–8. [PubMed] [Google Scholar]
- 17.Galvao TF, Khairallah RJ, Dabkowski ER, Brown BH, Hecker PA, O’Connell KA, et al. Marine n3 polyunsaturated fatty acids enhance resistance to mitochondrial permeability transition in heart failure but do not improve survival. American journal of physiology Heart and circulatory physiology. 2013;304:H12–21. doi: 10.1152/ajpheart.00657.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galvao TF, Brown BH, Hecker PA, O’Connell KA, O’Shea KM, Sabbah HN, et al. High intake of saturated fat, but not polyunsaturated fat, improves survival in heart failure despite persistent mitochondrial defects. Cardiovascular research. 2012;93:24–32. doi: 10.1093/cvr/cvr258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dabkowski ER, O’Connell KA, Xu W, Ribeiro RF, Jr, Hecker PA, Shekar KC, et al. Docosahexaenoic acid supplementation alters key properties of cardiac mitochondria and modestly attenuates development of left ventricular dysfunction in pressure overload-induced heart failure. Cardiovascular drugs and therapy / sponsored by the International Society of Cardiovascular Pharmacotherapy. 2013;27:499–510. doi: 10.1007/s10557-013-6487-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaha V, Grohmann J, Gobel H, Geibel A, Beyersdorf F, Doenst T. Experimental model for heart failure in rats–induction and diagnosis. The Thoracic and cardiovascular surgeon. 2003;51:211–5. doi: 10.1055/s-2003-42264. [DOI] [PubMed] [Google Scholar]
- 21.Duda MK, O’Shea KM, Tintinu A, Xu W, Khairallah RJ, Barrows BR, et al. Fish oil, but not flaxseed oil, decreases inflammation and prevents pressure overload-induced cardiac dysfunction. Cardiovascular research. 2009;81:319–27. doi: 10.1093/cvr/cvn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. The Journal of biological chemistry. 1977;252:8731–9. [PubMed] [Google Scholar]
- 23.Kerner J, Minkler PE, Lesnefsky EJ, Hoppel CL. Fatty acid chain elongation in palmitate-perfused working rat heart: mitochondrial acetyl-CoA is the source of two-carbon units for chain elongation. The Journal of biological chemistry. 2014;289:10223–34. doi: 10.1074/jbc.M113.524314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The Journal of biological chemistry. 1951;193:265–75. [PubMed] [Google Scholar]
- 25.Khairallah RJ, O’Shea KM, Brown BH, Khanna N, Des Rosiers C, Stanley WC. Treatment with docosahexaenoic acid, but not eicosapentaenoic acid, delays Ca2+-induced mitochondria permeability transition in normal and hypertrophied myocardium. The Journal of pharmacology and experimental therapeutics. 2010;335:155–62. doi: 10.1124/jpet.110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Shea KM, Khairallah RJ, Sparagna GC, Xu W, Hecker PA, Robillard-Frayne I, et al. Dietary omega-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. Journal of molecular and cellular cardiology. 2009;47:819–27. doi: 10.1016/j.yjmcc.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panchal AR, Stanley WC, Kerner J, Sabbah HN. Beta-receptor blockade decreases carnitine palmitoyl transferase I activity in dogs with heart failure. Journal of cardiac failure. 1998;4:121–6. doi: 10.1016/s1071-9164(98)90252-4. [DOI] [PubMed] [Google Scholar]
- 28.Price JC, Khambatta CF, Li KW, Bruss MD, Shankaran M, Dalidd M, et al. The effect of long term calorie restriction on in vivo hepatic proteostatis: a novel combination of dynamic and quantitative proteomics. Molecular & cellular proteomics : MCP. 2012;11:1801–14. doi: 10.1074/mcp.M112.021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doherty MK, Hammond DE, Clague MJ, Gaskell SJ, Beynon RJ. Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. Journal of proteome research. 2009;8:104–12. doi: 10.1021/pr800641v. [DOI] [PubMed] [Google Scholar]
- 30.Gonda DK, Bachmair A, Wunning I, Tobias JW, Lane WS, Varshavsky A. Universality and structure of the N-end rule. The Journal of biological chemistry. 1989;264:16700–12. [PubMed] [Google Scholar]
- 31.Dice JF. Molecular determinants of protein half-lives in eukaryotic cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1987;1:349–57. doi: 10.1096/fasebj.1.5.2824267. [DOI] [PubMed] [Google Scholar]
- 32.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiological reviews. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 33.Lesnefsky EJ, Gudz TI, Moghaddas S, Migita CT, Ikeda-Saito M, Turkaly PJ, et al. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. Journal of molecular and cellular cardiology. 2001;33:37–47. doi: 10.1006/jmcc.2000.1273. [DOI] [PubMed] [Google Scholar]
- 34.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, et al. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovascular research. 2012;93:79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kasumov T, Willard B, Li L, Li M, Conger H, Buffa JA, et al. 2H2O-based high-density lipoprotein turnover method for the assessment of dynamic high-density lipoprotein function in mice. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1994–2003. doi: 10.1161/ATVBAHA.113.301700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. 2002;106:606–12. doi: 10.1161/01.cir.0000023531.22727.c1. [DOI] [PubMed] [Google Scholar]
- 37.Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart failure reviews. 2013;18:607–22. doi: 10.1007/s10741-012-9340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovascular research. 2008;80:30–9. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, et al. PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circulation research. 2011;109:783–93. doi: 10.1161/CIRCRESAHA.111.243964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, et al. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circulation research. 2014;114:626–36. doi: 10.1161/CIRCRESAHA.114.302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. The Journal of clinical investigation. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes & development. 2008;22:1948–61. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10086–91. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovascular research. 2011;90:234–42. doi: 10.1093/cvr/cvr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. Journal of cell science. 2000;113(Pt 7):1189–98. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 46.Powell SR. The ubiquitin-proteasome system in cardiac physiology and pathology. American journal of physiology Heart and circulatory physiology. 2006;291:H1–H19. doi: 10.1152/ajpheart.00062.2006. [DOI] [PubMed] [Google Scholar]
- 47.Varshavsky A, Bachmair A, Finley D. The N-end rule of selective protein turnover: mechanistic aspects and functional implications. Biochemical Society transactions. 1987;15:815–6. doi: 10.1042/bst0150815. [DOI] [PubMed] [Google Scholar]
- 48.Tompa P, Prilusky J, Silman I, Sussman JL. Structural disorder serves as a weak signal for intracellular protein degradation. Proteins. 2008;71:903–9. doi: 10.1002/prot.21773. [DOI] [PubMed] [Google Scholar]
- 49.Lau E, Wang D, Zhang J, Yu H, Lam MP, Liang X, et al. Substrate- and isoform-specific proteome stability in normal and stressed cardiac mitochondria. Circulation research. 2012;110:1174–8. doi: 10.1161/CIRCRESAHA.112.268359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.