

Abstract
The XIII Banff meeting, held in conjunction the Canadian Society of Transplantation in Vancouver, Canada, reviewed the clinical impact of updates of C4d‐negative antibody‐mediated rejection (ABMR) from the 2013 meeting, reports from active Banff Working Groups, the relationships of donor‐specific antibody tests (anti‐HLA and non‐HLA) with transplant histopathology, and questions of molecular transplant diagnostics. The use of transcriptome gene sets, their resultant diagnostic classifiers, or common key genes to supplement the diagnosis and classification of rejection requires further consensus agreement and validation in biopsies. Newly introduced concepts include the i‐IFTA score, comprising inflammation within areas of fibrosis and atrophy and acceptance of transplant arteriolopathy within the descriptions of chronic active T cell–mediated rejection (TCMR) or chronic ABMR. The pattern of mixed TCMR and ABMR was increasingly recognized. This report also includes improved definitions of TCMR and ABMR in pancreas transplants with specification of vascular lesions and prospects for defining a vascularized composite allograft rejection classification. The goal of the Banff process is ongoing integration of advances in histologic, serologic, and molecular diagnostic techniques to produce a consensus‐based reporting system that offers precise composite scores, accurate routine diagnostics, and applicability to next‐generation clinical trials.
Keywords: clinical research/practice, translational research/science, kidney transplantation/nephrology, pathology/histopathology, organ transplantation in general, rejection, rejection: antibody‐mediated (ABMR), rejection: subclinical, rejection: T cell mediated (TCMR)
Short abstract
In this article, the Banff consortium presents the most updated version of the kidney, pancreas, and VCA transplant rejection classification and prospects for implementing intragraft molecular assessment. See the companion report on page 42.
Abbreviations
- aah
hyaline arteriolar thickening
- ah
arteriorlar hyalinosis
- ABMR
antibody‐mediated rejection
- ASHI
American Society for Histocompatibility and Immunogenetics
- BWG
Banff Working Groups
- cg
glomerular double contours
- ci
interstitial fibrosis
- ct
tubular atrophy
- cv
vascular fibrous intimal thickening
- DGF
delayed graft function
- DSA
donor‐specific antibody
- DSAST
donor‐specific antibody–specific transcript
- ELISA
enzyme‐linked immunosorbent assay
- EM
electron microscopy
- FDA
U.S. Food and Drug Administration
- FFPE
formalin‐fixed, paraffin‐embedded
- g
glomerulitis
- GBM
glomerular basement membrane
- HS
highly sensitized
- i
inflammation
- IFTA
interstitial fibrosis and tubular atrophy
- i‐IFTA
interstitial inflammation in areas of interstitial fibrosis and tubular atrophy
- IHC
immunohistochemistry
- IVIG
intravenous immunoglobulin
- mRNA
messenger RNA
- miRNA
microRNA
- MPGN
membranoproliferative glomerulonephritis
- MVI
microvascular invasion
- PAS
periodic acid–Schiff
- PCR
polymerase chain reaction
- ptc
peritubular capillaritis
- PTC
peritubular capillary
- t
tubulitis
- TCMR
T cell–mediated rejection
- TG
transplant glomerulopathy
- ti
total inflammation
- TMA
thrombotic microangiopathy
- v
intimal arteritis
- VCA
vascularized composite allograft
Introduction
The XIII Banff meeting was held October 5–10, 2015, in Vancouver, Canada, in conjunction with the annual meeting of the Canadian Society of Transplantation. A total of 451 delegates from 28 countries attended the conference, including pathologists, immunologists, physicians, surgeons, and immunogeneticists. The main aims of the 2015 conference were to review the clinical impact of the 2013 changes related to the new diagnostic criteria for antibody‐mediated rejection (ABMR) 1 and to identify the next set of challenges in transplant diagnostics. Given the limitations of the current Banff system, a need for a more integrated diagnostic system, including complementary approaches as a companion to the current morphologic gold standard, are needed. Consequently, the prospects for introducing molecular diagnostics into the Banff classification were a main focus. Accordingly, the Banff 2015 conference was preceded by a full‐day premeeting on “Precision Diagnostics” in transplantation. This included presentations from key opinion leaders of the American Society for Histocompatibility and Immunogenetics (ASHI) with the aim to foster collaboration between the societies in transplant diagnostics. This meeting report summarizes the main outcomes from the Banff kidney, pancreas, and vascularized composite allograft (VCA) sessions; the main conclusions from the 2015 Banff liver, heart, and lung sessions will be published elsewhere. The XIV Banff meeting will be held jointly with the Catalan Society of Transplantation in Barcelona, Spain, March 27–31, 2017.
Results From the Banff Working Groups and New Developments
Banff Working Groups (BWGs) have been formed at each of the last four Banff conferences to address and potentially modify specific aspects of the classification 2. Their activities are dynamic and goal directed; therefore, the Banff community decided during the 2015 conference to close or suspend working groups whose work has been completed and published, in press, and/or incorporated into the classification (isolated endarteritis, Banff Initiative for Quality Assurance in Transplantation, fibrosis, implantation biopsy, polyoma virus, C4d‐negative ABMR, and glomerular lesions BWGs) 1, 3, 4, 5, 6, 7. The BWG on highly sensitized patients presented the results of three surveys of pathologists, clinicians, and histocompatibility laboratory directors, comprising 193 centers from six continents, and revealed wide heterogeneity among participating centers regarding immune modulation/desensitization practices, timing of kidney allograft protocol biopsies, and testing and reporting of HLA antibody and donor‐specific antibody (DSA) levels. The TCMR working group's main aims and related ongoing studies are detailed in Table 1 and are expected to provide novel insights by the next Banff meeting.
Table 1.
