

Abstract
Tocilizumab is a humanized anti–interleukin‐6 receptor antibody for treating rheumatoid arthritis. Pharmacokinetic/pharmacodynamic analysis was performed on the 24‐week double‐blind parts of 2 randomized, controlled trials: SUMMACTA and BREVACTA. SUMMACTA compared subcutaneous tocilizumab 162 mg every week to intravenous tocilizumab 8 mg/kg every 4 weeks, whereas BREVACTA evaluated 162 mg subcutaneous tocilizumab every 2 weeks versus placebo. In addition to noncompartmental analysis, a 2‐compartment population pharmacokinetic model, with first‐order absorption (for subcutaneous) and linear and Michaelis–Menten elimination was used. Mean observed steady‐state predose tocilizumab concentrations in week 24 were 40 and 7.4 μg/mL for subcutaneous every‐week and every‐2‐week dosing, respectively, and 18 μg/mL for intravenous dosing. In the population PK model, body weight was an important covariate affecting clearance and volume of distribution. Mean ± SD population‐predicted predose concentration for patients ≥100 kg was 23.0 ± 13.5 μg/mL for subcutaneous tocilizumab every week and 1.0 ± 1.6 μg/mL for every 2 weeks. Efficacy was lowest with subcutaneous every‐2‐week dosing in patients > 100 kg, reflecting lower exposure. The subcutaneous every‐2‐week regimen is not recommended for these patients. Pharmacodynamic responses were comparable for the every‐week subcutaneous and every‐4‐week intravenous regimens and less pronounced with the every‐2‐week subcutaneous regimen. No trend was observed for increased adverse events with increasing tocilizumab exposure. The results of this analysis are consistent with the noninferiority of efficacy of the every‐week subcutaneous regimen to the every‐4‐week intravenous regimen and the superiority of the every‐2‐week subcutaneous regimen to placebo. These results support the label recommendations for subcutaneous dosing of tocilizumab in rheumatoid arthritis patients.
Keywords: rheumatoid arthritis, tocilizumab, subcutaneous, pharmacokinetics
Biologic disease‐modifying antirheumatic drugs (bDMARDs) are recommended for the treatment of patients with rheumatoid arthritis (RA) who do not respond to nonbiologic DMARDs (nbDMARDs), such as methotrexate, or as initial treatment in combination with methotrexate in patients with poor prognoses.1, 2 Currently available bDMARDs for the treatment of RA are administered either as intravenous infusion or subcutaneous injection.3, 4
Tocilizumab (Actemra, RoActemra) is a recombinant humanized monoclonal antibody that binds to the interleukin‐6 receptor (IL‐6R), blocking IL‐6 inflammatory signaling.5, 6 It is currently approved for the treatment of patients with RA who have had inadequate responses to DMARDs7, 8 or were not previously treated with methotrexate.8 Tocilizumab is available as intravenous infusion and subcutaneous injection.
The route of administration may be an important factor influencing adherence to bDMARDs.9 A subcutaneous formulation offers improved patient choice and potential for self‐administration,10, 11 and many patients prefer subcutaneous over intravenous administration.12, 13 The subcutaneous formulation of tocilizumab provides patients with RA with an alternative and more convenient route of administration, which could be self‐administered in a home setting, negating the requirement for and associated health care costs of intravenous access and frequent clinic visits.
The safety and efficacy of subcutaneous tocilizumab in combination with background nbDMARDs have been demonstrated in 2 randomized, double‐blind, controlled phase 3 trials, SUMMACTA and BREVACTA, which have been presented separately.14, 15 The 2 studies were designed to constitute a comprehensive strategy to bridge intravenous to subcutaneous dosing in the 2 main world regions, the European Union and the United States, where the approved intravenous tocilizumab starting doses are different. In European Union countries, the approved starting dose is 8 mg/kg every 4 weeks, which could be reduced to 4 mg/kg every 4 weeks to address safety concerns.8 The US label, on the other hand, recommends a starting dose of 4 mg/kg every 4 weeks, followed by an increase to 8 mg/kg every 4 weeks based on clinical response. In the SUMMACTA study, the efficacy and safety of tocilizumab 162 mg subcutaneously every week was compared with tocilizumab 8 mg/kg intravenously every 4 weeks, whereas in BREVACTA, tocilizumab 162 mg subcutaneously every 2 weeks were tested against placebo. SUMMACTA showed safety and noninferior efficacy comparable to tocilizumab 162 mg subcutaneously every week compared with tocilizumab 8 mg/kg intravenously every 4 weeks, whereas BREVACTA showed that tocilizumab 162 mg subcutaneously every 2 weeks was superior to placebo. The results of the 2 studies were the basis for approval of the subcutaneous formulation and the dosing recommendations in the European Union and the United States.7, 8 Consistent with the respective intravenous labels, the approved starting dose for subcutaneous tocilizumab is 162 mg every week in the European Union, with a possible decrease to every 2 weeks for safety, and 162 mg every 2 weeks in the United States, with a possible increase to every week based on clinical response.
The pharmacokinetics (PK) and pharmacodynamics (PD) of intravenous tocilizumab have been reviewed extensively,16 whereas the PK/PD of subcutaneous tocilizumab in patients with RA has only been described in a small open‐label study in 29 patients without a control treatment.17 Therefore, an analysis was performed to compare the PK, PD, and exposure‐efficacy and exposure‐safety relationships of subcutaneous tocilizumab 162 mg administered every week or every 2 weeks with those of tocilizumab 8 mg/kg intravenously every 4 weeks or placebo, using data from the SUMMACTA and BREVACTA studies.
