

Abstract
DNA methylation is an epigenetic modification that plays an important role in the regulation of gene expression. The function of RUNDC3B has yet to be determined, although its dysregulated expression has been associated with malignant potential of both breast and lung carcinoma. To elucidate the potential of using DNA methylation in RUNDC3B as a biomarker in lymphoid malignancies, the methylation status of six regions spanning the CpG island in the promoter region of RUNDC3B was determined in cancer cell lines. Lymphoid malignancies were found to have more prominent methylation and did not express RUNDC3B compared with myeloid malignancies and solid tumours, supporting the potential use of DNA methylation in this region as a biomarker for lymphoid malignancies. RUNDC3B contains a RUN domain in its N‐terminal region that mediates interaction with Rap2, an important component of the mitogen‐activated protein kinase (MAPK) cascade, which regulates cellular proliferation and differentiation. The protein sequence of RUNDC3B also contains characteristic binding sites for MAPK intermediates. Therefore, it is possible that RUNDC3B serves as a mediator between Rap2 and the MAPK signalling cascade. Three genes with MAPK‐inducible expression were downregulated in a methylated leukaemia cell line (HSPA5, Jun and Fos). Jun and Fos combine to form the activating protein 1 transcription factor, and loss of this factor is associated with the dysregulation of genes involved in differentiation and proliferation. We hypothesize that the loss of RUNDC3B secondary to aberrant hypermethylation of the early growth response 3 transcription factor binding site results in dysregulated MAPK signalling and carcinogenesis in lymphoid malignancies. © 2015 The Authors. Hematological Oncology published by John Wiley & Sons Ltd
Keywords: RUNDC3B, DNA methylation, lymphoma, leukaemia, B cell, gene expression
Introduction
DNA methylation is an epigenetic modification that can alter chromatin structure and physically block the access of transcriptional machinery. This modification most often occurs on cytosine residues followed by guanine residues (CpG dinucleotides) in mammals. CpG dinucleotides are not evenly distributed throughout the genome but occur in clusters known as CpG islands (CGIs). CGIs present within the promoter region of genes are normally unmethylated and actively transcribed. The disruption of normal methylation patterns is a hallmark of tumorigenesis, and the gain of methylation in gene promoters often leads to the silencing of tumour suppressor genes 1. These aberrant methylation events can be used as biomarkers and have proven useful in identifying cancer types, risk assessment, diagnosis and response to drug treatment.
Methylation of RUNDC3B has been reported in acute lymphoblastic leukaemia (ALL) and is associated with a reduction in gene expression 2. Interestingly, treatment of ALL cell lines with a demethylating agent restored expression of the RUNDC3B gene, suggesting that methylation within the promoter of this gene plays a role in the regulation of RUNDC3B expression in ALL. Furthermore, methylation of RUNDC3B was not observed in acute myeloid leukaemia (AML), suggesting the putative utility of RUNDC3B methylation as a biomarker in lymphoid malignancies 3.
RUNDC3B is expressed in many different tissues including brain, thymus, ovary, testis, leukocytes, liver, small intestines and prostate 4. It shares high homology with RUNDC3A, a Rap2 interacting protein, and is predicted to also interact with Rap2. The Rap protein family constitutes a subgroup of the Ras superfamily, which are small GTPases that work as molecular switches to regulate many cellular functions such as proliferation, differentiation and cell motility 5. The dysregulation of these cellular functions is commonly associated with cancer formation.
In this study, we sought to determine if RUNDC3B methylation was unique to lymphoid malignancies and to elucidate the molecular mechanisms that contribute to the silencing of the gene. To accomplish these goals, the methylation status of malignant and normal cell lines and of normal B cells isolated from healthy individuals was determined and correlated with expression data. The data suggest that RUNDC3B methylation may have a role in the pathogenesis of lymphoid malignancies.
Materials and methods
Sample preparation
Cell lines comprised nine lymphoid malignancies (MHHCall 3, Nalm 6, Jurkat, Mec‐1, DB, Granta‐519, RL, Raji and Daudi), two myeloid malignancies (U266B1 and KG‐1), five solid tumours (MDA‐MB 231, Hela, A431, A549 and WiDr) and two control lymphoid cell lines (GM06990 and GM00536). One cord blood and one bone marrow sample were also included (Table 1). DNA and RNA extractions were performed with the DNeasy Blood & Tissue Kit and the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). All samples were collected in accordance with approval from the University of Missouri's Institutional Review Board.
Table 1.