Summary of active Banff 2015 working groups
| Leaders | Issues to address | Group findings/plans | |
|---|---|---|---|
| TCMR | V. Nickeleit, P. Randhawa | Possible incorporation of i‐IFTA into classification; possible elimination of borderline category; reevaluate thresholds for inflammation and t and possible addition of other findings (e.g. edema) to TCMR diagnostic criteria | Group currently collecting cases of “pure” TCMR (no DSA or C4d) for pathologic evaluation and clinicopathologic correlation |
| Sensitized | L. Cornell, E. Kraus, S. Bagnasco, C. Schinstock, D. Dadhania | Define criteria for HS patients, determine consensus for what personnel and facilities are needed for centers to perform transplantation in HS recipients, standardize the definitions related to management of sensitized transplant recipients | Survey results presented by L. Cornell at 2015 Banff conference; expanded survey, future discussions to address core issues; prepare consensus paper for publication |
| Molecular | M. Mengel, B. Sis | Develop consensus guidelines for circumstances under which it is advisable to apply molecular analysis to renal biopsy tissue and/or serum/urine collected at the time of biopsy; determine the best molecular studies to perform with the aim of generating the needed evidence for adoption of molecular diagnostics into the Banff classification; standardize diagnostic criteria for molecular microscope | Single‐center data using the NanoString method on FFPE tissue presented by Banu Sis at the Banff 2015 conference; validation needed of biopsies from additional centers |
| Electron microscopy | C. Roufosse, H.K. Singh | Interobserver variability and clinical correlations in cg1a lesions and ptcml scoring; potential refinement of ptcml scoring criteria; criteria for amount of GBM reduplication and immune complex‐type deposits allowable in cg1a; multicenter study of the natural history, associations, and predictive value of cg1a and ptcml using consensus criteria | Survey of current practice completed June 2016; circulation of images for interobserver reproducibility, fall 2016; multicenter study 2017–2018 |
| TMAa | M. Afrouzian, J. Becker, H. Liapis, S. Seshan | Generate consensus regarding diagnostic criteria for TMA in renal allografts using histopathology/laboratory data/molecular genetics correlation |
Survey 1 circulated in January 2016; results have been shared with the working group participants. Plan: TMA experts defined and identified; will collect ≈30 cases; generate virtual slides and run digital evaluation |
| Recurrent glomerular diseasea | N. Alachkar | Focus on glomerulopathies: IgA nephropathy, FSGS, MPGN/C3 glomerulopathy; what are frequencies, clinical manifestations, and pathologic characteristics of recurrent/de novo disease? Can any of these predict recurrence and/or graft outcomes? | New working group |
| Composite surrogate end pointsa | A. Loupy, B. Orandi | Respond to the unmet need raised by the FDA meeting held in Arlington, Virginia, in 2015: Build a validated multicenter composite scoring system integrating histopathology with other relevant allograft biomarkers to predict long‐term allograft outcome | New working group |
cg, glomerular double contours; DSA, donor‐specific antibody; FDA, U.S. Food and Drug Administration; FFPE, formalin‐fixed, paraffin‐embedded; FSGS, focal segmental glomerulosclerosis; GBM, glomerular basement membrane; HS, highly sensitized; i‐IFTA, interstitial inflammation in areas of interstitial fibrosis and tubular atrophy; MPGN, membranoproliferative glomerulonephritis; ptcml, peritubular capillary basement membrane multilayering; t, tubulitis; TCMR, T cell–mediated rejection; TMA, thrombotic microangiopathy.
New working group.
Four new BWGs have been formed: (i) thrombotic microangiopathy, (ii) recurrent glomerular diseases, (iii) diagnostic electron microscopy, and (iv) composite surrogate end points. The aim of the latter BWG is to build and validate a composite scoring system integrating histopathology with other relevant allograft biomarkers to predict long‐term allograft outcome as a potential end point for next‐generation clinical trials in the area. The currently active and new working groups and their aims, leaders, initial findings (if appropriate), and ongoing work are listed in Table 1. As an outlook on future challenges, the Banff process founder Kim Solez gave a keynote address on tissue engineering pathology, a new pathology discipline that will likely play an increasing role in future Banff meetings, as transplant pathologists need to embrace tissue engineering pathology in the era of regenerative medicine 8.
New Challenges in Rejection Diagnosis and Classification
During the 2015 Banff conference, there was lively discussion about diagnostic concerns regarding ABMR, T cell–mediated rejection (TCMR), and mixed rejection in renal allografts. Important new data were presented revealing the heterogeneity of clinical expression of ABMR with consequent difficulties for diagnosis. In addition, important insights were presented by ASHI members on how testing for DSAs and interpretation of results should be included in the Banff classification (Table 2).
Table 2.
Key points addressed by the American Society for Histocompatibility and Immunogenetics expert panel during the Banff 2015 conference for improving the current diagnostic system
| Key points |
|---|
| Comprehensive typing of recipient and donor is required to determine presence of HLA‐DSA (HLA‐A, ‐B, ‐C, ‐DRB1, ‐DRB3/4/5, ‐DQA1, ‐DQB1, ‐DPA1, ‐DPB1). |
| Determine DSA specificity at the allelic level (including DQA and DQB and for other loci when allelic‐specific antibodies are present). |
Recognize the assay limitations and minimize the inherent issues with reagents and patient sera when DSA specificity and level are considered:
|
| Correlation of DSA with biopsy findings including molecular data should incorporate some quantitation of antibody level to better estimate DSA burden. |
DSA, donor‐specific antibody.
Recent evidence indicates that subclinical ABMR has important clinical implications, even in non–highly sensitized patients with de novo DSAs 9. As noted by Orandi et al, “Increasing numbers of transplant physicians are encountering this problem, which may become more common given new therapeutic agents and new organ allocation policies” 10.
A growing number of centers perform high‐risk renal transplants, thereby intensifying the need for improved assessment of subclinical ABMR 11 and the clinical implications of its kinetics and response to therapy 10. Advances in antibody testing by multiplex bead array assays have greatly enhanced the sensitivity and precision of detection of circulating DSAs 12. Accumulating evidence supports the concept that not all DSAs are equivalent and that DSA properties (ability to bind complement or IgG subclass), beyond simple positivity and mean fluorescence intensity, are associated with distinct outcomes and injury phenotypes in preexisting or recurrent as well as de novo DSAs 13, 14, 15, 16, 17, 18, 19, 20. These distinct DSA properties and their relationship with distinct allograft injury patterns is also increasingly demonstrated in other solid organ transplants such as liver 21 and heart 22. It was also noted that time course, kinetics, and properties of DSA fluctuate 15, 23. Consequently, interpretation of studies evaluating sera at a single time point, especially late after transplantation, should be interpreted with caution because of potential selection bias 24, 25. Despite the usefulness of multiplex bead array assays, inherent limitations, technical issues, and lack of available DSA data at the time of biopsy make diagnoses complex. It was reemphasized that non–anti‐HLA DSAs can produce allograft injury alone or together with anti‐HLA DSAs 26, 27, 28. These observations raise the question of whether ABMR can be diagnosed in the absence of documented DSAs based on ABMR‐related pathology only, namely, microcirculation inflammation, C4d deposition, and vasculitis with or without increased expression of DSA‐associated gene sets 29, 30.
Furthermore, many cases of ABMR in renal allografts, particularly late ABMR associated with de novo DSAs, can present as mixed ABMR and TCMR 31. Renal allograft biopsies with microvascular inflammation plus intimal arteritis also frequently show tubulointerstitial TCMR changes 9, 32. These cases likely represent mixed ABMR and TCMR and, not surprisingly, are often not responsive to treatment for either ABMR or TCMR alone 32, 33. This may be related in large part to the fact that many cases of late ABMR are associated with nonadherence 34. TCMR is also a documented predisposing factor for the future development of de novo DSAs, as demonstrated in two recent studies 9, 11. More data are needed regarding transplant glomerulopathy (TG) or double contours with or without microcirculation inflammation in terms of disease activity and progression and thus necessity of treatment. A key question discussed during the meeting was whether patients with TG should be treated for active ABMR or whether it should be accepted that these patients will progress to graft loss regardless of treatment. A study by Kahwaji et al 35 showed in a small cohort of patients, all with TG, that those with active microvascular invasion (MVI) were significantly more likely to show stabilization of graft function with intravenous immunoglobulin (IVIG) and rituximab than patients with similar histology who were not treated, whereas patients with TG without active MVI did not benefit from treatment with IVIG and rituximab. The findings suggest that the decision as to whether to treat patients with TG, particularly those with DSAs, should depend on whether there is concurrent active MVI. More recently, a pilot randomized control trial showed that patients with chronic ABMR that responded to complement blockade eculizumab by improved GFR were the ones that had complement (C1q binding) circulating anti‐HLA DSAs at the time of diagnosis 36. This important issue will be addressed further at Banff 2017.