Methods
All patients signed informed consent documents that were approved by an independent ethics committee or institutional review board, and the studies were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Data were pooled from SUMMACTA (NCT01194414) and BREVACTA (NCT01232569), 2 phase 3 randomized, controlled, parallel‐arm trials in patients with RA who had inadequate responses to DMARDs (this may have included ≥1 antitumor necrosis factor agent, but the proportion of patients who had an inadequate response to these agents was capped at 20%). Both trials had a 24‐week double‐blind period followed by a 72‐week open‐label extension. PK/PD results are presented for the 24‐week double‐blind periods.
Inclusion and exclusion criteria for the studies were published previously.14, 15 Briefly, the studies included adult patients (≥ 18 years old) with moderate to severe RA of at least 6 months’ duration who had been receiving nbDMARDs at a stable dose for at least 8 weeks before baseline and had discontinued bDMARDs for a suitable washout period before randomization.
Tocilizumab was administered as a subcutaneous injection of 162 mg via a prefilled syringe or by intravenous infusion at 8 mg/kg. Patients in SUMMACTA received tocilizumab 162 mg subcutaneously every week plus placebo intravenously every 4 weeks or placebo subcutaneously every week plus tocilizumab 8 mg/kg intravenously every 4 weeks for 24 weeks in combination with nbDMARDs. Per US and EU labels, patients weighing ≥ 100 kg received a flat dose of 800 mg every 4 weeks. Patients in BREVACTA received tocilizumab 162 mg subcutaneously or placebo subcutaneously every 2 weeks for 24 weeks in combination with nbDMARDs; during this period, patients with <20% improvement from baseline in swollen joint count and tender joint count could escape to open‐label rescue therapy with tocilizumab 162 mg subcutaneously every week from week 12.
In both studies, predose serum samples were taken from all patients in the PK/PD population to measure PK and PD parameters (patients from whom at least 1 blood sample was collected: SUMMACTA, n = 1262; BREVACTA, n = 437; total number of samples, 13 642). Patients could also enter an optional PK/PD substudy in which more intense serum samples were collected. Noncompartmental analysis, in addition to the population PK analysis, was also performed on samples from patients in the substudies. The serum samples for PK/PD analysis (tocilizumab, IL‐6, and soluble IL‐6R [sIL‐6R] for SUMMACTA; tocilizumab and sIL‐6R for BREVACTA) were collected before tocilizumab administration at baseline; in weeks 1, 2, and 4; and then every 4 weeks thereafter until week 24 in the double‐blind period of SUMMACTA, and at baseline and every 2 weeks until week 8 and then every 4 weeks until week 24 in the double‐blind period of BREVACTA. Patients’ C‐reactive protein (CRP) and erythrocyte sedimentation rate (ESR) were assessed at baseline, in weeks 2 and 4, and every 4 weeks thereafter until week 24 in the double‐blind period of both studies. Antitocilizumab antibodies were assessed at baseline and in week 12 and week 24 in the double‐blind period of both studies. Additional sequential serum samples were taken for the PK/PD substudy following the day 1 and week 20 doses for SUMMACTA and following the day 1 and week 12 doses for BREVACTA.
The effect of body weight on systemic drug exposure at steady state was assessed using noncompartmental analysis and confirmed by the population PK analysis. Patients from BREVACTA who escaped to rescue therapy were included up to the time of escape, after which they were classed as withdrawn and excluded. Nonlinear mixed‐effects modeling (NONMEM 7.2)18 was used to analyze the concentration–time data for tocilizumab from intravenous and subcutaneous dosing simultaneously. The population PK model previously developed using data from the intravenous formulation was used as a starting point of the analysis.19 Structural model refinement was driven by the data and was based on various goodness‐of‐fit indicators, plausibility of the parameter estimates, precision of the parameter estimates, the minimum objective function value, and the number of parameters. Covariates such as body weight, sex, age, creatinine clearance, and rheumatoid factor were tested. Steady‐state secondary PK parameters (area under the concentration curve [AUC], maximum concentration [Cmax], trough concentration [Ctrough]) were computed using Bayesian post hoc estimates obtained for each patient. The PD analysis included IL‐6 levels (SUMMACTA only) and sIL‐6R, CRP, and ESR levels (both studies). PD markers are presented using descriptive summary statistics.
Immunogenicity was assessed as serum levels of antitocilizumab antibodies measured using a bridging enzyme‐linked immunosorbent assay (ELISA), as described previously,20 in samples collected at baseline, every 12 weeks, and at study completion or early withdrawal. Samples that tested positive in an initial screening assay were analyzed by a confirmation assay to confirm specificity. If the confirmation assay was positive, an inhibition ELISA was performed to evaluate the neutralizing potential of the antitocilizumab antibody.14, 15 The effect of antitocilizumab antibodies on the PK of tocilizumab and the effect of neutralizing antitocilizumab antibodies on the PK/PD relationship between tocilizumab concentration and 28‐joint Disease Activity Score (DAS28) was investigated.