Description of cell lines and healthy control samples
| Sample | Description | |
|---|---|---|
| L | MHH Call 3 | B‐acute lymphoblastic leukaemia cell line |
| Nalm 6 | B‐acute lymphoblastic leukaemia cell line | |
| Jurkat | T‐acute lymphoblastic leukaemia cell line | |
| Mec‐1 | Chronic lymphocytic leukaemia cell line | |
| DB | Diffuse large B‐cell lymphoma cell line | |
| Granta‐519 | Mantle cell lymphoma cell line | |
| RL | Follicular lymphoma cell line | |
| Raji | Burkitt's cell line | |
| Daudi | Burkitt's cell line | |
| M | U226B1 | Multiple myeloma cell line |
| KG‐1 | Acute myeloid leukaemia cell line | |
| S | MDA‐MB 231 | Breast carcinoma cell line |
| Hela | Cervical carcinoma cell line | |
| A431 | Epidermal carcinoma cell line | |
| A549 | Lung carcinoma cell line | |
| WiDr | Colorectal adenocarcinoma cell line | |
| C | GM06990 | Lymphoblast cell line |
| GM00536 | Fibroblast cell line | |
| BM | Bone marrow B cells | |
| CB | Cord blood B cells |
Samples are grouped by tissue type.
L, lymphoid; M, myeloid; S, solid tumour; C, healthy controls.
Methylation assays
Bisulfite treatment was performed on 1 µg of DNA using the ZYMO EZ DNA Methylation Gold kit (Zymo Research, Irvine, CA, USA). CT conversion reagent was added to each sample and incubated at 98 °C for 10 min followed by 64 °C for 2.5 h. Each sample was then desulfonated and eluted in a final volume of 50 µl.
MethPrimer (http://www.urogene.org/methprimer/) was utilized to design combined bisulfite and restriction analysis (COBRA) and methylation‐specific polymerase chain reaction (MSP) primers (Table 2). The 25.0 µl reaction included 2.5 µl of 25.0 mM MgCl2, 2.5 µl of PCR Gold Buffer, 0.5 µl of 10 mM dNTPs, 0.75 µl of 10 μM forward and reverse primers, 0.125 µl of AmpliTaq Gold, and 3.0 µl DNA template. Thermal cycling was performed with a 10‐min hot start at 95 °C, followed by four touchdown cycles of 95 °C for 15 s, 60 °C (regions 1, 2 and 6) or 64 °C (region 5) decreasing 1 °C each cycle for 30 s and 68 °C for 30 s. This was followed by 32 cycles of 95 °C for 15 s, 56 °C (regions 1, 2 and 5) or 60 °C (region 4) for 30 s and 68 °C for 30 s. A final extension at 68 °C for 7 min was included. All PCR amplifications were visualized on a 1.5% agarose gel.
Table 2.
RUNDC3B primer sequences and amplicon characteristics
| Primer | Forward sequence/reverse sequence | Amplicon size (bp) | COBRA digest sizesa |
|---|---|---|---|
| Region 1 | 5′‐GTTTTAGGATTTTGAGGGAGTAGTTTAG‐3′ | 301 | 125, 107 and 69 bp |
| 5′‐CCCAAAAACTAATAAACAACAACAC‐3′ | |||
| Region 2 | 5′‐TTGTTGTTTATTAGTTTTTGGGAGG‐3′ | 176 | 93 and 83 bp |
| 5′‐CCCCTTACCTATAACCAAACTTTAAC‐3′ | |||
| Region 3 | 5′‐GTGGTTATTGGCGGTAGTTAGC‐3′ | 104 | MSP |
| Methylated | 5′‐GCGAACCTTTTAAAACAACGA‐3′ | ||
| Region 3 | 5′‐GTGTGGTTATTGGTGGTAGTTAGTG‐3′ | 107 | MSP |
| Unmethylated | 5′‐CACAAACCTTTTAAAACAACAAA‐3′ | ||
| Region 4 | 5′‐GGGTTTTGTCGTTGTTTTTC‐3′ | 374 | MSP |
| Methylated | 5′‐CTTAAAAAAATTCTCGCTCGA‐3′ | ||
| Region 4 | 5′‐GGGTTTTGTTGTTGTTTTTT‐3′ | 378 | MSP |
| Unmethylated | 5′‐CCTACTTAAAAAAATTCTCACTCAA‐3′ | ||
| Region 5 | 5′‐GAGAATTTTTTTAAGTAGGTGTGG‐3′ | 249 | 143 and 106 bp |
| 5′‐AAAACCCAAAACTCTCAACCC‐3′ | |||
| Region 6 | 5′‐GTGGAGAGGAGGAATTTGATTAT‐3′ | 236 | 93, 80 and 63 bp |
| 5′‐AAACTAACACAAAATCCAAAACTAC‐3′ |
The restriction enzyme BstUI was used in all COBRA reactions. COBRA reactions were performed for regions 1, 2, 5 and 6. MSP reactions were performed for regions 3 and 4.