DSA Against HLA or Other Antigens in the Diagnosis of ABMR
The Banff 2013 classification requires the presence of “serologic evidence of DSA against HLA or other antigens” (criterion 3) for diagnosis of both acute/active and chronic active ABMR; however, peritubular capillary C4d deposition is highly specific for DSA and potentially identifies antibodies against endothelial antigens and DSA currently not tested for in many laboratories (e.g. antibodies to HLA DP, non‐HLA antigens). Furthermore, a recent study showed similar graft outcomes, at least in chronic active ABMR, in cases with C4d or DSA and those with C4d and DSA 37. The attendees of kidney‐specific sessions at Banff 2015 were polled as to whether the requirement for DSA for diagnosis of ABMR can be waived in biopsies showing both morphologic evidence of acute or chronic tissue injury (as defined in criterion 1 of the Banff 2013 classification for acute/active and chronic active ABMR, respectively) and C4d staining in peritubular capillaries; however, the opinion of the majority of the Banff panel (with some dissenters) was that this was not warranted by the current data. It was instead decided to add the following phrase to the classification for both acute/active and chronic active ABMR, as a corollary to criterion 3: “Biopsies meeting the above histologic criteria and showing diffuse or focal linear peritubular capillary C4d staining on frozen or paraffin sections are associated with a high probability of ABMR and should [undergo] prompt expedited DSA testing.”. Table 3 summarizes this new addition to the classification, and the complete and most updated Banff classifications for renal allograft diagnoses are shown in Table 3.
Table 3.
Updated 2015 Banff classification categories
| Category 1: Normal biopsy or nonspecific changes | |
| Category 2: Antibody‐mediated changes | |
| Acute/active ABMR | All three features must be present for diagnosis. Biopsies showing histological features plus evidence of current/recent antibody interaction with vascular endothelium or DSA, but not both, may be designated as suspicious for acute/active ABMR. Lesions may be clinically acute or smoldering or may be subclinical; it should be noted if the lesion is C4d‐positive or C4d‐negative, based on the following criteria:
|
| Chronic active ABMRb | All three features must be present for diagnosis. As with acute/active ABMR, biopsies showing histological features plus evidence of current/recent antibody interaction with vascular endothelium or DSA, but not both, may be designated as suspicious, and it should be noted if the lesion is C4d‐positive or C4d‐negative, based on the criteria listed:
|
| C4d staining without evidence of rejection | All three features must be present for diagnosisd
|
| Category 3: Borderline changes | |
| Suspicious for acute TCMR |
|
| Category 4: TCMR | |
| Acute TCMR Grade |
|
| Chronic active TCMR | Chronic allograft arteriopathy (arterial intimal fibrosis with mononuclear cell infiltration in fibrosis, formation of neointima); note that such lesions may represent chronic active ABMR as well as TCMR; the latter may also be manifest in the tubulointerstitial compartment |
| Category 5: Interstitial fibrosis and tubular atrophy | |
| Grade |
|
| Category 6: Other changes not considered to be caused by acute or chronic rejection | |
|
|
ABMR, antibody‐mediated rejection; cg, glomerular double contours; DSA, donor‐specific antibody; EM, electron microscopy; g, glomerulitis; i, inflammation; IF, immunofluorescence; IHC, immunohistochemistry; ptc, peritubular capillaritis; t, tubulitis; TCMR, T cell–mediated rejection; TG, transplant glomerulopathy; TMA, thrombotic microangiopathy; v, intimal arteritis.
It should be noted that these arterial lesions may be indicative of ABMR, TCMR, or mixed ABMR/TCMR. The v lesions are only scored in arteries having a continuous media with two or more smooth muscle layers.
Lesions of chronic, active ABMR can range from primarily active lesions with early TG evident only by EM (cg1a) to those with advanced TG and other chronic changes in addition to active microvascular inflammation. In the absence of evidence of current/recent antibody interaction with the endothelium (those features in the second section of Table 3), the term “active” should be omitted; in such cases, DSAs may be present at the time of biopsy or at any previous time after transplantation.
Seven or more layers in one cortical peritubular capillary and five or more in two additional capillaries, avoiding portions cut tangentially.
The clinical significance of these findings may be quite different in grafts exposed to anti–blood group antibodies (ABO‐incompatible allografts), in which they do not appear to be injurious to the graft and may represent accommodation; however, with anti‐HLA antibodies, such lesions may progress to chronic ABMR and more outcome data are needed.
A set of transcripts (DSA‐specific transcripts [DSASTs]) was determined to be differentially expressed in renal allograft biopsies from DSA‐positive versus ‐negative patients 29, a finding that was later confirmed independently at a different center 30. Consequently, DSASTs have the potential to identify cases of ABMR in patients with non‐detectable HLA DSA. It is not clear to what extent, if any, transcript patterns will be affected by prognostically different DSAs, including anti‐HLA class I versus class II; antibodies with high versus low mean fluorescence intensity; complement‐binding versus non–complement‐binding antibodies 15, 16, 17, 19; and antibodies of different IgG subclasses 24. Further prospective validation is required.
Chronic Active TCMR and Interstitial Inflammation in Areas of Interstitial Fibrosis and Tubular Atrophy
The most recent Banff criteria for chronic active TCMR 38 list only vascular lesions (arterial intimal fibrosis with mononuclear cell infiltration within the sclerotic intima; transplant arteriopathy) (Table 3). This is likely neither complete nor fully accurate; however, sufficient data are currently not available to properly define this diagnosis. Interstitial inflammation in areas of interstitial fibrosis and tubular atrophy (i‐IFTA) was discussed among participants of the Banff meeting as a potential lesion of chronic active TCMR. Although the association of i‐IFTA with decreased graft survival is well documented 39, 40, 41, the pathogenesis of i‐IFTA and to what extent this represents a manifestation of TCMR is much less clear. Similarly, the significance of tubulitis in atrophic tubules is unclear. Gene expression studies on microdissected foci of i‐IFTA might help assess this. In light of the established deleterious effect on graft survival of i‐IFTA and IFTA with Banff inflammation (i) score >0, it was agreed that i‐IFTA should be included as part of the Banff lesion scoring. Moreover, i‐IFTA should be graded as mild, moderate, or severe based on whether it involves 10–25%, 26–50%, or >50%, respectively, of the scarred cortical tissue (Table 4, and supplementary material for scoring criteria). Note that the extent of i‐IFTA is not analogous to the Banff total inflammation score, the latter representing the sum of inflammation in scarred and nonscarred areas of the cortex. Consequently, it was decided to modify the Banff 2007 criteria by adding a statement (Table 3, category 4), reflecting findings that lesions of transplant arteriopathy may represent chronic active ABMR 42 as well as TCMR—also shown in experimental studies 43—and that chronic active TCMR may also be manifest in the tubulointerstitial compartment.
Table 4.