The relationship between exposure to tocilizumab and efficacy was assessed using American College of Rheumatology (ACR) 20/50/70 responses in week 24, summarized by tocilizumab observed Ctrough exposure quartiles in week 24. Graphical analyses using logistic regression were also performed for ACR20/50/70. Patients from BREVACTA who escaped to rescue therapy after week 12 were treated as ACR20/50/70 nonresponders, and the last Ctrough value before escape was used in the analysis. The relationship between exposure to tocilizumab and safety was assessed by summarizing laboratory abnormalities and adverse events (AEs) up to week 24 by tocilizumab observed Ctrough exposure quartiles in week 24. Patients from BREVACTA who escaped to rescue therapy were not included in the exposure–safety analysis.
Results
Baseline demographics and disease characteristics were similar for patients in the safety populations (all patients who received ≥1 dose of the study drug and had ≥1 postdose safety assessment) across the 2 studies and across treatment arms, except for a slightly heavier mean weight and larger proportion ≥100 kg for patients in SUMMACTA and longer disease duration in BREVACTA (11.1 years) than in SUMMACTA (8.7 years); see Supplementary Table S1. The data set used for the population PK analysis and noncompartmental analysis included 1759 patients from both SUMMACTA and BREVACTA.
Pharmacokinetics
Based on observed data in the PK/PD population, the mean predose tocilizumab concentrations with 8 mg/kg intravenous every‐4‐week administration increased from baseline to week 16 and appeared to reach steady state thereafter (Figure 1). There was an approximately 2‐fold increase in mean predose concentration from week 4 to week 24. Steady‐state predose tocilizumab concentration in week 24 was approximately 18 μg/mL, with every‐4‐week intravenous dosing. Following subcutaneous every‐week administration, tocilizumab concentrations increased from baseline to week 12 and appeared to reach steady state thereafter. There was approximately a 5‐fold increase in mean predose tocilizumab concentration from week 1 to week 24. Steady‐state predose tocilizumab concentration in week 24 was approximately 40 μg/mL. Subcutaneous dosing of tocilizumab 162 mg every 2 weeks resulted in increasing predose concentrations from baseline to week 20 and appeared to reach steady state thereafter (Figure 1). There was an approximately 4‐fold increase in mean predose tocilizumab concentrations from week 2 to week 24. Steady‐state predose tocilizumab concentration in week 24 was approximately 7.4 μg/mL. Results from the PK substudies are also included in Supplementary Tables S2 and S3.
Figure 1.
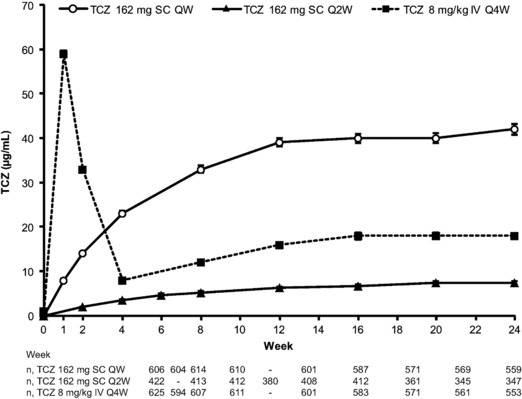
Mean observed predose tocilizumab concentrations for SC QW, SC Q2W, or IV Q4W dosing. Error bars show standard error. IV, intravenous; Q2W, every 2 weeks; Q4W, every 4 weeks; QW, weekly; SC, subcutaneous; TCZ, tocilizumab.
The population PK model developed for this analysis had a structure similar to the model previously developed based only on intravenous data19; it was a 2‐compartment PK model with parallel linear and Michaelis–Menten eliminations. In addition, the model contained a first‐order absorption process to describe subcutaneous administration. Diagnostic plots for the model are provided in Supplementary Figure 1, and the parameter estimates are listed in Table 1A, along with their corresponding relative standard errors and 95% confidence intervals (CI). The absolute bioavailability of tocilizumab following subcutaneous administration was estimated to be 79.5%, with a 95% confidence interval of 77.9%–81.1%. The following parameters estimates (95% CI) were obtained for tocilizumab, which were in good agreement with those of the previous model19: linear clearance, 216 mL/day (95%CI, 211–221 mL/day); central volume of distribution, 4.5 L (94%CI, 4.4–4.7 L); peripheral volume of distribution, 2.8 L (95%CI, 2.7–2.9 L); KM (Michaelis–Menten constant), 0.34 μg/mL (95%CI, 0.33–0.36 μg/mL); and VM (maximum target‐mediated elimination rate), 1.9 mg/L/day (95%CI, 1.8–1.9 mg/L/day). Model‐predicted secondary PK parameters for the every‐week, every‐2‐week, and intravenous regimens are shown in Table 1B. The predicted mean ± SD steady‐state AUC within the dosing interval (AUCτ) values for tocilizumab 162‐mg subcutaneous every‐week and every‐2‐week dosing were 8254 ± 3833 and 3460 ± 2530 μg·h/mL, respectively. The mean steady‐state predose Ctrough and Cmax were 7.7‐fold and 3.9‐fold higher, respectively, for every‐week versus every‐2‐week dosing (Table 1 and Figure 2).
Table 1.