MSP, methylation‐specific polymerase chain reaction.
Restriction enzyme digest reactions were performed to determine whether the amplified region contained methylated cytosines. Each amplicon generated for regions 1, 2, 5 and 6 contained a BstUI restriction site, CG*CG. The 25.0 µl reaction included 6.0 µl of PCR product, 1.0 µl of BstUI enzyme, and 2.5 µl of NEB2 buffer. The reactions were incubated for 4 h at 60 °C and visualized on a 2.5% agarose gel. Samples were categorized as methylated if they exhibited the expected enzyme digest banding pattern (Table 2). If the PCR product was present and no enzyme digest banding pattern was observed, the sample was categorized as unmethylated. Finally, samples that exhibited the digested banding pattern and the undigested PCR product were considered partially methylated.
No acceptable COBRA primers were generated for the interval between regions 2 and 5; therefore, MSP primers were developed for regions 3 and 4. Thermal cycling was performed with a 10‐min hot start at 95 °C, followed by four touchdown cycles of 95 °C for 15 s, 60 °C for 30 s, decreasing 1 °C each cycle, and 68 °C for 30 s. This was followed by 32 cycles of 95 °C for 15 s, 56 °C for 30 s and 68 °C for 30 s. A final extension of 68 °C for 7 min was included. All PCR amplifications were visualized on a 1.5% agarose gel. Samples were categorized as methylated if there was an appropriately sized amplicon produced by the methylated primer pair. Likewise, if the unmethylated primer pair amplified the sample, it was categorized as unmethylated. Finally, if amplicons were visible for both primer sets, the samples were considered partially methylated.
Expression assays
Quantitative real‐time PCR using Taqman primer/probe sets (Applied Biosystems RUNDC3B Hs00379227_m1 and GAPDH Hs0392909_g1) was performed to detect mRNA expression (Life Technologies, Grand Island NY, USA). The 20 µl reactions included 40.0 ng RNA template, 4.0 µl of Taqman reaction buffer, 2.4 µl of magnesium acetate, 0.6 µl of each 10 mM dNTP, 0.8 µl of rth polymerase and 0.2 µl Uracil‐N‐Glycosylase (UNG). All reactions were conducted on the iCycler iQ and processed by icycler software v 3.1 (Bio‐Rad, Hercules, CA, USA). Thermal cycling began with 50 °C for 2 min to activate UNG, 60 °C for 30 min to perform the reverse transcription and 95 °C for 5 min to deactivate UNG. This was followed by 40 cycles of 20 s at 94 °C for 20 s and 62 °C for 1 min. All reactions were performed in triplicate. Cycle thresholds (C T) were established for each gene in each sample. If the C T value was N/A or greater than 35, the value was recorded as 35. The ΔCt value was calculated for each sample by subtracting the C T from GAPDH from the C T for RUNDC3B.
Statistical methods
The methylation status for each sample was recorded in the six PCR regions as unmethylated, partially methylated or methylated. An ad hoc methylation density score was attributed to each sample to describe the methylation across the CGI. Each region was given a numerical value (1 = methylated, 0.5 = partially methylated, and 0 = unmethylated), and the methylation density was calculated by averaging the numerical values across all regions for each sample.
Odds ratios were calculated based on dichotomous criteria for PCR region methylation and RUNDC3B expression status. The methylation data were categorized as either a methylated region (which also included partially methylated) or an unmethylated region. The expression data were defined as RUNDC3B expressing (Ct < 35) and RUNDC3B non‐expressing (Ct > 35).
The odds ratios including 95% confidence intervals and p‐values were calculated using medcalc software v 12.4 (MedCalc Software, Ostend, Belgium). In instances with zero observations for any cell in the contingency table, a constant equal to 0.5 was added to each cell 6. Spearman's rank correlation was performed using spss software (IBM, Armonk, NY, USA). Fisher's exact test and exact Mann–Whitney were computed in R 3.0.1 using the coin package.
MAPK signalling pathway PCR array
The relative expression of 84 genes related to the mitogen‐activated protein kinase (MAPK) pathway was determined by comparing expression levels in the methylated Nalm 6 cell line versus unmethylated healthy bone marrow using RT2 Profiler PCR Arrays (Qiagen, Valencia, CA, USA). RNA was converted to cDNA using the Superscript III First‐Strand Synthesis System following the protocol provided (Life Technologies, Grand Island, NY, USA). The cDNA was added to the qPCR master mix provided with MAPK PCR array and aliquoted into the plate containing optimized RT primers for genes involved in the MAPK pathway and housekeeping genes. The plates were run on a Bio‐Rad iCycler iQ with a 10‐min hot start at 95 °C followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The data were analysed using the ΔΔC T method.