Banff lesion grading system
| Lesions | |
|---|---|
| Quantitative criteria for inflammation: i score | |
| i0 | No inflammation or in <10% of unscarred cortical parenchyma |
| i1 | Inflammation in 10–25% of unscarred cortical parenchyma |
| i2 | Inflammation in 26–50% of unscarred cortical parenchyma |
| i3 | Inflammation in >50% of unscarred cortical parenchyma |
| Quantitative criteria for tubulitis: t score | |
| t0 | No mononuclear leukocytes in tubules |
| t1 | Foci with one to four leukocytes per tubular cross‐section (or 10 tubular cells) |
| t2 | Foci with five to 10 leukocytes per tubular cross‐section (or 10 tubular cells) |
| t3 | Foci with >10 leukocytes per tubular cross‐section or the presence of two or more areas of tubular basement membrane destruction accompanied by i2/i3 inflammation and t2 elsewhere |
| Quantitative criteria for intimal arteritis: v score | |
| v0 | No arteritis |
| v1 | Mild to moderate intimal arteritis in at least one arterial cross‐section |
| v2 | Severe intimal arteritis with at least 25% luminal area lost in at least one arterial cross‐section |
| v3 | Transmural arteritis and/or arterial fibrinoid change and medial smooth muscle necrosis with lymphocytic infiltrate in vessel |
| Quantitative criteria for glomerulitis: g score | |
| g0 | No glomerulitis |
| g1 | Glomerulitis in <25% of glomeruli |
| g2 | Segmental or global glomerulitis in 25–75% of glomeruli |
| g3 | Glomerulitis in >75% of glomeruli |
| Quantitative criteria for peritubular capillaritis: ptc score | |
| ptc0 | At least one leukocyte in <10% of cortical PTCs and/or maximum number of leukocytes <3 |
| ptc1 | At least one leukocyte cell in ≥10% of cortical PTCs with three or four leukocytes in most severely involved PTC |
| ptc2 | At least one leukocyte in ≥10% of cortical PTCs with five to 10 leukocytes in most severely involved PTC |
| ptc3 | At least one leukocyte in ≥10% of cortical PTCs with >10 leukocytes in most severely involved PTC |
| Quantitative criteria for total inflammation: ti score | |
| ti0 | No or trivial interstitial inflammation (<10% of total cortical parenchyma) |
| ti1 | 10–25% of total cortical parenchyma inflamed |
| ti2 | 26–50% of total cortical parenchyma inflamed |
| ti3 | >50% of total cortical parenchyma inflamed |
| Quantitative criteria for inflammation in area of interstitial fibrosis and tubular atrophy: i‐IFTA score | |
| i‐IFTA0 | No inflammation or <10% of scarred cortical parenchyma |
| i‐IFTA1 | Inflammation in 10–25% of scarred cortical parenchyma |
| i‐IFTA2 | Inflammation in 26–50% of scarred cortical parenchyma |
| i‐IFTA3 | Inflammation in >50% of scarred cortical parenchyma |
| Quantitative criteria for C4d score | |
| C4d0 | No staining of PTCs (0%) |
| C4d1 | Minimal C4d staining (>0 but <10% of PTCs) |
| C4d2 | Focal C4d staining (10–50% of PTCs) |
| C4d3 | Diffuse C4d staining (>50% of PTCs) |
| Quantitative criteria for double contour: cg score | |
| cg0 | No GBM double contours by light microscopy or EM |
| cg1a | No GBM double contours by light microscopy but GBM double contours (incomplete or circumferential) in at least three glomerular capillaries by EM, with associated endothelial swelling and/or subendothelial electron‐lucent widening |
| cg1b | Double contours of the GBM in 1–25% of capillary loops in the most affected nonsclerotic glomerulus by light microscopy; EM confirmation is recommended if EM is available |
| cg2 | Double contours affecting 26–50% of peripheral capillary loops in the most affected glomerulus |
| cg3 | Double contours affecting >50% of peripheral capillary loops in the most affected glomerulus |
| Quantitative criteria for mesangial matrix expansion: mm score | |
| mm0 | No more than mild mesangial matrix increase in any glomerulus |
| mm1 | At least moderate mesangial matrix increase in up to 25% of nonsclerotic glomeruli |
| mm2 | At least moderate mesangial matrix increase in 26–50% of nonsclerotic glomeruli |
| mm3 | At least moderate mesangial matrix increase in >50% of nonsclerotic glomeruli |
| Quantitative criteria for arteriolar hyalinosis: ah score | |
| ah0 | No PAS‐positive hyaline arteriolar thickening |
| ah1 | Mild to moderate PAS‐positive hyaline thickening in at least one arteriole |
| ah2 | Moderate to severe PAS‐positive hyaline thickening in more than one arteriole |
| ah3 | Severe PAS‐positive hyaline thickening in many arterioles |
| Alternative quantitative criteria for hyaline arteriolar thickening: aah score | |
| aah0 | No typical lesions of calcineurin inhibitor–related arteriolopathy |
| aah1 | Replacement of degenerated smooth muscle cells by hyaline deposits in only one arteriole, without circumferential involvement |
| aah2 | Replacement of degenerated smooth muscle cells by hyaline deposits in more than one arteriole, without circumferential involvement |
| aah3 | Replacement of degenerated smooth muscle cells by hyaline deposits with circumferential involvement, independent of the number of arterioles involved. |
| Quantitative criteria for vascular fibrous intimal thickening: cv score | |
| cv0 | No chronic vascular changes |
| cv1 | Vascular narrowing of up to 25% luminal area by fibrointimal thickening |
| cv2 | Vascular narrowing of 26–50% luminal area by fibrointimal thickening |
| cv3 | Vascular narrowing of >50% luminal area by fibrointimal thickening |
| Quantitative criteria for interstitial fibrosis: ci score | |
| ci0 | Interstitial fibrosis in up to 5% of cortical area |
| ci1 | Interstitial fibrosis in 6–25% of cortical area (mild interstitial fibrosis) |
| ci2 | Interstitial fibrosis in 26–50% of cortical area (moderate interstitial fibrosis) |
| ci3 | Interstitial fibrosis in >50% of cortical area (severe interstitial fibrosis) |
| Quantitative criteria for tubular atrophy: ct score | |
| ct0 | No tubular atrophy |
| ct1 | Tubular atrophy involving up to 25% of the area of cortical tubules (mild tubular atrophy) |
| ct2 | Tubular atrophy involving 26–50% of the area of cortical tubules (moderate tubular atrophy) |
| ct3 | Tubular atrophy involving in >50% of the area of cortical tubules (severe tubular atrophy) |
aah, hyaline arteriolar thickening; ah, arteriorlar hyalinosis; cg, glomerular double contours; ci, interstitial fibrosis; ct, tubular atrophy; cv, vascular fibrous intimal thickening; EM, electron microscopy; g, glomerulitis; GBM, glomerular basement membrane; i, inflammation; i‐IFTA, interstitial inflammation in areas of interstitial fibrosis and tubular atrophy; mm, mesangial matrix expansion; PAS, periodic acid–Schiff; ptc, peritubular capillaritis; PTC, peritubular capillary; t, tubulitis; v, intimal arteritis.
During the postmeeting discussion, it was clearly articulated that further studies are needed to understand the significance of i‐IFTA in the context of chronic active TCMR before i‐IFTA can be included as a diagnostic criterion. In particular, the ongoing work of the borderline/TCMR BWG is expected to generate relevant data in this context.