Parameter Estimates From the Final PK Model: (A) Primary Parameters and (B) Secondary Parameters
| A. Primary Parameters | ||||
|---|---|---|---|---|
| Parameter | Estimate | % RSE | 95%CI | |
| CL (mL/day) | θ1 | 216 | 1.2 | 211–221 |
| V2 (L) | θ2 | 4.5 | 1.6 | 4.4–4.7 |
| Q (mL/day) | θ3 | 274 | 2.2 | 262–285 |
| V3 (L) | θ4 | 2.8 | 1.7 | 2.7–2.9 |
| VM (mg/L/day) | θ5 | 1.9 | 1.0 | 1.8–1.9 |
| KM (μg/mL) | θ6 | 0.34 | 2.5 | 0.33–0.36 |
| ka (1/day) | θ7 | 0.23 | 2.7 | 0.22–0.25 |
| FSC | θ8 | 0.80 | 1.1 | 0.79–0.81 |
| B. Secondary Parametersa (Mean ± SD) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 162 mg SC QW | 162 mg SC Q2W | 8 mg/kg IV Q4W | |||||||
| Tocilizumab Dose | First Dosing Interval | Steady State | RAC | First Dosing Interval | Steady State | RAC | First Dosing Interval | Steady State | RAC |
| AUCτ, μg·h/mL | 1243 ± 689 | 8254 ± 3833 | 6.8 | 1210 ± 940 | 3460 ± 2530 | 2.7 | 28 824 ± 7704 | 39 216 ± 14 304 | 1.36 |
| Cmax, μg/mL | 9.3 ± 5.1 | 51.3 ± 23.2 | 5.5 | 5.8 ± 4.1 | 13 ± 8.3 | 2.1 | 136 ± 34 | 154 ± 42 | 1.13 |
| Ctrough, μg/mL | 7.0 ± 4.1 | 45.3 ± 22.2 | 6.4 | 1.0 ± 1.5 | 5.9 ± 6.3 | 5.6 | 7.7 ± 6.7 | 18.7 ± 15.3 | 2.43 |
CL, linear clearance; V2, central volume of distribution; Q, intercompartmental clearance; V3, peripheral volume of distribution; VM, maximum target‐mediated elimination rate; KM, Mechaelis–Menten constant; Ka, absorption rate constant; FSC, absolute bioavailability following SC dosing; RSE, relative standard error, RSE, 100·SE/parameter estimate; 95%CI, 95% confidence interval; AUCτ, area under the concentration curve within the dosing interval; Cmax, maximum concentration; Ctrough, predose trough concentration; PK, pharmacokinetic; Q2W, every 2 weeks; QW, weekly; RAC, accumulation ratio; SC, subcutaneous.
Data are from simulations performed for 5000 subjects.
Figure 2.
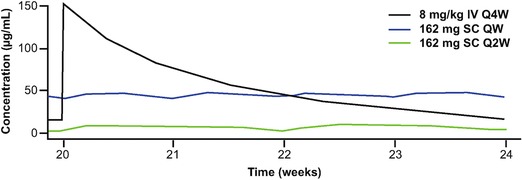
Simulated tocilizumab serum concentrations over time at steady state. Black line, tocilizumab 8 mg/kg IV Q4W; blue line, tocilizumab 162 mg SC QW; green line, tocilizumab 162 mg SC Q2W. IV, intravenous; Q2W, every 2 weeks; Q4W, every 4 weeks; QW, weekly; SC, subcutaneous; TCZ, tocilizumab.
Effect of Body Weight on Tocilizumab Exposure and PK Parameters
Based on observed data in the PK/PD population, increase in body weight was associated with lower tocilizumab predose concentration with both every‐week and every‐2‐week subcutaneous dosing (Table 2 and Supplementary Figure S2).
Table 2.
Tocilizumab Exposure in Week 24 (Observed) and at Steady State (Predicted) by Baseline Body Weight Categories
| TCZ 162 mg SC QW | TCZ 162 mg SC Q2W | TCZ 8 mg/kg IV Q4W | ||||
|---|---|---|---|---|---|---|
| Body Weight | Observed | Model Predicted | Observed | Model Predicted | Observed | Model Predicted |
| Ctrough (μg/mL) | ||||||
| <60 kg |
|
|
|
|
|
|
| 60 to <100 kg |
|
|
|
|
|
|
| ≥100 kg |
|
|
|
|
|
|
| AUCτ (μg·day/mL) | ||||||
| <60 kg |
|
|
|
|||
| 60 to <100 kg |
|
|
|
|||
| ≥100 kg |
|
|
|
|||
| Cmean (μg/mL) | ||||||
| <60 kg |
|
|
|
|||
| 60 to <100 kg |
|
|
|
|||
| ≥100 kg |
|
|
|
|||
| Cmax (μg/mL) | ||||||
| <60 kg |
|
|
|
|||
| 60 to <100 kg |
|
|
|
|||
| ≥100 kg |
|
|
|
|||
Data are mean ± SD (median: min–max). AUCτ, area under the concentration curve within the dosing interval; Cmax, maximum observed concentration; Cmean, average observed concentration; Ctrough, predose trough concentration; IV, intravenous; Q2W, every 2 weeks; Q4W, every 4 weeks; QW, weekly; SC, subcutaneous; TCZ, tocilizumab.
Among all identified covariate relationships in the population PK analysis, the only strong covariate dependence was the influence of weight on tocilizumab clearance and volume parameters. Increase of clearances and volumes with weight was described by the power function CL ∼ (WT/70)CLWT and V ∼ (WT/70)VWT, with power coefficients estimated at 0.51 (95%CI, 0.47–0.56) and 0.68 (95%CI, 0.63–0.74), respectively. Clearance terms of patients with weight of 40 and 140 kg (values close to the extreme values of weight in the study population) decreased and increased, respectively, by 25% and 47%, and volumes decreased and increased, respectively, by 32% and 61% compared with the CL of a patient weighing 70 kg.