Results
Methylation of RUNDC3B is prevalent in lymphoid malignancies
In our previous work, we showed that RUNDC3B promoter methylation is present in lymphoid cell lines and ALL patient samples but not in myeloid cell lines 2, 3. To ascertain the extent of methylation across the RUNDC3B (NM_138290.2) promoter and to determine if methylation is unique to lymphoid malignancies, primers were developed to encompass the CGI that spans the transcriptional start site (Figure 1). This region was assayed for methylation using COBRA (regions 1, 2, 5 and 6) and MSP (regions 3 and 4). Higher levels of methylation were observed in the lymphoid malignancies than in the myeloid malignancies and normal controls consistent with our previous findings (Figure 2). Methylation was also observed in some solid tumours with the greatest amount in lung, colon and squamous cell carcinoma. The overall density of methylation was higher in lymphoid malignancies averaging 0.69 across the six regions analysed, while solid tumours, myeloid malignancies and healthy tissues had average methylation densities of 0.22, 0.08 and 0.0, respectively. Using an exact Mann–Whitney test on the methylation density average score, higher levels of methylation were observed in the lymphoid malignancies than in the solid tumours (p = 0.029), normal controls (p = 4.99 × 10−4) and myeloid malignancies (p = 0.109).
Figure 1.
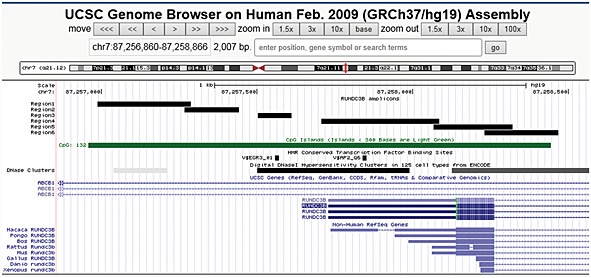
RUNDC3B promoter and amplicons. The RUNDC3B promoter is present within an intron of the ABCB1 gene and contains an annotated CGI (green bar). The methylation status of regions 1–6 was determined. A conserved early growth response 3 transcription factor binding site is present within region 3, and a conserved activating protein 2 transcription factor binding site is located within region 4. Each of these transcription factor binding sites is located within a region of DNaseI hypersensitivity, further supporting the regulatory potential of this region
Figure 2.
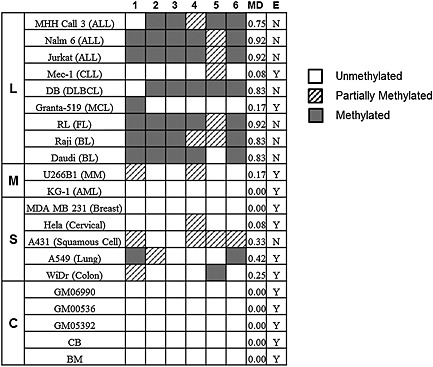
RUNDC3B methylation and expression in cell lines and controls. The methylation status of regions 1–6 was determined. Samples are organized based on tissue type: L = lymphoid, M = myeloid, S = solid tumour, C = control tissue. Methylation density (MD) was determined for the entire region and given a numeric value. The expression status for each sample was also determined, Y = C T < 35, N = C T ≥ 35. ALL, acute lymphoblastic leukaemia; CLL, chronic lymphocytic leukaemia; DLBCL, diffuse large B‐cell lymphoma; MCL, Mantle cell lymphoma; AML, acute myeloid leukaemia; BL, Burkitt's lymphoma; FL, follicular lymphoma; MM, multiple myeloma
Methylation density impacts RUNDC3B expression
Real‐time PCR was performed to determine the effect of methylation on RUNDC3B expression. In the lymphoid malignancies, no expression was observed in cell lines with a high methylation density score (Figure 2). With the exception of A431, all solid tumour cell lines expressed RUNDC3B regardless of methylation density. Expression data for RUNDC3B (ΔCt values) were ranked and correlated to ranked methylation density scores using Spearman's rank correlation. A significant correlation between a decrease in expression and increasing methylation density (ρ = 0.77, p < 0.001, df = 19) was observed.