Prospects for Adopting Molecular Pathology in Renal Allograft Diagnosis
As part of the 2013 revision of the Banff classification for diagnosing ABMR, molecular assessment of transcripts indicative of endothelial injury in the renal allograft biopsy was added as a potential diagnostic criterion 1; however, there is no consensus on which transcripts are diagnostic or on the criteria for positivity. Standards for platforms, methods, and reproducibility for such molecular diagnostic assays have not yet been set; such standards are a requirement for robust clinical validation and adoption in diagnostic pathology laboratories. During the 2015 Banff premeeting on “Precision Diagnostics in Transplantation,” current knowledge in the area of molecular transplant diagnostics was reviewed. State‐of‐the‐art presentations on molecular diagnostics in allograft biopsies and body fluids revealed that significant commonalities exist with regard to the molecular phenotype in transplant biopsies from different organ types 44. In addition, overlap exists with molecular signatures found in body fluids 45. In contrast, there is considerable heterogeneity among published studies with regard to how the molecular phenotype has been assessed and applied as a potential diagnostic and/or predictive tool 46. Over the last decade, transplant biopsies, blood, and urine have been studied comprehensively, primarily using transcriptomics, and have led to novel insights into the molecular phenotypes of organ transplants 47, 48, 49, 50, 51, 52, 53. Current ongoing studies—for example, the INTERCOM studies 47, 48—are assessing a molecular microscope approach in real time for examining kidney allograft biopsies and comparing the gene expression classifiers and diagnosis to the current gold standard histopathology. This represents a step forward and will generate important results to help guide integration of molecular analysis with morphology. Accordingly, at the 2015 Banff meeting, converging opinion was supported by recent data 50, 53 that molecular transplantation pathology is at the point where it can be translated into clinically relevant and applicable diagnostic tools. The obstacles to be overcome lie in (i) the lack of a true diagnostic gold standard against which new molecular diagnostics can be compared and calibrated (there is no gold standard for serology or histology either); (ii) the fact that data have been generated from heterogeneous cohorts with diagnostic labels assigned based on different iterations of the Banff classification; (iii) the absence of completed prospective, controlled, randomized validation studies; and (iv) the lack of agreement on the transcripts to be measured and how to measure them.
Most disease processes operating in organ transplants represent a spectrum of certain biological processes. Accordingly, our current diagnostic criteria (e.g. for TCMR and ABMR) are built on semiquantitative diagnostic thresholds of lesions associated with a certain phenotype. Such thresholds aim to represent the optimal trade‐off between side effects of enhanced treatment (i.e. overimmunosuppression) and the detrimental impact of further disease progression (i.e. underimmunosuppression). A potential path forward would be to generate consensus in molecular transplant diagnostics regarding which molecules are assessed or quantified in what settings (Table 5) and then to develop clinically relevant diagnostic thresholds through retrospective and prospective multicenter validation studies based on standardized assessment of the same molecular lesions in the same clinical context. This approach would be analogous to the Banff consensus process in 1991 for morphologic lesions. Previous research revealed strong associations between certain molecular pathways and well‐established Banff histologic lesions (Figure 1). These key molecular pathways can be represented and thus assessed by relatively few molecules from each pathway, either through quantification of respective gene sets or through summarizing such genes in weighted equations as diagnostic classifiers. Generating consensus for sets of molecules or classifiers reflecting certain biological or disease processes related to the established histologic Banff lesions would enable us to assess and validate their clinical value. In this regard, the most robust evidence is currently available for the association among antibody‐mediated injury; microcirculation inflammation; and increased expression of endothelial, NK cell, and inflammation‐associated transcripts in the allograft 29, 54, 55.
Table 5.
Key areas for which consensus needs to be generated and validated to adopt molecular diagnostics into the Banff classification
| Indication | Applications | Methods |
|---|---|---|
| Diagnosis | Tissue/biopsy | Targets |
|
TCMR ABMR Injury, acute Injury, chronic |
Biopsies for cause Protocol biopsies |
mRNA miRNA Free DNA Proteins Metabolites |
| Body fluids | ||
|
Urine Blood Bile | ||
| Prediction (prognosis) | ||
|
Failure Initial function/DGF Response to treatment (companion diagnostic) |
Platforms | |
|
PCR Microarrays ELISA Flow NanoString Luminex IHC Other |
||
| Treatment monitoring | ||
|
Response to treatment (after treatment) Side effects/dosing Trial end point |
||
ABMR, antibody‐mediated rejection; DGF, delayed graft function; ELISA, enzyme‐linked immunosorbent assay; IHC, immunohistochemistry; mRNA, messenger RNA; miRNA, microRNA; PCR, polymerase chain reaction; TCMR, T cell–mediated rejection.
Figure 1.
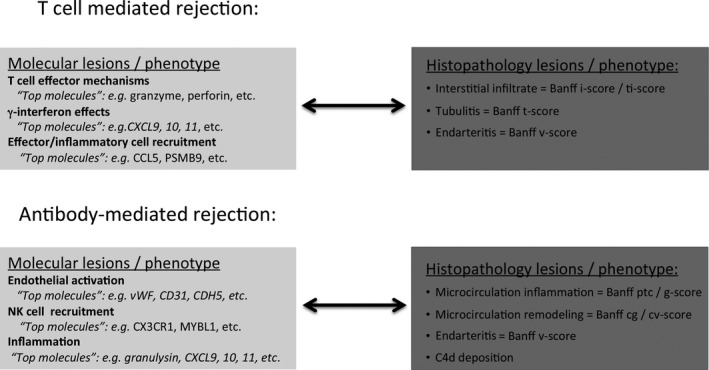
Molecular lesions and their corresponding histologic lesions in T cell–mediated rejection and antibody‐mediated rejection in kidney allografts. cg, glomerular double contours; cv, vascular fibrous intimal thickening; i, inflammation; ptc, peritubular capillaritis; ti, total inflammation; v, intimal arteritis.
Discussion of the above approach took place at the 2015 Banff meeting and continued afterward via e‐mail exchange among the key opinion leaders. From these interactions, key next steps toward adopting molecular diagnostics into transplantation pathology were identified and are summarized (Table 6):
Table 6.
Identified knowledge gap in the adoption process for molecular transplant diagnostics
| ABMR |
| Comparison of subclinical ABMR versus clinical ABMR |
|
Comparison of DSA‐negative biopsies versus DSA‐positive biopsies in sequence from the same patient |
|
Comparison of matched biopsies from adherent versus nonadherent patients |
|
Comparison of histologically similar biopsies from patients with anti‐HLA versus non‐HLA antibody ligands mediating ABMR; define the molecular and histologic phenotypes of ABMR mediated by non‐HLA antibodies |
|
Comparison of ABMR biopsies with TMA to TMA in native kidneys |
|
Comparison of consensus gene sets to diagnostic ABMR classifiers |
| TCMR |
|
Comparison of TCMR with and without DSA but no glomerulitis or TG (note: ptc is often seen with TCMR) |
| Comparison of early versus late TCMR with different levels of Banff i, t, and i‐IFTA scores |
|
Define the molecular phenotype of borderline cases in the current clinical context, (i.e. after elimination of ABMR and mixed cases) |
|
Comparison of consensus gene sets to diagnostic TCMR classifiers |
| Mixed rejection |
|
Should be a focus because recent data suggest that most cases of ABMR (at least in nonsensitized, nonadherent patients) are mixed rejection |
|
Testing the utility of one common rejection gene signature or classifier versus two separate classifiers for ABMR and TCMR in mixed cases |
ABMR, antibody‐mediated rejection; DSA, donor‐specific antibody; i, inflammation; i‐IFTA, interstitial inflammation in areas of interstitial fibrosis and tubular atrophy; ptc, peritubular capillaritis; t, tubulitis; TCMR, T cell–mediated rejection; TG, transplant glomerulopathy; TMA, thrombotic microangiopathy.
