

Abstract
Protein‐templated reactions enable the target‐guided formation of protein ligands from reactive fragments, ideally with no background reaction. Herein, we investigate the templated formation of amides. A nucleophilic fragment that binds to the coagulation factor Xa was incubated with the protein and thirteen differentially activated dipeptides. The protein induced a non‐catalytic templated reaction for the phenyl and trifluoroethyl esters; the latter was shown to be a completely background‐free reaction. Starting from two fragments with millimolar affinity, a 29 nm superadditive inhibitor of factor Xa was obtained. The fragment ligation reaction was detected with high sensitivity by an enzyme activity assay and by mass spectrometry. The reaction progress and autoinhibition of the templated reaction by the formed ligation product were determined, and the structure of the protein–inhibitor complex was elucidated.
Keywords: amide bond formation, enzyme inhibitors, fragment-based drug discovery, peptide bond formation, protein-templated reactions
Over the past years, fragment‐based drug discovery has been recognized as a powerful bottom‐up approach towards potent, selective, and efficient protein ligands.1 The development of fragment‐based ligands, however, is challenging for several reasons. Bioactive fragments are usually difficult to identify owing to their low, often millimolar affinities.2 In most cases, biophysical methods such as NMR spectroscopy,3 protein crystallography,4 or surface plasmon resonance5 have been employed to solve this “detection problem” of fragment‐based drug discovery. Once two fragments with adjacent, non‐overlapping binding sites have been identified, a suitable linker has to be found to prepare a bioactive fragment combination with increased affinity. This challenge can be denominated as the “linkage problem” and usually requires considerable chemical variation to be solved. The covalent assembly of protein ligands on the protein target, also referred to as protein‐templated or target‐guided formation of protein ligands, can be a powerful solution for both of these challenges.6
The templated formation of protein ligands from fragments has been realized for both reversible and irreversible ligation reactions. Whereas there are numerous examples of reversible ligation reactions,6, 7 the investigation of irreversible ligation reactions has been limited to a few reaction types, mostly involving dipolar cycloaddition reactions.8, 9, 10 In addition, larger protein ligands have been constructed from peptides by native chemical ligation11 and alkylation reactions12 but these were usually accompanied by considerable background reactions. An extension of the repertoire of protein‐templated reactions would be especially desirable for linkages that are found in many bioactive ligands for diverse protein targets. The amide bond belongs to this class of privileged fragment linkages, as revealed by an analysis of the World Drug Index, a database of bioactive molecules.13 Our goal was to establish protein‐templated amidation reactions that allow for direct, sensitive, and rapid detection of biological activity (not only binding) of the formed fragment combination products in a biochemical assay.
Amide linkages are generally formed from activated carboxylic acid derivatives (“active esters”) and amine nucleophiles.14 A clean templated amidation reaction requires the reversible binding of both reactive fragments to the protein and has to proceed from the bound state only, with no background reaction of non‐bound fragments (Scheme 1).
Scheme 1.

Protein‐templated amide bond formation. The reaction of an activated carboxylic acid derivative (yellow) with a nucleophilic amine fragment (green) occurs only in the presence of the protein target by a non‐catalytic mechanism (templated reaction, bottom) whereas the non‐templated background reaction (top) does not proceed.
We used the protein factor Xa as a template, a serine protease of the blood coagulation cascade and target of antithrombotic drugs.15 The dipeptide O‐benzyl‐N‐benzylsulfonyl‐d‐serinyl‐glycine (1; Scheme 2) was selected as a small‐molecule fragment of a reported inhibitor and was expected to bind to the S2–S4 pockets of the enzyme.16 To screen active esters for their suitability as substrates in protein‐templated amidation reactions, carboxylic acid derivatives 1–13 were prepared, which cover a broad range of reactivity towards a nucleophilic amine fragment. 4‐Aminomethylbenzamidine (14) was employed as the nucleophile and typical S1‐binding fragment of trypsin‐like serine proteases.17 The inhibition of factor Xa was determined in an enzyme activity assay using the fluorogenic substrate 7‐(N‐Boc‐leucinyl‐glycinyl‐arginyl)‐7‐amino‐4‐methylcoumarin (K M=76 μm) for detection.18 Carboxylic acid 1 was found to be a moderately active competitive inhibitor of factor Xa with a binding affinity (K I) of 5.5 mm, and the K I value of 14 was 0.68 mm. Ligation product 15, however, was highly active with a K I value of 29 nm. Biolayer interferometry (BLI) was used as an independent biophysical method to confirm the binding kinetics of the inhibitors (see the Supporting Information).19
Scheme 2.