Odds ratios were also calculated for each region of interest to assess the strength of the inverse association between RUNDC3B expression and the presence of methylation. Regions 2, 3, 4, 5 and 6 showed a significant inverse association between CGI methylation and RUNDC3B expression: region 1, OR: 6.75, 95% CI: 0.93–49.23, p = 0.06; region 2, OR: 84, 95% CI: 4.51–1564.34, p = 0.003; region 3, OR: 135, 95% CI: 4.87–3744.64, p = 0.004; region 4, OR: 78.2, 95% CI: 3.31–1849.13, p = 0.007; region 5, OR: 38.5, 95% CI: 2.92–508.49, p = 0.006; region 6, OR: 141.67, 95% CI: 5.14–3907.44, p = 0.004. In the lymphoid malignancies, regions 2, 3, 4 and 6 were methylated in each cell line that did not express RUNDC3B and were not methylated in the cell lines that did express RUNDC3B. The relationship between methylation and gene expression in the solid tumours was less pronounced, suggesting that different regulatory mechanisms may be accountable in these tumours.
The RUNDC3B CGI lies within an intron of ATP‐binding cassette sub‐family B member 1 (ABCB1), which encodes a P‐glycoprotein that is responsible for transporting molecules, including therapeutic drugs, across cellular membranes 7. Methylation of the ABCB1 promoter has been shown to contribute to progression in prostate cancer and AML 8, 9. To determine if methylation of the RUNDC3B promoter also affected regulation of ABCB1, quantitative PCR for ABCB1 was also performed. Expression of ABCB1 was consistent across samples regardless of methylation status.
Methylation and expression trends in cell lines are representative of patient samples
To determine if the methylation and expression patterns present in cell lines also hold true in patient samples, the methylation and expression present in ALL, AML, chronic lymphocytic leukaemia (CLL) and diffuse large B‐cell lymphoma (DLBCL) patients were explored. Methylation profiles generated in our laboratory by enriching for methylated DNA using the Methylated CpG Island Recovery Assay (MIRA) followed by next‐generation sequencing (unpublished data) in 20 ALL patients and healthy cord blood revealed a peak that spans the six amplicons within the RUNDC3B CGI, chr7:87256845‐87258468 (Figure 3). This peak was present in 17 of 20 ALL patients and in only 1 of 10 healthy cord blood samples (p = 1.34 × 10−4, Fisher's exact test). RNA‐seq data for the same 20 patients revealed an average fragments per kilobase of exon per million fragments mapped for the group of 0.22 (range 0–1.19), indicating that there is virtually no expression of RUNDC3B in these samples. Methylation profiles have also been generated for 11 CLL patients using reduced representation bisulfite sequencing 10. The methylation values were low across CLL patients with an average methylation score of 0.096 (Figure 3, Table 3). In some cases, high methylation values were observed at a particular CpG site; however, the average methylation level remained low across the region. No expression data were available for these patients; therefore, to determine the expression of RUNDC3B in CLL patient samples, a publically available data set comprising 68 CLL patients (GSE10138) was utilized 11. The average expression value across all patients fell within the second quartile, confirming that RUNDC3B is expressed in these patients (Figure 4A). Two additional lymphoma data sets were analysed from the Cancer Genome Atlas (TCGA) using the UCSC Cancer Genomics Browser representing AML patients (194 samples; LAML DNA methylation data; LAML gene expression pancan normalized) and DLBCL patients (48 samples; DLBC DNA methylation data; DLBC gene expression pancan normalized). The methylation and expression profiles were consistent with data from the AML and DLBCL cell lines and showed lower levels of DNA methylation and higher levels of expression in AML patients when compared with DLBCL patients (Figure 4B, C). Although this does not represent all of the diseases included in our study, in the cases of ALL, AML, CLL and DLBCL, the patient data are consistent with our findings in cell lines.
Figure 3.
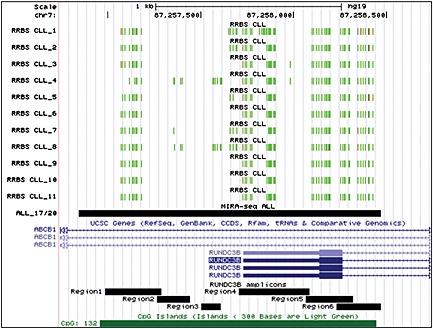
RUNDC3B methylation in acute lymphoblastic leukaemia (ALL) and chronic lymphocytic leukaemia (CLL) patients. RUNDC3B‐associated methylation data were extracted from previously generated ALL (unpublished data) and CLL 10 methylation profiles. Reduced representation bisulfite sequencing (RRBS) data are shown for each CLL patient. Green and red bars represent unmethylated and methylated CpG sites, respectively. MIRA‐seq data were utilized for peak identification in ALL patient samples. The location of a peak spanning the RUNDC3B CGI and present in 17 out of 20 ALL samples is shown
Table 3.
RRBS data for RUNDC3B in CLL patients 10
| Patient ID | CpG sites | Average | Median | Low | High |
|---|---|---|---|---|---|
| CLL_1 | 61 | 0.13 | 0.04 | 0 | 0.96 |
| CLL_2 | 55 | 0.09 | 0.02 | 0 | 0.54 |
| CLL_3 | 52 | 0.12 | 0.04 | 0 | 0.73 |
| CLL_4 | 60 | 0.06 | 0.02 | 0 | 0.52 |
| CLL_5 | 52 | 0.11 | 0.03 | 0 | 0.68 |
| CLL_6 | 51 | 0.10 | 0.03 | 0 | 0.50 |
| CLL_7 | 54 | 0.08 | 0 | 0 | 0.58 |
| CLL_8 | 66 | 0.10 | 0.02 | 0 | 0.65 |
| CLL_9 | 51 | 0.07 | 0 | 0 | 0.48 |
| CLL_10 | 51 | 0.08 | 0 | 0 | 0.56 |
| CLL_11 | 51 | 0.12 | 0.08 | 0 | 0.44 |
For each patient, the number of CpG sites analysed is provided. The average and median are in reference to the total number of CpG sites within the RUNDC3B CGI. Low and high represent the range of values across all CpG sites.
RRBS, reduced representation bisulfite sequencing; CLL, chronic lymphocytic leukaemia.
Figure 4.
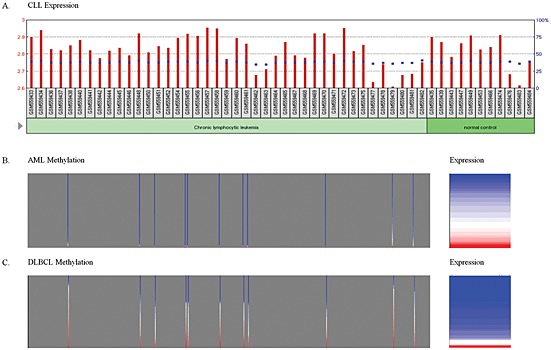
Methylation and expression of RUNDC3B in patient samples. The results shown here are based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. (A) Expression of RUNDC3B in 41 chronic lymphocytic leukaemia (CLL) patients and 11 normal controls. The red bars and the left axis represent normalized counts. The blue squares and the right axis represent the percentile rank within the sample. (B) Methylation and expression in acute myeloid leukaemia (AML) patients. Methylation shown in grey box (left panel) for 11 cytosines (vertical bars) present in the RUNDC3B CGI. Proportional expression is shown in the right panel. (C) Methylation and expression in diffuse large B‐cell lymphoma (DLBCL) patients. Proportional methylation shown in grey box (left panel) for 11 cytosines (vertical bars) present in the RUNDC3B CGI. Proportional expression is shown in the right panel. Blue to red scale represents lowest to highest methylation and expression values
The MAPK pathway is dysregulated in the Nalm 6 cell line
It is hypothesized that RUNDC3B is an effector protein of the Rap2 GTPase, and activation of Rap2 has been shown to regulate the MAPK pathway. In addition to a RUN domain, six MAPK D docking domains were identified in the RUNDC3B protein sequence, suggesting a potential role for RUNDC3B as a mediator between MAPK genes and Rap2. MAPK signalling pathway PCR arrays were utilized to determine whether key genes were upregulated or downregulated in the Nalm 6 cell line, which was methylated in each region of the RUNDC3B promoter. A total of 23 genes were upregulated, and six genes were downregulated in the Nalm 6 cell line, including genes with MAPK‐induced expression and transcription factors with MAPK‐dependent activation (Table 4).
Table 4.
Differential expression of MAPK pathway genes in a RUNDC3B methylated leukaemia cell line versus a RUNDC3B unmethylated control bone marrow sample
| Upregulated genes | Downregulated genes | ||
|---|---|---|---|
| Symbol | Fold change | Symbol | Fold change |
| CCND3 | 37.79 | BRAF | −5.5 |
| CDC42 | 8.82 | CCNA1 | −71.51 |
| CDK6 | 46.53 | CCND2 | −82.14 |
| CDKN1A | 23.26 | FOS | −17.88 |
| CDKN2C | 10.13 | HSPA5 | −8.94 |
| CREB1 | 13.36 | JUN | −44.12 |
| ETS1 | 4.72 | ||
| ETS2 | 10.85 | ||
| GRB2 | 18.89 | ||
| HRAS | 10.13 | ||
| HSPB1 | 93.05 | ||
| KRAS | 10.85 | ||
| MAP2K3 | 6.68 | ||
| MAP3K1 | 5.06 | ||
| MAP4K1 | 7.16 | ||
| MAPK13 | 8.82 | ||
| MAPK6 | 9.45 | ||
| MAPKAPK2 | 13.36 | ||
| MAPKAPK3 | 4.41 | ||
| MAX | 10.85 | ||
| NRAS | 7.67 | ||
| RB1 | 37.79 | ||
| SMAD4 | 7.67 | ||
MAPK, mitogen‐activated protein kinase.