The overwhelming majority of those who commented support pursuing the generation of molecular consensus gene sets (or classifiers) from the overlap between published gene lists, adding key genes based on pathogenesis‐based association with the main clinical indications and phenotypes (TCMR, ABMR).
More collaborative multicenter studies are needed (Table 6) to close existing knowledge gaps before Banff can “officially” adopt specific molecular diagnostics as part of the classification.
Consensus must be generated on gene sets, which can be studied further in a multicenter setting.
Results from such studies should be reviewed at future Banff meetings as part of an ongoing consensus process for molecular diagnostics.
Once consensus for gene sets and/or classifiers for molecular biopsy assessment is achieved, prospective and retrospective validation trials can be initiated. Similar to the validation of histologic Banff lesions and diagnostic rules established in 1991, only multicenter validation of different diagnostic approaches with hard clinical end points (e.g. allograft survival, response to treatment) can establish clinically useful diagnostic thresholds. In the absence of a true diagnostic gold standard, adoption of molecular diagnostics can only be accomplished in a stepwise and iterative approach over time including constant revisiting and refinement of current molecular consensus as new knowledge emerges.
Summary of the Banff Pancreas Session
Three main topics (Table 7) were emphasized at the pancreas transplant session: (i) discussion of controversial morphologic aspects, (ii) progress made with the working groups since the 2013 meeting, and (iii) encouragement of data regarding the utility of endoscopic duodenal cuff biopsies as surrogates of biopsies of the pancreas transplant. Data were presented showing that a normal duodenal cuff biopsy accurately predicts absence of TCMR in the pancreas parenchyma. A study of duodenal cuff biopsies showed a high incidence of cytomegalovirus infection in these samples that we do not know how to interpret at this stage. Furthermore, data from detailed morphologic studies on pale acinar nodules in native and transplant pancreas biopsies, which are still of unclear etiology and clinical significance, were presented 57. A study was presented at the Banff session that showed simultaneous pancreas and kidney transplant biopsies demonstrating high concurrence between acute ABMR in both organs and significant discrepancy between organs for TCMR. Modifications to the Banff pancreas allograft pathology schema were made after consensus was reached following e‐mail circulation to the BWG for Pancreas Pathology and via discussions during the meeting. Main updates include incorporation of acute ABMR grading, improved definitions for TCMR and ABMR, specification of vascular lesions, and inclusion of β cell islet toxicity in the category of islet pathology. In the second part of the session, key opinion leaders discussed morphologic and clinical aspects of graft loss in whole pancreas transplants as well as islet transplantation. It was concluded that better understanding of the etiology of graft loss represents an unmet need. This will require systematic integration of morphologic (pathology), serologic (DSAs and autoantibodies), and clinical–functional (e.g. oral glucose tolerance test) parameters for studying the cause and incidence of pancreas transplant failure.
Table 7.
Updated Banff pancreas allograft rejection grading schema
| 1. Normal |
|
| 2. Indeterminate |
|
| 3. Acute TCMR |
|
| 4. Acute/active ABMR |
|
| 5. Chronic active ABMR |
|
| 6. Chronic arteriopathyc |
|
| 7. Chronic graft fibrosis |
|
| 8. Islet pathology |
|
| 9. Other histologic diagnosis |
|
Categories 2 to 9 may be diagnosed concurrently and should be listed in the diagnosis in the order of their clinicopathologic significance. See Drachenberg et al (61) for morphologic definition of lesions of cell‐mediated rejection and for a list of other histologic diagnoses. ABMR, antibody‐mediated rejection; DSA, donor‐specific antibody; TCMR, T cell–mediated rejection. Histologic features of stereotypical TCMR and ABMR, see Table 3 in Drachenberg et al (60).
Arteritis is not required for the diagnosis of ABMR but can be seen in ABMR as well as TCMR.
Inactive chronic arteriopathy can also be included if there is evidence to suggest it is of new onset.
The pathology report should specify how many medium and large arteries were sampled.
Summary of the VCA Session
The VCA session included speaker presentations and discussion. Focal points of the former were ABMR after face transplantation 58, graft vasculopathy in the skin 59, cutaneous changes among transplant patients, and the expansion of the Banff VCA scoring system. The discussion included challenges to the Banff VCA system, immunohistochemistry markers, specimen adequacy, and differential diagnoses. Collaborative efforts were discussed, and the working group concentrated on the standardization of a document for the retrospective and prospective collection of data. The group will reconvene at an international workshop on VCA histopathology with the goals of continuing discussions of the refinement of the Banff VCA system, the standardized form, and the development of a consensus document that would be accessible worldwide. The goal is to compile data and to review it at the Banff 2017 meeting.
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting information
Data S1: The Banff Manual: Definitions and Rules.
Acknowledgments
We would like to acknowledge the instrumental support from the Roche Organ Transplantation Research Foundation Grant 608390948 awarded to Dr. Kim Solez, which allowed establishing the Banff Foundation for Allograft Pathology. The joint 2015 Banff and Canadian Transplant Society meeting acknowledges the receipt of sponsorship from Astellas, Alexion, Novartis, One Lambda, Renal Pathology Society, American Society of Transplantation, Wiley, Qiagen, Canadian Institute for Health Research, Immucor, Bridge to Life, Organ Recovery Systems, Transplant Connect, Glycorex Transplantation, Transpath Inc., and the University of Alberta.
Loupy A, Haas M, Solez K, Racusen L, Glotz D, Seron D, Nankivell BJ, Colvin RB, Afrouzian M, Akalin E, Alachkar N, Bagnasco S, Becker JU, Cornell L, Drachenberg C, Dragun D, De Kort H, Gibson IW, Kraus ES, Lefaucheur C, Legendre C, Liapis H, Muthukumar T, Nickeleit V, Orandi B, Park W, Rabant M, Randhawa P, Reed EF, Roufosse C, Seshan SV, Sis B, Singh HK, Schinstock C, Tambur A, Zeevi A & Mengel M. The Banff 2015 Kidney Meeting Report: Current Challenges in Rejection Classification and Prospects for Adopting Molecular Pathology. Am J Transplant 2017; 17: 28–41
See also: Bruneval et al.