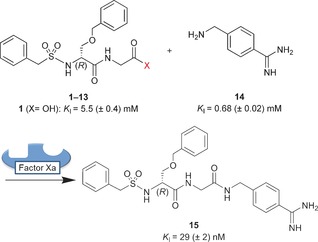
Dipeptide derivatives 1–13 and 4‐aminomethylbenzamidine (14) were incubated in the presence of the protein target factor Xa to observe the protein‐templated formation of inhibitor 15.
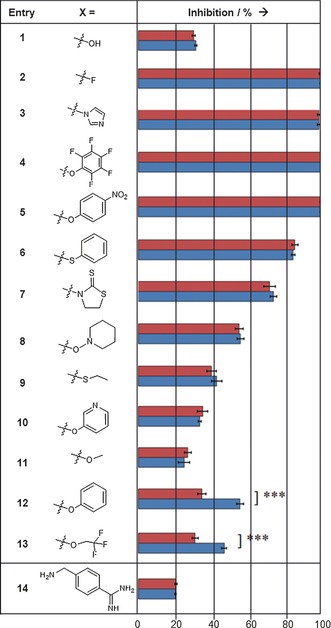
For the sensitive detection of templated inhibitor formation, each of the carboxylic acid derivatives 1–13 and the nucleophilic fragment 14 was incubated at room temperature with protein factor Xa (14.5 nm) under “templating conditions” for two hours. Then, the fluorogenic substrate was added, and the relative inhibition of the enzyme was determined (Table 1). For comparison, carboxylic acid derivatives 1–13 were incubated for the same time only with the nucleophilic fragment 14 but without the protein under “non‐templating conditions”. The inhibition was recorded after adding equal concentrations of substrate and enzyme as before. Nucleophile 14 at a concentration of 0.285 mm and a mixture of 1 (5 mm) and 14 (0.285 mm) were employed as negative controls, resulting in 20 % and 32 % inhibition, respectively, both under templating and non‐templating conditions. Complete inhibition of factor Xa was observed for 14 and 0.285 mm of either acyl fluoride 2, acyl imidazole 3, pentafluorophenyl ester 4, and 4‐nitrophenyl ester 5, indicating the formation of inhibitor 15 with and without the protein and the high reactivity of these activated acid derivatives. Reactions with phenyl thioester 6, 3‐acyl‐2‐thiono‐1,3‐thiazolidine 7, and piperidinyl‐1‐ester 8 afforded partial inhibition of factor Xa but no templating effect was detected. The corresponding ethyl thioester 9, pyridin‐3‐yl ester 10, and methyl ester 11 displayed only weak partial inhibition even at 5 mm when incubated with nucleophile 14, again without any signs of a templating effect. A clear, statistically significant, and reproducible templating effect was observed for two dipeptide derivatives, namely phenyl ester 12 and 2,2,2‐trifluoroethyl ester 13.
Table 1.
Inhibition of factor Xa through the formation of inhibitor 15.[a]
|
[a] In the fragment ligation assay, the dipeptide derivatives 1–13 were incubated with 4‐aminomethylbenzamidine (14) in the presence (blue) or absence (red) of the target protein (pH 8, 2 h, 20 °C; see the Supporting Information for details). The active esters 12 and 13 displayed a statistically significant templating effect as determined with an unpaired t‐test yielding p=0.0002.