Discussion
The human RUNDC3B CGI is 1486 base pairs, contains 132 CpG sites and harbours two conserved transcription factor binding sites (TFBS), early growth response 3 (EGR‐3) and activating protein 2 (AP‐2). Interestingly, EGR‐3 is a zinc finger protein implicated in neuronal, muscle and lymphocyte development 12. The EGFR‐3 TFBS also lies within a region of DNase hypersensitivity, which is associated with active transcription. A survey of publically available DNA methylation data and DNase hypersensitivity data revealed a 1:1 correlation between the absence of methylation and the presence of a DNase hypersensitivity site and vice versa in the RUNDC3B CGI 13. It is likely that the direct methylation of the EGR‐3 TFBS interferes with the trans‐acting regulatory interaction between EGR‐3 and the RUNDC3B promoter.
Considering that RUNDC3B is a putative Rap‐2 binding protein and because it contains characteristic structural MAPK binding domains, we hypothesize that it is an effector of the MAPK signalling cascade (Figure 5). Rap2A interacts directly with upstream MAPK signalling element MAP4K4, and thus, increased Rap2A activity can perpetuate downstream signalling 14. If not properly regulated, Rap2B activity can increase phospholipase C, epsilon 1 activity, which can lead to the activation of Ras signalling, which may lead to increased cell growth and differentiation, and ultimately, tumorigenesis 15. Downregulation of c‐Jun and c‐Fos, downstream effectors of Rap2 proteins, was observed in Nalm 6. Together, Jun and Fos form the AP‐1 transcription factor, which is activated by the c‐Jun N‐terminal kinase pathway to regulate transcriptional activation of genes. Loss of AP‐1 expression results in deregulated transcription of genes necessary for differentiation and proliferation. As a Rap2‐interacting protein, RUNDC3B may play a role in the activation of c‐Jun and c‐Fos, and the loss of RUNDC3B may lead to the downregulation of these genes.
Figure 5.
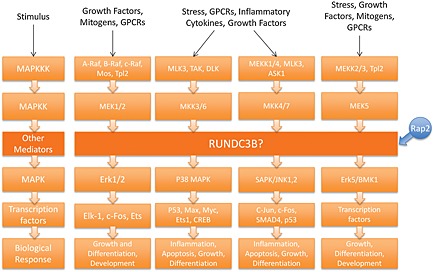
Proposed role of RUNDC3B in the MAPK signalling pathway. RUNDC3B is hypothesized to interact with Rap2 based on homology with RUNDC3A. In this model, Rap2 activates RUNDC3B, which then interacts with MAP kinases to influence downstream responses
An alternative function for RUNDC3B may also be inferred based on recent reported interactions between RUNDC3B and casein kinase 1 gamma 1 and 2 (CSNK1G1/2) 16. Both CSNK1G1 and CSNK1G2 are serine and threonine kinases involved in canonical Wnt signalling. A clear role has been established between abnormal Wnt signalling and the development of lymphoid cancers 17, 18. Specifically, the casein kinases are responsible for phosphorylating the transmembrane proteins Lrp5 and Lrp6 19. This enables the formation of the signalosome that perpetuates Wnt signalling and β‐catenin to transcribe Wnt genes. Therefore, the absence of RUNDC3B expression may alter the ability of CSNKIG1/2 to contribute to the formation of the signalosome, resulting in the aberrant regulation of Wnt signalling.
In summary, aberrant DNA methylation of the RUNDC3B CGI results in the repression of the gene. Methylation was observed in several solid tumour cell lines, and in samples representing lymphoid malignancies, with higher methylation densities being observed in lymphoid samples that are derived from the bone marrow and the germinal centre. Other lymphoid malignancies such as CLL and MCL expressed RUNDC3B and were not methylated. Therefore, RUNDC3B methylation may prove to be a useful biomarker for diagnosis and prognosis and in tracking minimal residual disease in malignancies of the bone marrow and germinal centre, which include ALL, Burkitt's lymphoma (BL), follicular lymphoma (FL) and DLBCL.
Conflict of interest
The authors have no potential conflicts to disclose.
Acknowledgements
This work was supported by the National Institutes of Health NCI R00 CA132784 (K. H. Taylor). We kindly thank Darren Hawkins, Emily Shank and Md Almamun for generating the figures included in this manuscript.