References
- 1. Haas M, Sis B, Racusen LC, et al. Banff 2013 meeting report: Inclusion of c4d‐negative antibody‐mediated rejection and antibody‐associated arterial lesions. Am J Transplant 2014; 14: 272–283. [DOI] [PubMed] [Google Scholar]
- 2. Sis B, Mengel M, Haas M, et al. Banff ‘09 meeting report: Antibody mediated graft deterioration and implementation of Banff working groups. Am J Transplant 2010; 10: 464–471. [DOI] [PubMed] [Google Scholar]
- 3. Sis B, Bagnasco SM, Cornell LD, et al. Isolated endarteritis and kidney transplant survival: A multicenter collaborative study. J Am Soc Nephrol 2015; 26: 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mengel M, Chan S, Climenhaga J, et al. Banff initiative for quality assurance in transplantation (BIFQUIT): Reproducibility of C4d immunohistochemistry in kidney allografts. Am J Transplant 2013; 13: 1235–1245. [DOI] [PubMed] [Google Scholar]
- 5. Farris AB, Chan S, Climenhaga J, et al. Banff fibrosis study: Multicenter visual assessment and computerized analysis of interstitial fibrosis in kidney biopsies. Am J Transplant 2014; 14: 897–907. [DOI] [PubMed] [Google Scholar]
- 6. Liapis H, Gaut JP, Klein C, et al. Banff histopathological consensus criteria for pre‐implantation kidney biopsies. Am J Transplant 2016. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adam B, Randhawa P, Chan S, et al. Banff Initiative for Quality Assurance in Transplantation (BIFQUIT): Reproducibility of polyomavirus immunohistochemistry in kidney allografts. Am J Transplant 2014; 14: 2137–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Solez K. Multibiomarker article gives a taste of what the era of regenerative medicine/tissue engineering pathology will be like. Crit Care Med 2015; 43: e599–e600. [DOI] [PubMed] [Google Scholar]
- 9. Wiebe C, Gibson IW, Blydt‐Hansen TD, et al. Rates and determinants of progression to graft failure in kidney allograft recipients with de novo donor‐specific antibody. Am J Transplant 2015; 15: 2921–2930. [DOI] [PubMed] [Google Scholar]
- 10. Orandi BJ, Luo X, Massie AB, et al. Survival benefit with kidney transplants from hla‐incompatible live donors. N Engl J Med 2016; 374: 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loupy A, Vernerey D, Tinel C, et al. Subclinical rejection phenotypes at 1 year post‐transplant and outcome of kidney allografts. J Am Soc Nephrol 2015; 26: 1721–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tait BD, Susal C, Gebel HM, et al. Consensus guidelines on the testing and clinical management issues associated with HLA and non‐HLA antibodies in transplantation. Transplantation 2013; 95: 19–47. [DOI] [PubMed] [Google Scholar]
- 13. Chen G, Sequeira F, Tyan DB. Novel C1q assay reveals a clinically relevant subset of human leukocyte antigen antibodies independent of immunoglobulin G strength on single antigen beads. Hum Immunol 2011; 72: 849–858. [DOI] [PubMed] [Google Scholar]
- 14. Yabu JM, Higgins JP, Chen G, Sequeira F, Busque S, Tyan DB. C1q‐fixing human leukocyte antigen antibodies are specific for predicting transplant glomerulopathy and late graft failure after kidney transplantation. Transplantation 2011; 91: 342–347. [DOI] [PubMed] [Google Scholar]
- 15. Loupy A, Lefaucheur C, Vernerey D, et al. Complement‐binding anti‐HLA antibodies and kidney‐allograft survival. N Engl J Med 2013; 369: 1215–1226. [DOI] [PubMed] [Google Scholar]
- 16. Sicard A, Ducreux S, Rabeyrin M, et al. Detection of C3d‐binding donor‐specific anti‐HLA antibodies at diagnosis of humoral rejection predicts renal graft loss. J Am Soc Nephrol 2015; 26: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Susal C, Wettstein D, Dohler B, et al. Association of kidney graft loss with de novo produced donor‐specific and non‐donor‐specific HLA antibodies detected by single antigen testing. Transplantation 2015; 99: 1976–1980. [DOI] [PubMed] [Google Scholar]
- 18. Yamamoto T, Watarai Y, Takeda A, et al. De novo anti‐HLA DSA characteristics and subclinical antibody‐mediated kidney allograft injury. Transplantation 2015. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 19. Fichtner A, Susal C, Hocker B, et al. Association of C1q‐fixing DSA with late graft failure in pediatric renal transplant recipients. Pediatr Nephrol 2016; 31: 1157–1166. [DOI] [PubMed] [Google Scholar]
- 20. Bamoulid J, Roodenburg A, Staeck O, et al. Clinical outcome of patients with de novo C1q‐binding donor‐specific HLA antibodies after renal transplantation. Transplantation 2016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21. O'Leary JG, Kaneku H, Banuelos N, Jennings LW, Klintmalm GB, Terasaki PI. Impact of IgG3 subclass and C1q‐fixing donor‐specific HLA alloantibodies on rejection and survival in liver transplantation. Am J Transplant 2015; 15: 1003–1013. [DOI] [PubMed] [Google Scholar]
- 22. Zeevi A, Lunz J, Feingold B, et al. Persistent strong anti‐HLA antibody at high titer is complement binding and associated with increased risk of antibody‐mediated rejection in heart transplant recipients. J Heart Lung Transplant 2013; 32: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tambur AR, Herrera ND, Haarberg KM, et al. Assessing antibody strength: Comparison of MFI, C1q, and titer information. Am J Transplant 2015; 15: 2421–2430. [DOI] [PubMed] [Google Scholar]
- 24. Lefaucheur C, Viglietti D, Bentlejewski C, et al. IgG donor‐specific anti‐human HLA antibody subclasses and kidney allograft antibody‐mediated injury. J Am Soc Nephrol 2016; 27: 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Calp‐Inal S, Ajaimy M, Melamed ML, et al. The prevalence and clinical significance of C1q‐binding donor‐specific anti‐HLA antibodies early and late after kidney transplantation. Kidney Int 2016; 89: 209–216. [DOI] [PubMed] [Google Scholar]
- 26. Dragun D, Muller DN, Brasen JH, et al. Angiotensin II type 1‐receptor activating antibodies in renal‐allograft rejection. N Engl J Med 2005; 352: 558–569. [DOI] [PubMed] [Google Scholar]
- 27. Reinsmoen NL, Lai CH, Mirocha J, et al. Increased negative impact of donor HLA‐specific together with non‐HLA‐specific antibodies on graft outcome. Transplantation 2014; 97: 595–601. [DOI] [PubMed] [Google Scholar]
- 28. Zhang Q, Reed EF. The importance of non‐HLA antibodies in transplantation. Nat Rev Nephrol 2016; 12: 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hidalgo LG, Sis B, Sellares J, et al. NK cell transcripts and NK cells in kidney biopsies from patients with donor‐specific antibodies: Evidence for NK cell involvement in antibody‐mediated rejection. Am J Transplant 2010; 10: 1812–1822. [DOI] [PubMed] [Google Scholar]
- 30. Gupta A, Broin PO, Bao Y, et al. Clinical and molecular significance of microvascular inflammation in transplant kidney biopsies. Kidney Int 2016; 89: 217–225. [DOI] [PubMed] [Google Scholar]
- 31. Nickeleit V, Zeiler M, Gudat F, Thiel G, Mihatsch MJ. Detection of the complement degradation product C4d in renal allografts: Diagnostic and therapeutic implications. J Am Soc Nephrol 2002; 13: 242–251. [DOI] [PubMed] [Google Scholar]