Thus the templated amidation reactions of these two ester fragments were investigated in greater detail. Reactions of 12 or 13 with 14 in the presence of the protein target, factor Xa, were analyzed after 2, 5, and 10 h at 20 °C (RT) or 37 °C (Figure 1). Both the extension of the reaction time and the higher temperature led to markedly increased inhibition for both esters. With phenyl ester 12, inhibition was slightly enhanced in the non‐templated reaction at 37 °C and after prolonged reaction times. For ester 13, the inhibition remained the same for the non‐templated reaction, both at room temperature and at 37 °C, indicating the absence of a non‐templated background reaction.
Figure 1.
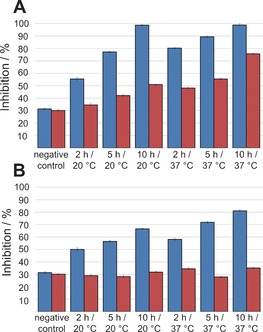
Inhibition of factor Xa by the ligation product of phenyl ester 12 (A) or 2,2,2‐trifluoroethyl ester 13 (B; both 5 mm) with 4‐aminomethylbenzamidine (14; 0.285 mm). Blue: templated reaction; red: non‐templated reaction. A mixture of carboxylic acid 1 and amine 14 was used as negative control.
The templated formation of protein ligand 15 was validated by reverse‐phase HPLC‐MS. After 2 h incubation with the protein, significant amounts of amidation product 15 were detected for both active esters 12 and 13 in the templated reaction at 20 °C, whereas no product was detectable without the protein (see the Supporting Information, Figure S1). To quantify the amount of product formed by the templated reaction, a high‐resolution quadrupole time of flight (QTOF) mass spectrometer was employed as a detector (Figure 2). Phenyl ester 12 furnished 26 nm of the inhibitor 15 with a half saturation time (t 1/2) of 1.4 h; trifluoroethyl ester 13 yielded 10 nm of 15 with t 1/2=1.7 h. Without the protein template, only 4 and 0.15 nm of 15 were detected with no time‐dependent background reaction. The templated reactions of both esters were clearly autoinhibited as the highly active product 15 prevented the binding and templated reaction of further fragments.
Figure 2.
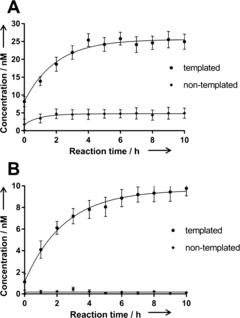
Formation of the factor Xa inhibitor 15 in the templated amidation reaction of 14 (0.285 mm) and phenyl ester 12 (A) or 2,2,2‐trifluoroethyl ester 13 (5 mm; B) at room temperature. The concentration of 15 was quantified by extracted ion chromatography HPLC/QTOF‐MS, yielding a saturation curve with c max=25.6/9.6 nm and t 1/2=1.4/1.7 h, respectively.
Finally, it was our goal to rationalize the observed templated reaction on the basis of the protein structure (Figure 3). Crystals of factor Xa in complex with inhibitor 15 were generated by back‐soaking (see the Supporting Information for details), and the crystal structure of the complex was solved at 2.2 Å resolution (PDB No. 5K0H). As expected, the benzamidine occupies the S1 pocket while the O‐benzyl and N‐benzylsulfonyl residues are found in adjacent hydrophobic pockets. Starting from the crystal structure, the binding modes of both ester fragments 12 and 13 and benzamidine 14 were derived. The energy‐minimized model of the protein complex with ester 12/13 and the benzamidine fragment reveals a distance of 3.4 Å between the amine N atom of 14 and the ester carbonyl C atom of 12 and 13 while the van der Waals radii of nitrogen and carbon add up to 3.25 Å. The N‐C‐O angle between the N nucleophile and the attacked carbonyl double bond amounts to 82° for 12 and 83° for 13. Thus the structure model supports the feasibility of a templated reaction between ester fragments and aminomethylbenzamidine 14 to inhibitor 15.