The predicted mean ± SD tocilizumab Ctrough values for patients weighing ≥100 kg were lower with tocilizumab subcutaneous every‐2‐week dosing (1.0 ± 1.6 μg/mL) than with subcutaneous every‐week dosing (23 ± 14 μg/mL) or intravenous every‐4‐week dosing (31 ± 24 μg/mL); a similar trend was observed for predicted AUCτ, average observed concentration, and Cmax in patients ≥100 kg (Table 2).
Pharmacodynamics
IL‐6 and sIL‐6R levels were comparable with tocilizumab 162‐mg subcutaneous every‐week and tocilizumab 8 mg/kg intravenous every‐4‐week dosing in SUMMACTA (Figure 3A,B). In the BREVACTA study, tocilizumab 162‐mg subcutaneous every‐2‐week dosing also resulted in rapid increases in sIL‐6R, whereas sIL‐6R values were unchanged with placebo (Figure 3B). CRP and ESR levels were comparable with tocilizumab 162‐mg subcutaneously every‐week and tocilizumab 8‐mg/kg intravenous every‐4‐week dosing (Figure 3C,D). CRP and ESR levels decreased rapidly following tocilizumab 162‐mg subcutaneous every‐2‐week dosing and continued to drop to week 24, remaining below the upper limit of normal (ULN). CRP and ESR levels also decreased slightly following treatment with placebo subcutaneously, but were higher than in patients treated with tocilizumab and did not fall below the ULN.
Figure 3.
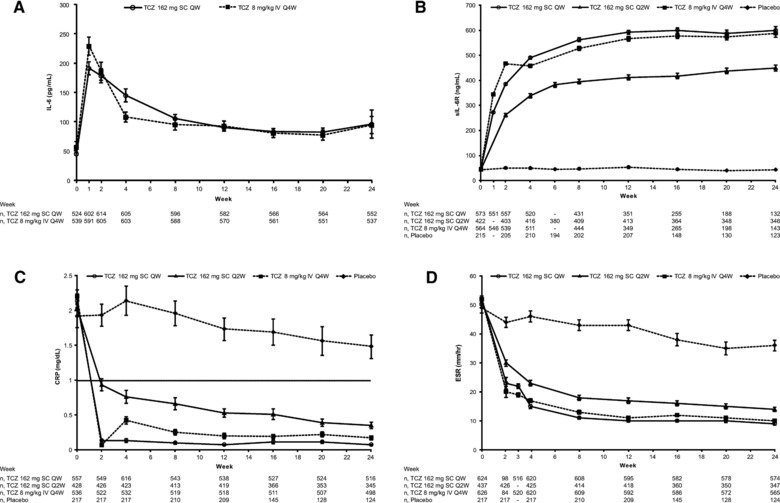
Mean IL‐6 (A), sIL‐6R (B), CRP (C), and ESR (D) levels following treatment with SC or IV tocilizumab. Error bars show standard error. CRP, C‐reactive protein; ESR, erythrocyte sedimentation rate; IL‐6, interleukin 6; IV, intravenous; Q2W, every 2 weeks; Q4W, every 4 weeks; QW, weekly; SC, subcutaneous; sIL‐6R, soluble IL‐6 receptor; TCZ, tocilizumab.
Immunogenicity
In the safety populations, 5 of 631 patients (0.8%) receiving tocilizumab 162 mg subcutaneously every week, 5 of 631 patients (0.8%) receiving tocilizumab 8 mg/kg intravenously every 4 weeks, 7 of 437 patients (1.6%) receiving tocilizumab 162 mg every 2 weeks, and 3 of 218 patients (1.4%) receiving placebo developed antitocilizumab antibodies in the confirmation assay by week 24. Of these patients, all 5 receiving tocilizumab 162 mg subcutaneously every week, all 5 receiving tocilizumab 8 mg/kg intravenously every 4 weeks, 6 of 7 patients receiving tocilizumab 162 mg every 2 weeks, and 1 of 3 patients receiving placebo also developed antibodies with neutralizing potential. None of the patients with positive antitocilizumab antibodies in the confirmation assay had serious or clinically significant hypersensitivity reactions or withdrew because of insufficient therapeutic response. Overall, antitocilizumab antibodies had no impact on the PK of tocilizumab. In the limited numbers of patients who had confirmed antitocilizumab antibodies, there was no apparent trend for reduced tocilizumab concentrations when evaluating their PK profiles with individual dosing records and actual sampling times (data not shown). Furthermore, in the population PK analysis, antitocilizumab antibodies were not identified as a covariate that influenced the PK of tocilizumab.
Exposure–Efficacy and Exposure–Safety Relationships
Clinical response (proportions of patients with ACR20/50/70 responses) increased with increasing tocilizumab Ctrough exposure quartiles after subcutaneous administration, but this was not observed with intravenous administration (Table 3). Exposure quartiles showed a clearer gradation in response with the subcutaneous every‐2‐week regimen compared with the subcutaneous every‐week regimen, in which the efficacy parameters showed a plateau past the first quartile. Logistic regressions for the subcutaneous every‐2‐week regimen and for the combined every‐2‐week and every‐week regimens demonstrated almost identical relationships between tocilizumab exposure and ACR responses (Supplementary Figure S3).
Table 3.