- 32. Lefaucheur C, Loupy A, Vernerey D, et al. Antibody‐mediated vascular rejection of kidney allografts: A population‐based study. Lancet 2013; 381: 313–319. [DOI] [PubMed] [Google Scholar]
- 33. Haas M, Kraus ES, Samaniego‐Picota M, Racusen LC, Ni W, Eustace JA. Acute renal allograft rejection with intimal arteritis: Histologic predictors of response to therapy and graft survival. Kidney Int 2002; 61: 1516–1526. [DOI] [PubMed] [Google Scholar]
- 34. Sellares J, de Freitas DG, Mengel M, et al. Understanding the causes of kidney transplant failure: The dominant role of antibody‐mediated rejection and nonadherence. Am J Transplant 2012; 12: 388–399. [DOI] [PubMed] [Google Scholar]
- 35. Kahwaji J, Najjar R, Kancherla D, et al. Histopathologic features of transplant glomerulopathy associated with response to therapy with intravenous immune globulin and rituximab. Clin Transplant 2014; 28: 546–553. [DOI] [PubMed] [Google Scholar]
- 36. Kulkarni S, Kirkiles‐Smith NC, Deng YH, et al. Eculizumab therapy for chronic antibody‐mediated injury in kidney transplant recipients: A pilot randomized controlled trial. Am J Transplant 2016. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 37. Lesage J, Noel R, Lapointe I, et al. Donor‐specific antibodies, C4d and their relationship with the prognosis of transplant glomerulopathy. Transplantation 2015; 99: 69–76. [DOI] [PubMed] [Google Scholar]
- 38. Solez K, Colvin RB, Racusen LC, et al. Banff 07 classification of renal allograft pathology: Updates and future directions. Am J Transplant 2008; 8: 753–760. [DOI] [PubMed] [Google Scholar]
- 39. Mengel M, Reeve J, Bunnag S, et al. Scoring total inflammation is superior to the current Banff inflammation score in predicting outcome and the degree of molecular disturbance in renal allografts. Am J Transplant 2009; 9: 1859–1867. [DOI] [PubMed] [Google Scholar]
- 40. Mannon RB, Matas AJ, Grande J, et al. Inflammation in areas of tubular atrophy in kidney allograft biopsies: A potent predictor of allograft failure. Am J Transplant 2010; 10: 2066–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hueso M, Navarro E, Moreso F, et al. Intragraft expression of the IL‐10 gene is up‐regulated in renal protocol biopsies with early interstitial fibrosis, tubular atrophy, and subclinical rejection. Am J Pathol 2010; 176: 1696–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Loupy A, Vernerey D, Viglietti D, et al. Determinants and outcomes of accelerated arteriosclerosis: Major impact of circulating antibodies. Circ Res 2015; 117: 470–482. [DOI] [PubMed] [Google Scholar]
- 43. Hirohashi T, Uehara S, Chase CM, et al. Complement independent antibody‐mediated endarteritis and transplant arteriopathy in mice. Am J Transplant 2010; 10: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sarwal M, Sigdel T. A common blood gene assay predates clinical and histological rejection in kidney and heart allografts. Clin Transpl 2013: 241–247. [PubMed] [Google Scholar]
- 45. Muthukumar T, Lee JR, Dadhania DM, et al. Allograft rejection and tubulointerstitial fibrosis in human kidney allografts: Interrogation by urinary cell mRNA profiling. Transplant Rev 2014; 28: 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Halloran PF, de Freitas DG, Einecke G, et al. An integrated view of molecular changes, histopathology and outcomes in kidney transplants. Am J Transplant 2010; 10: 2223–2230. [DOI] [PubMed] [Google Scholar]
- 47. Halloran PF, Pereira AB, Chang J, et al. Potential impact of microarray diagnosis of T cell‐mediated rejection in kidney transplants: The INTERCOM study. Am J Transplant 2013; 13: 2352–2363. [DOI] [PubMed] [Google Scholar]
- 48. Halloran PF, Pereira AB, Chang J, et al. Microarray diagnosis of antibody‐mediated rejection in kidney transplant biopsies: An international prospective study (INTERCOM). Am J Transplant 2013; 13: 2865–2874. [DOI] [PubMed] [Google Scholar]
- 49. Halloran PF, Reeve JP, Pereira AB, Hidalgo LG, Famulski KS. Antibody‐mediated rejection, T cell‐mediated rejection, and the injury‐repair response: New insights from the Genome Canada studies of kidney transplant biopsies. Kidney Int 2014; 85: 258–264. [DOI] [PubMed] [Google Scholar]
- 50. Loupy A, Lefaucheur C, Vernerey D, et al. Molecular microscope strategy to improve risk stratification in early antibody‐mediated kidney allograft rejection. J Am Soc Nephrol 2014; 25: 2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li L, Khush K, Hsieh SC, et al. Identification of common blood gene signatures for the diagnosis of renal and cardiac acute allograft rejection. PLoS ONE 2013; 8: e82153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khatri P, Roedder S, Kimura N, et al. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J Exp Med 2013; 210: 2205–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Halloran PF, Famulski KS, Reeve J. Molecular assessment of disease states in kidney transplant biopsy samples. Nat Rev Nephrol 2016; 12: 534–548. [DOI] [PubMed] [Google Scholar]
- 54. Sis B, Jhangri GS, Bunnag S, Allanach K, Kaplan B, Halloran PF. Endothelial gene expression in kidney transplants with alloantibody indicates antibody‐mediated damage despite lack of C4d staining. Am J Transplant 2009; 9: 2312–2323. [DOI] [PubMed] [Google Scholar]
- 55. Hayde N, Broin PO, Bao Y, et al. Increased intragraft rejection‐associated gene transcripts in patients with donor‐specific antibodies and normal biopsies. Kidney Int 2014; 86: 600–609. [DOI] [PubMed] [Google Scholar]
- 56. Halloran PF, Famulski K, Reeve J. The molecular phenotypes of rejection in kidney transplant biopsies. Curr Opin Organ Transplant 2015; 20: 359–367. [DOI] [PubMed] [Google Scholar]
- 57. Troxell ML, Drachenberg C. Allograft pancreas: Pale acinar nodules. Hum Pathol 2016; 54: 127–133. [DOI] [PubMed] [Google Scholar]
- 58. Chandraker A, Arscott R, Murphy GF, et al. The management of antibody‐mediated rejection in the first presensitized recipient of a full‐face allotransplant. Am J Transplant 2014; 14: 1446–1452. [DOI] [PubMed] [Google Scholar]
- 59. Kanitakis J, Petruzzo P, Gazarian A, et al. Capillary thrombosis in the skin: A pathologic hallmark of severe/chronic rejection of human vascularized composite tissue allografts? Transplantation 2016; 100: 954–957. [DOI] [PubMed] [Google Scholar]
- 60. Drachenberg CB, Torrealba JR, Nankivell BJ, et al. Guidelines for the diagnosis of antibody‐mediated rejection in pancreas allografts—Updated Banff grading schema. Am J Transplant 2011; 11: 1792–1802. [DOI] [PubMed] [Google Scholar]
- 61. Drachenberg CB, Odorico J, Demetris AJ, et al. Banff schema for grading pancreas allograft rejection: Working proposal by a multi‐disciplinary international consensus panel. Am J Transplant 2008; 8: 1237–1249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: The Banff Manual: Definitions and Rules.