Figure 3.
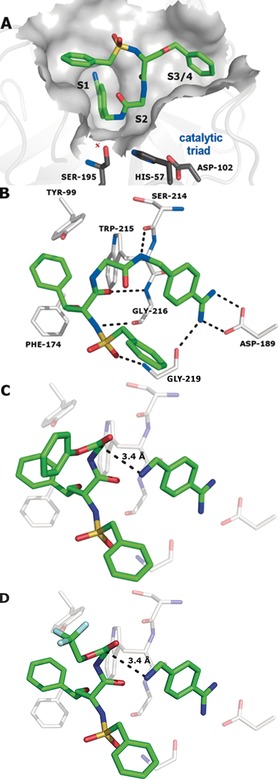
Structure of factor Xa in complex with inhibitor 15 (PDB No. 5K0H) displaying the protein surface (A) and the key interacting amino acids (B). C, D) Calculated complexes of the protein with benzamidine 14 and phenyl ester 12 or 2,2,2‐trifluoroethyl ester 13, respectively.
In summary, we have described the first examples of a background‐free protein‐templated amidation reaction. Templated formation of the factor Xa inhibitor 15 (K I=29 nm) was shown to occur from phenyl ester 12 and 2,2,2‐trifluorethyl ester 13 by direct determination of the enzymatic activity in a kinetic assay and by analysis of the autoinhibited ligation reaction by HPLC‐MS. While phenyl ester 12 showed nearly no background reaction with amine 14 at room temperature (20 °C) after two hours, 2,2,2‐trifluoroethyl ester 13 was completely unreactive without the protein template even at 37 °C and after ten hours. Applying the equation ΔG=−RT lnKI, the KI values of the fragments correspond to free binding energies of −12.7 and −17.8 kJ mol−1 for 1 and 14, respectively.
Assuming that the free binding energies of the fragments are additive in the ligation product, a free binding energy of −30.4 kJ mol−1 would be expected for inhibitor 15, corresponding to a K I value of 3.7 μm. Thus the fragment ligation product 15 with an inhibition constant of 29 nm providing a free binding energy of −42.3 kJ mol−1 constitutes an example for a strongly superadditive enhancement effect.20 One possible reason for this effect of fragment linking is that the linker between the fragments, in this case the amide bond, is more favorable for binding than the respective functional groups in the starting fragments. To investigate this possibility, the primary amide derivative of 1 (→compound 16) and the N‐acetylated derivative of 14 (→compound 17) were synthesized and tested in the inhibition assay (see the Supporting Information). As 16 and 17 showed slightly reduced affinities to the protein target (16: K I=6 mm; 17: 1.1 mm) compared with their parent compounds, it can be concluded that the linker does not contribute to the superadditive effect of inhibitor 15. Instead, the results suggest that the binding entropy of the fragment ligation product 15 is less negative than that of the fragments 1 and 14, giving rise to the increased affinity of the inhibitor.
It should be noted that the reported templated amidation reactions of an amine with stable amino acid esters demonstrate an alternative, non‐catalytic mechanism of peptide bond formation without a covalent acyl–RNA or acyl–protein intermediate as in ribosomal and non‐ribosomal amidation reactions. It remains to be investigated whether the reported mechanism is relevant in living systems. In principle, however, it should be functional on all protein surfaces that are capable of binding two fragments in an orientation that favors a subsequent ligation reaction. Therefore, the reported results suggest a systematic search for protein sites that are capable of inducing ligand‐molding reactions to yield bioactive protein ligands independent of the catalytic function of the protein.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the DFG (SFB 765 (B9)). We thank Pall ForteBio and its field application scientist Dr. Hendrik Wünsche for helping us with the biolayer interferometry (BLI) measurements.
M. Jaegle, T. Steinmetzer, J. Rademann, Angew. Chem. Int. Ed. 2017, 56, 3718.
Contributor Information
Mike Jaegle, http://www.bcp.fu‐berlin.de/ag‐rademann.