Burmeister, D. W. , Smith, E. H. , Cristel, R. T. , McKay, S. D. , Shi, H. , Arthur, G. L. , Davis, J. W. , and Taylor, K. H. (2017) The expression of RUNDC3B is associated with promoter methylation in lymphoid malignancies. Hematological Oncology, 35: 25–33. doi: 10.1002/hon.2238.
References
- 1. Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet 2000; 1: 11–19. [DOI] [PubMed] [Google Scholar]
- 2. Taylor KH, Pena‐Hernandez KE, Davis JW, et al. Large‐scale CpG methylation analysis identifies novel candidate genes and reveals methylation hotspots in acute lymphoblastic leukemia. Cancer Res 2007; 67: 2617–2625. [DOI] [PubMed] [Google Scholar]
- 3. Wang MX, Wang HY, Zhao X, et al. Molecular detection of B‐cell neoplasms by specific DNA methylation biomarkers. Int J Clin Exp Pathol 2010; 3: 265–279. [PMC free article] [PubMed] [Google Scholar]
- 4. Raguz S, De Bella MT, Slade MJ, Higgins CF, Coombes RC, Yague E. Expression of RPIP9 (Rap2 interacting protein 9) is activated in breast carcinoma and correlates with a poor prognosis. Int J Cancer 2005; 117: 934–941. [DOI] [PubMed] [Google Scholar]
- 5. Rountree MR, Bachman KE, Herman JG, Baylin SB. DNA methylation, chromatin inheritance, and cancer. Oncogene 2001; 20: 3156–3165. [DOI] [PubMed] [Google Scholar]
- 6. Walter SD. Point estimation of the odds ratio in sparse 2 × 2 contingency tables In Biostatistics. MacNeill IB, Umphrey GJ, Reidel D. (eds). Springer: Netherlands, 1987; 71–102. [Google Scholar]
- 7. Fardel O, Lecureur V, Guillouzo A. The P‐glycoprotein multidrug transporter. Gen Pharmacol 1996; 27: 1283–1291. [DOI] [PubMed] [Google Scholar]
- 8. Enokida H, Shiina H, Igawa M, et al. CpG hypermethylation of MDR1 gene contributes to the pathogenesis and progression of human prostate cancer. Cancer Res 2004; 64: 5956–5962. [DOI] [PubMed] [Google Scholar]
- 9. Toyota M, Kopecky KJ, Toyota MO, Jair KW, Willman CL, Issa JP. Methylation profiling in acute myeloid leukemia. Blood 2001; 97: 2823–2829. [DOI] [PubMed] [Google Scholar]
- 10. Pei L, Choi JH, Liu J, et al. Genome‐wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics 2012; 7(6): 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedman DR, Weinberg JB, Barry WT, et al. A genomic approach to improve prognosis and predict therapeutic response in chronic lymphocytic leukemia. Clin Cancer Res 2009; 15(22): 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patwardhan S, Gashler A, Siegel MG, et al. EGR3, a novel member of the Egr family of genes encoding immediate‐early transcription factors. Oncogene 1991; 6: 917–928. [PubMed] [Google Scholar]
- 13. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Machida N, Umikawa M, Takei K, et al. Mitogen‐activated protein kinase kinase kinase kinase 4 as a putative effector of Rap2 to activate the c‐Jun N‐terminal kinase. J Biol Chem 2004; 279: 15711–15714. [DOI] [PubMed] [Google Scholar]
- 15. Evellin S, Nolte J, Tysack K, et al. Stimulation of phospholipase C‐epsilon by the M3 muscarinic acetylcholine receptor mediated by cyclic AMP and the GTPase Rap2B. J Biol Chem 2002; 277: 16805–16813. [DOI] [PubMed] [Google Scholar]
- 16. Vinayagam A, Stelzl U, Foulle R, et al. A directed protein interaction network for investigating intracellular signal transduction. Sci Signal 2011; 4: rs8. [DOI] [PubMed] [Google Scholar]
- 17. Khan NI, Bradstock KF, Bendall LJ. Activation of Wnt/beta‐catenin pathway mediates growth and survival in B‐cell progenitor acute lymphoblastic leukaemia. Br J Haematol 2007; 138: 338–348. [DOI] [PubMed] [Google Scholar]
- 18. Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self‐renewal of haematopoietic stem cells. Nature 2003; 423: 409–414. [DOI] [PubMed] [Google Scholar]
- 19. Bilic J, Huang YL, Davidson G, et al. Wnt induces LRP6 signalosomes and promotes dishevelled‐dependent LRP6 phosphorylation. Science 2007; 316: 1619–1622. [DOI] [PubMed] [Google Scholar]