Proportion of Patients With ACR Responses in Week 24 by Observed Tocilizumab Ctrough Exposure Quartiles
| TCZ 162 mg SC QW | TCZ 162 mg SC Q2W | TCZ 8 mg/kg IV Q4W | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q1 (n = 131) | Q2 (n = 129) | Q3 (n = 130) | Q4 (n = 129) | Q1 (n = 98) | Q2 (n = 98) | Q3 (n = 98) | Q4 (n = 97) | Q1 (n = 134) | Q2 (n = 135) | Q3 (n = 134) | Q4 (n = 131) | |
| ACR20 responders, n (%) | 82 (63) | 99 (76) | 95 (73) | 106 (82) | 41 (43) | 63 (64) | 70 (71) | 71 (73) | 101 (75) | 110 (81) | 108 (81) | 102 (78) |
| ACR50 responders, n (%) | 51 (39) | 67 (52) | 69 (53) | 69 (53) | 24 (24) | 40 (41) | 48 (49) | 50 (52) | 78 (58) | 69 (51) | 77 (57) | 59 (45) |
| ACR70 responders, n (%) | 26 (20) | 37 (28) | 36 (28) | 35 (27) | 8 (8) | 16 (16) | 29 (30) | 25 (26) | 46 (34) | 40 (30) | 43 (32) | 33 (25) |
| Mean (median) Ctrough, μg/mL | 14.6 (15.4) | 32.7 (33.1) | 47.9 (47.7) | 80.1 (72.5) | 0.1 (0.1) | 2.6 (2.4) | 7.6 (7.5) | 17.4 (16.5) | 4.1 (4.1) | 12.2 (12.0) | 20.2 (19.8) | 38.9 (36.9) |
Q1–Q4 refer to Ctrough exposure quartiles (lowest to highest). ACR, American College of Rheumatology; Ctrough, predose trough concentration; IV, intravenous; Q2W, every 2 weeks; Q4W, every 4 weeks; QW, weekly; SC, subcutaneous; TCZ, tocilizumab.
There was no apparent association between increasing tocilizumab exposure and occurrence of AEs, including infections and infestations, for any of the dosing schedules (Table 4). The occurrence of serious AEs (SAEs) was low and similar with tocilizumab subcutaneous every‐week (26 of 631, 4.1%), subcutaneous every‐2‐week (15 of 437, 3.4%), and intravenous every‐4‐week (26 of 631, 4.1%) dosing. However, quartile analysis was not feasible because of the low numbers of SAEs. Neutrophil counts decreased with increasing tocilizumab exposure quartiles; there was increased incidence of grade 1 and grade 2 neutropenia with increasing tocilizumab exposure (Table 4), which was not temporally associated with serious infections. There was no clear trend for increased alanine aminotransferase elevations with increasing tocilizumab exposure quartiles (Table 4).
Table 4.
Neutropenia, ALT Elevations, and Adverse Events to Week 24 by Observed Tocilizumab Ctrough Exposure Quartiles
| TCZ 162 mg SC QW | TCZ 162 mg SC Q2W | TCZ 8 mg/kg IV Q4W | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q1 (n = 131) | Q2 (n = 129) | Q3 (n = 130) | Q4 (n = 129) | Q1 (n = 81) | Q2 (n = 80) | Q3 (n = 80) | Q4 (n = 80) | Q1 (n = 134) | Q2 (n = 135) | Q3 (n = 134) | Q4 (n = 131) | |
| Total patient‐years | 63.3 | 62.1 | 62.6 | 62.2 | 37.5 | 37.0 | 37.1 | 37.0 | 64.7 | 65.0 | 64.7 | 63.3 |
| Total AEs, n (n per 100 PY: 95%CI) | 388 (613: 553–677) | 292 (471: 418–528) | 373 (596: 537–660) | 346 (556: 499–618) | 177 (472: 405–547) | 136 (367: 308–434) | 139 (375: 315–442) | 158 (427: 363–499) | 349 (539: 484–599) | 345 (531: 476–590) | 367 (567: 510– 628) | 356 (563: 506– 624) |
| Infection and infestation AEs, n (n per 100 PY: 95%CI) | 68 (107: 83–136) | 62 (100: (77–128) | 75 (120: 94–150) | 72 (116: 91–146) | 40 (107: 76–145) | 30 (81: 55–116) | 25 (67: 44–99) | 26 (70: 46–103) | 61 (94: 72–121) | 80 (123: 98–153) | 76 (117: 93–147) | 78 (123: 97–154) |
| Neutropenia grade 1, n (%) | 20 (15) | 24 (19) | 33(26) | 26 (20) | 5 (6) | 3 (4) | 12 (15) | 18 (23) | 5 (4) | 9 (7) | 19 (14) | 17 (13) |
| Neutropenia grade 2, n (%) | 9 (7) | 17 (13) | 12 (9) | 27 (21) | 0 (0) | 4 (5) | 5 (6) | 9 (11) | 5 (4) | 9 (7) | 19 (14) | 17 (13) |
| Neutropenia grade 3, n (%) | 1 (1) | 2 (2) | 5 (4) | 2 (2) | 1 (1) | 2 (3) | 4 (5) | 1 (1) | 3 (2) | 2 (2) | 3 (2) | 7 (5) |
| ALT elevation grade 1, n (%) | 69 (59) | 59 (50) | 68 (57) | 49 (40) | 24 (31) | 28 (38) | 32 (44) | 30 (38) | 55 (45) | 52 (44) | 65 (50) | 44 (37) |
| ALT elevation grade 2, n (%) | 4 (3) | 8 (7) | 4 (3) | 1 (1) | 1 (1) | 3 (4) | 2 (3) | 0 (0) | 5 (4) | 5 (4) | 5 (4) | 4 (3) |
| ALT elevation grade 3, n (%) | 2 (2) | 0 (0) | 1 (0.8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (2) | 1 (1) | 0 (0) | 0 (0) |
| Mean (median) Ctrough, μg/mL | 14.6 (15.4) | 32.7 (33.0) | 47.9 (47.7) | 80.1 (72.5) | 0.2 (0.1) | 3.5 (3.6) | 8.2 (8.1) | 17.9 (16.6) | 4.1 (4.1) | 12.2 (12.0) | 20.2 (19.8) | 38.9 (36.9) |
Q1–Q4 refer to Ctrough exposure quartiles (lowest to highest). There were no cases of grade 4 neutropenia observed. AE, adverse event; ALT, alanine aminotransferase; Ctrough, predose trough concentration; IV, intravenous; PY, patient‐years; Q2W, every 2 weeks; Q4W, every 4 weeks; QW, weekly; SC, subcutaneous; TCZ, tocilizumab.