Prof. Dr. Jörg Rademann, Email: joerg.rademann@fu-berlin.de.
References
- 1.
- 1a. Murray C. W., Rees D. C., Nat. Chem. 2009, 1, 187–192; [DOI] [PubMed] [Google Scholar]
- 1b. Scott D. E., Coyne A. G., Hudson S. A., Abell C., Biochemistry 2012, 51, 4990–5003; [DOI] [PubMed] [Google Scholar]
- 1c. Erlanson D. A., Fesik S. W., Hubbard R. E., Jahnke W., Jhoti H., Nat. Rev. Drug Discovery 2016, 15, 605–619. [DOI] [PubMed] [Google Scholar]
- 2. Davis B. J., Erlanson D. A., Bioorg. Med. Chem. Lett. 2013, 23, 2844–2852. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Lepre C. A., Moore J. M., Peng J. W., Chem. Rev. 2004, 104, 3641–3675; [DOI] [PubMed] [Google Scholar]
- 3b. Klages J., Coles M., Kessler H., Analyst 2007, 132, 692–705. [DOI] [PubMed] [Google Scholar]
- 4. Chilingaryan Z., Yin Z., Oakley A. J., Int. J. Mol. Sci. 2012, 13, 12857–12879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neumann T., Junker H.-D., Schmidt K., Sekul R., ACS Med. Chem. Lett. 2007, 7, 1630–1642. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Ramström O., Lehn J.-M., Nat. Rev. Drug Discovery 2002, 1, 26–36; [DOI] [PubMed] [Google Scholar]
- 6b. Corbett P. T., Leclaire J., Vial L., West K. R., Wietor J.-L., Sanders J. K. M., Otto S., Chem. Rev. 2006, 106, 3652–3711; [DOI] [PubMed] [Google Scholar]
- 6c. Sharpless K. B., Manetsch R., Expert Opin. Drug Discovery 2006, 1, 525–538; [DOI] [PubMed] [Google Scholar]
- 6d. Schmidt M. F., Rademann J., Trends Biotechnol. 2009, 27, 512–521; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Mondal M., Hirsch A. K. H., Chem. Soc. Rev. 2015, 44, 2455–2488; [DOI] [PubMed] [Google Scholar]
- 6f. Jaegle M., Nawrotzky E., Wong E. L., Arkona C., Rademann J. in Fragment-based Drug Discovery Lessons and Outlook (Eds.: D. A. Erlanson, W. Jahnke), Wiley-VCH, Weinheim, 2016, pp. 293–326. [Google Scholar]
- 7.
- 7a. Schmidt M. F., Isidro-Llobet A., Lisurek M., El-Dahshan A., Tan J., Hilgenfeld R., Rademann J., Angew. Chem. Int. Ed. 2008, 47, 3275–3278; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3319–3323; [Google Scholar]
- 7b. Schmidt M. F., El-Dahshan A., Keller S., Rademann J., Angew. Chem. Int. Ed. 2009, 48, 6346–6349; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6464–6467; [Google Scholar]
- 7c. Schmidt M. F., Groves M. R., Rademann J., ChemBioChem 2011, 12, 2640–2646; [DOI] [PubMed] [Google Scholar]
- 7d. Fernández-Bachiller I., Horatscheck A., Lisurek M., Rademann J., ChemMedChem 2013, 8, 1041–1056; [DOI] [PubMed] [Google Scholar]
- 7e. Burda E., Rademann J., Nat. Commun. 2014, 5, 5170; [DOI] [PubMed] [Google Scholar]
- 7f. Becker D., Kaczmarska Z., Arkona C., Schulz R., Tauber C., Wolber G., Hilgenfeld R., Coll M., Rademann J., Nat. Commun. 2016, 7, 12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Hu X., Manetsch R., Chem. Soc. Rev. 2010, 39, 1316–1324; [DOI] [PubMed] [Google Scholar]
- 8b. Oueis E., Sabot C., Renard P.-Y., Chem. Commun. 2015, 51, 12158–12169; [DOI] [PubMed] [Google Scholar]
- 8c. Bosc D., Jakhlal J., Deprez-Poulain B., Future Med. Chem. 2016, 8, 381–404. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Lewis W. G., Green L. G., Grynszpan F., Radic Z., Carlier P. R., Taylor P., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 1053–1057; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1095–1099; [Google Scholar]