Discussion
As expected, fluctuation of tocilizumab concentration at steady state was small for the subcutaneous every‐week and every‐2‐week regimens compared with intravenous administration over the dosing interval. Mean Ctrough values were higher with 162‐mg subcutaneous every‐week dosing than with 8 mg/kg intravenous every‐4‐week dosing. Tocilizumab 162‐mg subcutaneous every‐2‐week dosing consistently resulted in the lowest Ctrough values. Tocilizumab 162 mg subcutaneous every week showed noninferior efficacy and comparable safety to the 8 mg/kg intravenous every‐4‐week regimen. Consistent with the clinical results, the 2 regimens exhibited similar PD responses (IL‐6, sIL‐6R, CRP, and ESR). CRP and ESR suppression compared with the 162‐mg subcutaneous every‐2‐week regimen, suggesting greater inhibition of IL‐6‐mediated activation of the acute‐phase response.
A 2‐compartment PK model with parallel linear and Michaelis–Menten elimination and first‐order absorption could best describe the PK of the tocilizumab subcutaneous formulation. Body weight had the most significant influence on tocilizumab clearance and volume parameters. The power coefficients of the effect of body weight on clearance and volume terms of the structural model (0.51 and 0.68, respectively) were lower than the allometric coefficients of 0.75 for clearance and 1 for volume,21 but consistent with the previous analysis for tocilizumab (in which CL depended on BSA with a power coefficient of 0.67 approximately corresponding to a coefficient of 0.50 for dependence on weight).19
Unlike intravenous dosing, the tocilizumab subcutaneous dosing regimens use a fixed dose (162 mg) over a wide range of body weights. It was important, therefore, to investigate the influence of body weight on tocilizumab PK. In both SUMMACTA and BREVACTA, patients were stratified by body weight category (<60, 60 to <100, and ≥100 kg) for efficacy and safety analysis. These analyses are summarized elsewhere.14, 15 We summarize here observed and model‐predicted PK parameters using the same body weight categories (Table 2). Tocilizumab exposure provided by the subcutaneous regimens was inversely correlated with body weight. Patients in the highest body weight category (≥100 kg) obtained little or no benefit from the every‐2‐week subcutaneous regimen,15 which resulted in Ctrough concentrations considered subefficacious.6 In these patients, the ACR response scores were numerically similar to placebo. The every‐week regimen resulted in clinical response (ACR scores) in patients weighing >100 kg, comparable to the 8 mg/kg every‐4‐weel intravenous regimen.14 On the other hand, patients in the lightest body weight category (<60 kg), who typically show the highest exposure from the subcutaneous every‐week regimen, did not show increased incidence of AEs. Examination of total AEs, infections, neutropenia, and liver enzyme elevation by exposure quartiles (Table 4) reinforced the safety profile of subcutaneous tocilizumab. Although there was a higher incidence of grade 1 or 2 neutropenia with higher tocilizumab exposure, the incidence of grade 3 or 4 neutropenia was low and comparable for the tocilizumab subcutaneous every‐week and intravenous regimens, and there was no association between grade 3 or 4 neutropenia and the development of serious infections.
Although the starting‐dose recommendation for tocilizumab is different in the United States (162 mg subcutaneously every 2 weeks) and the European Union (162 mg subcutaneous every week), regulatory authorities in both regions allow for dose adjustment up (United States) or down (European Union) to optimize the risk:benefit ratio for individual patients, based on well‐established efficacy scores, such as ACR and DAS28, and objective laboratory tests, such as liver enzymes and neutrophil count.7, 8
Conclusions
The results of this PK/PD analysis of subcutaneous and intravenous administration of tocilizumab in patients with RA demonstrate that the higher exposure obtained with subcutaneous every‐week dosing compared with subcutaneous every‐2‐week dosing is associated with more pronounced PD effects and with greater clinical efficacy that is comparable to tocilizumab intravenous 8 mg/kg every‐4‐week dosing. Tocilizumab exposure was lowest in patients weighing >100 kg who received subcutaneous every‐2‐week dosing, supporting recommendations for subcutaneous every‐week dosing in this body weight group. There was no trend for increasing AEs with increasing tocilizumab exposure.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Table S1. Baseline demographics and disease characteristics (safety populations)
Supplementary Table S2. TCZ PK Parameters for SUMMACTA
Supplementary Table S3. TCZ PK Parameters for BREVACTA
Supplementary Figure S1. Goodness‐of‐fit plots
Supplementary Figure S2. Observed Ctrough at week 24 versus body weight at week 20 with tocilizumab SC QW dosing (A), IV Q4W dosing (A) and SC Q2W dosing (B)
Supplementary Figure S3. Logistic regression analysis of ACR response
Declaration of Conflicting Interests
Hisham Abdallah, Peng Lu, Joy C. Hsu, Scott Fettner, Xiaoping Zhang, Wendy Douglass, and Lucy Rowell are employees of Roche. Min Bao is an employee of Genentech. Gerd R. Burmester has received honoraria for consulting and lectures from Roche. Alan Kivitz has received research grants and consulted for Genentech. Sara Duggan, PhD, and Meryl Mandle of Apothecom provided writing services on behalf of F Hoffmann‐La Roche Ltd.
References
- 1. Singh JA, Furst DE, Bharat A, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease‐modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(5):625–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smolen JS, Landewe R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gartlehner G, Hansen RA, Jonas BL, Thieda P, Lohr KN. The comparative efficacy and safety of biologics for the treatment of rheumatoid arthritis: a systematic review and metaanalysis. J Rheumatol. 2006;33(12):2398–2408. [PubMed] [Google Scholar]
- 4. Scott DL. Biologics‐based therapy for the treatment of rheumatoid arthritis. Clin Pharmacol Ther. 2012;91(1):30–43. [DOI] [PubMed] [Google Scholar]
- 5. Mihara M, Kasutani K, Okazaki M, et al. Tocilizumab inhibits signal transduction mediated by both mIL‐6R and sIL‐6R, but not by the receptors of other members of IL‐6 cytokine family. Int Immunopharmacol. 2005;5(12):1731–1740. [DOI] [PubMed] [Google Scholar]
- 6. Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin‐6 (IL‐6) and soluble IL‐6 receptor after administration of an anti‐IL‐6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112(10):3959–3964. [DOI] [PubMed] [Google Scholar]
- 7. ACTEMRA (tocilizumab) injection, for intravenous use injection, for subcutaneous use [package insert]. South San Francisco, CA: Genentech, Inc; 2014.
- 8. RoActemra (tocilizumab 20 mg/ml concentrate for solution for infusion) [summary of product characteristics]. Welwyn Garden City, UK: Roche Registration Limited; 2015.
- 9. Malaviya AP, Ostor AJ. Drug adherence to biologic DMARDS with a special emphasis on the benefits of subcutaneous abatacept. Patient Prefer Adherence. 2012;6:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwartzman S, Morgan GJ Jr. Does route of administration affect the outcome of TNF antagonist therapy? Arthritis Res Ther. 2004;6(suppl 2):S19–S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barton JL. Patient preferences and satisfaction in the treatment of rheumatoid arthritis with biologic therapy. Patient Prefer Adherence. 2009;3:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Williams EL, Edwards CJ. Patient preferences in choosing anti‐TNF therapies‐R1. Rheumatology (Oxford). 2006;45(12):1575–1576. [DOI] [PubMed] [Google Scholar]
- 13. Chilton F, Collett RA. Treatment choices, preferences and decision‐making by patients with rheumatoid arthritis. Musculoskeletal Care. 2008;6(1):1–14. [DOI] [PubMed] [Google Scholar]
- 14. Burmester GR, Rubbert‐Roth A, Cantagrel A, et al. A randomised, double‐blind, parallel‐group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease‐modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study). Ann Rheum Dis. 2014;73(1):69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kivitz A, Olech E, Borofsky M, et al. Subcutaneous tocilizumab versus placebo in combination with disease‐modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(11):1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang X, Chen YC, Fettner S, et al. Pharmacokinetics and pharmacodynamics of tocilizumab after subcutaneous administration in patients with rheumatoid arthritis. Int J Clin Pharmacol Ther. 2013;51(8):620–630. [DOI] [PubMed] [Google Scholar]
- 17. Zhang X, Peck R. Clinical pharmacology of tocilizumab for the treatment of patients with rheumatoid arthritis. Expert Rev Clin Pharmacol. 2011;4(5):539–558. [DOI] [PubMed] [Google Scholar]
- 18. Beal S, Sheiner L, eds. NONMEM User's Guide. San Francisco, CA: NONMEM Project Group, University of California, San Francisco; 1992. [Google Scholar]
- 19. Frey N, Grange S, Woodworth T. Population pharmacokinetic analysis of tocilizumab in patients with rheumatoid arthritis. J Clin Pharmacol. 2010;50(7):754–766 [DOI] [PubMed] [Google Scholar]
- 20. Stubenrauch K, Wessels U, Birnboeck H, Ramirez F, Jahreis A, Schleypen J. Subset analysis of patients experiencing clinical events of a potentially immunogenic nature in the pivotal clinical trials of tocilizumab for rheumatoid arthritis: evaluation of an antidrug antibody ELISA using clinical adverse event‐driven immunogenicity testing. Clin Ther. 2010;32(9):1597–1609. [DOI] [PubMed] [Google Scholar]
- 21. Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Table S1. Baseline demographics and disease characteristics (safety populations)
Supplementary Table S2. TCZ PK Parameters for SUMMACTA
Supplementary Table S3. TCZ PK Parameters for BREVACTA
Supplementary Figure S1. Goodness‐of‐fit plots
Supplementary Figure S2. Observed Ctrough at week 24 versus body weight at week 20 with tocilizumab SC QW dosing (A), IV Q4W dosing (A) and SC Q2W dosing (B)
Supplementary Figure S3. Logistic regression analysis of ACR response