- 9b. Mocharla V. P., Colasson B., Lee L. V., Röper S., Sharpless K. B., Wong C., Kolb H. C., Angew. Chem. Int. Ed. 2005, 44, 116–120; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 118–122; [Google Scholar]
- 9c. Whiting M., Muldoon J., Lin Y., Silverman S. M., Lindstrom W., Olson A. J., Kolb H. C., Finn M. G., Sharpless K. B., Elder J. H., Fokin V. V., Angew. Chem. Int. Ed. 2006, 45, 1435–1439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1463–1467; [Google Scholar]; Suzuki T., Ota Y., Kasuya Y., Mutsuga M., Kawamura Y., Tsumoto H., Nakagawa H., Finn M. G., Miyata N., Angew. Chem. Int. Ed. 2010, 49, 6817–6820; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6969–6972. [Google Scholar]
- 10. Hu X., Sun J., Wang H. G., Manetsch R., J. Am. Chem. Soc. 2008, 130, 13820–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vázquez O., Seitz O., J. Pept. Sci. 2014, 20, 78–86. [DOI] [PubMed] [Google Scholar]
- 12. Brauckhoff N., Hahne G., Yeh J. T., Grossmann T. N., Angew. Chem. Int. Ed. 2014, 53, 4337–4340; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4425–4429. [Google Scholar]
- 13. Lisurek M., Rupp B., Wichard J., Neuenschwander M., von Kries J. P., Frank R., Rademann J., Kühne R., Mol. Diversity 2010, 14, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montalbetti C. A. G. N., Falque V., Tetrahedron 2005, 61, 10827–10852. [Google Scholar]
- 15.
- 15a. Kamata K., Kawamoto H., Honma T., Iwama T., Kim S.-H., Proc. Natl. Acad. Sci. USA 1998, 95, 6630–6635; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Straub A., Roehrig S., Hillisch A., Angew. Chem. Int. Ed. 2011, 50, 4574–4590; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4670–4686. [Google Scholar]
- 16. Schweinitz A., Stürzebecher A., Stürzebecher U., Schuster O., Stürzebecher J., Steinmetzer T., Med. Chem. 2006, 2, 349–361. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Wiley M. R., Chirgadze N. Y., Clawson D. K., Craft T. J., Gifford-Moore D. S., Jones N. D., Olkowski J. L., Weir L. C., Smith G. F., Bioorg. Med. Chem. Lett. 1996, 6, 2387–2392; [Google Scholar]
- 17b. Gustafsson D., Antonsson T., Bylund R., Eriksson U., Gyzander E., Nilsson I., Elg M., Mattsson C., Deinum J., Pehrsson S., Karlsson O., Nilsson A., Sörensen H., Thromb. Haemostasis 1998, 79, 110–118. [PubMed] [Google Scholar]
- 18. Morita T., Kato H., Iwanaga S., Takada K., Kimura T., Sakakibara S., J. Biochem. 1977, 82, 1495–1498. [DOI] [PubMed] [Google Scholar]
- 19. Wartchow C. A., Podlaski F., Li S., Rowan K., Zhang X., Mark D., Huang K. S., J. Comput.-Aided Mol. Des. 2011, 25, 669–676. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Nazaré M., Matter H., Will D. W., Wagner M., Urmann M., Czech J., Schreuder H., Bauer A., Ritter K., Wehner V., Angew. Chem. Int. Ed. 2012, 51, 905–911; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 929–935; [Google Scholar]
- 20b. Baum B., Muley L., Smolinski M., Heine A., Hangauer D., Klebe G., J. Mol. Biol. 2010, 397, 1042–1054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary