

Abstract
Amyotrophic lateral sclerosis (ALS) is a rapidly progressing neurodegenerative disease in which the majority of upper and lower motor neurons are degenerated. Despite intensive efforts to identify drug targets and develop neuroprotective strategies, effective therapeutics for ALS remains unavailable. The identification and characterization of novel targets and pathways remain crucial in the development of ALS therapeutics. Adenosine is a major neuromodulator that actively regulates synaptic transmission. Interestingly, adenosine levels are significantly elevated in the cerebrospinal fluid (CSF) of progressing human ALS patients. In the current study, we showed that adenosine 2a receptor (A2aR), but not adenosine 1 receptor (A1R), is highly enriched in spinal (motor) neurons. A2aR expression is also selectively increased at the symptomatic onset in the spinal cords of SOD1G93A mice and end-stage human ALS spinal cords. Interestingly, we found that direct adenosine treatment is sufficient to induce embryonic stem cell-derived motor neuron (ESMN) cell death in cultures. Subsequent pharmacological inhibition and partial genetic ablation of A2aR (A2aR+/−) significantly protect ESMN from SOD1G93A+ astrocyte-induced cell death and delay disease progression of SOD1G93A mice. Taken together, our results provide compelling novel evidence that A2aR-mediated adenosine signaling contributes to the selective spinal motor neuron degeneration observed in the SOD1G93A mouse model of ALS.
Keywords: Adenosine, A2a receptor, KW6002, Astrocyte, Motor neuron, Amyotrophic lateral sclerosis (ALS)
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressing neurodegenerative disease in which the majority of upper and lower motor neurons are degenerated. Although the exact cause of the majority of sporadic ALS remains unknown, the identification of genetic mutations in superoxide dismutase 1 (SOD1) (Rosen et al., 1993), TAR DNA-binding protein (TDP 43) (Neumann et al., 2006), the open reading frame 72 on chromosome 9 (C90RF72) (DeJesus-Hernandez et al., 2011; Renton et al., 2011), and several other genes have provided critical insights about the possible pathogenic mechanisms of ALS (Renton et al., 2014). In particular, SOD1 mouse models that faithfully recapitulate characteristic human clinical symptoms remain the most widely used in vivo model of ALS (Bruijn et al., 1998; Gurney et al., 1994). By employing these models, previous studies have characterized multiple pathways that contribute to motor neuron cell death, including glutamate excitotoxicity, abnormal protein aggregation, oxidative stress, and deficit in axon transport (Ilieva et al., 2009). Importantly, both intrinsic (from motor neurons) and extrinsic (from surrounding non-neuronal glial cells) pathways are implicated in motor neuron cell death in ALS (Boillee et al., 2006; Clement et al., 2003; Kang et al., 2013; Yamanaka et al., 2008).
Adenosine is a major neuromodulator that actively regulates synaptic transmission through the activation of its high affinity receptors (mainly adenosine receptors 1 and 2A, A1R and A2aR) (Brundege and Dunwiddie, 1997). Consequently, adenosine signaling plays important roles in many physiological and pathological conditions. Extracellular adenosine is mainly derived from the rapid hydrolysis of ATP, presumably released from both neurons and non-neuronal glial cells (Blutstein and Haydon, 2013; Latini and Pedata, 2001). Although pre- and post-synaptic activation of A1R leads to inhibitory neuromodulation and generally neuroprotective effects (Lupica et al., 1992; Trussell and Jackson, 1985), activation of A2aR increases neuronal excitability (Burnstock, 1997). Adenosine signaling plays important roles in many physiological and pathological conditions, including sleep and arousal, nociception, seizure susceptibility, drug addiction, and neurodegeneration (Armentero et al., 2011; Boison et al., 2010; Halassa et al., 2009; Nam et al., 2013; Sawynok, 1998). Interestingly, genetic deletion of A2aR in A2aR−/− (knock-out) mice significantly attenuates the volume of cerebral infarction and associated neurological deficits in a focal transient ischemia model (Chen et al., 1999), suggesting a potentially neurotoxic effect of A2aR activation. In vitro pharmacological inhibition of A2aR also significantly protects motor neurons from excitatory insult-induced cell death (Mojsilovic-Petrovic et al., 2006). Moreover, selective inhibition of A2aR activation prevents loss of nigral dopaminergic neurons induced by neurotoxin exposure and potentiates striatal dopamine transmission (Armentero et al., 2011).
We have previously observed elevated release of ATP from cultured SOD1G93A astrocytes that is inhibited by dnSNARE expression (Kawamata et al., 2014). Adenosine levels are also significantly elevated in the cerebrospinal fluid (CSF) of progressing human ALS patients (Yoshida et al., 1999). Here we investigated the role of A2aR-mediated adenosine signaling in motor neuron cell death and ALS-related motor phenotypes using both in vitro and in vivo models with complementary genetic and pharmacological approaches.
Materials and methods
Animals and drug treatment
Transgenic SOD1G93A+ (#002726), A2aR (#010685, C129 strain) knock-out (−/−) and F1 hybrid wild-type (#100012) mice were obtained from the Jackson Laboratory (Bar Harbor, ME). This SOD1G93A mouse strain is in the mixed B6SJL background with high copy numbers of SOD1G93A transgene. For each established colony, only 5–6 generations of breeding were performed to maintain high copy numbers of SOD1G93A transgene. SOD1G93A mice were bred to F1 hybrid mice to maintain the colony, as suggested by the Jackson labs. The SOD1G93A+ and A2aR−/− mice were bred to generate A2aR+/−SOD1G93A+ mice and subsequently littermates A2aR+/−SOD1G93A+, A2aR−/− SOD1G93A+, and A2aR+/+SOD1G93A+ mice. Both male and female mice were used in all experiments. Mice were housed in humidity-, temperature- and light-controlled environment and provided with food and water ad libitum in accordance with the procedures of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Guidelines for the Use of Animals in Neuroscience Research and were approved by the Animal Care and Use Committee of Tufts University. For in vivo KW6002 administration, sterile KW6002 (3 mg/kg, permeable to the blood–brain barrier) or PBS (sham control) was administered to SOD1G93A mice starting at P90–95 by daily intraperitoneal injection (i.p.).
Primary astrocyte cultures
Cortices were dissected, and the meninges were removed from the cortices under the dissecting microscope. The tissue was then minced with a razor blade and trypsinized (0.05% trypsin; Sigma-Aldrich) for 10 min in a 37 °C water bath and then dissociated gently by trituration with a fire-polished Pasteur pipette. Dissociated cells were filtered through a 70 μm strainer to collect the clear astrocyte cell suspension. For setting up astrocyte and motor neuron co-cultures, 50,000 astrocytes per well were directly seeded on eight-well culture slides (BD Biosciences).
Differentiation and purification of motor neurons from mouse ES cells
Mouse ES cells (a kind gift from Dr. Kevin Eggan, Harvard University, Cambridge, MA) were derived from Hb9-driven eGFP transgenic mice. ES cells were grown on top of mouse embryonic fibroblasts to 70–80% confluence in ES cell medium (knock-out DMEM, 10% fetal calf serum, 5% knock-out serum, 1% Glutamax, 1% nonessential amino acids, 1% penicillin/streptomycin, 0.14% 2-mercaptoethanol, and 1000 U of leukemia inhibitory factor). ES cells were then dissociated to induce the formation of embryoid bodies (EBs) in EB medium (DMEM/F-12, 10% knock-out serum, 1% Glutamax, 1% penicillin/streptomycin, and 0.1% 2-mercaptoethanol). To differentiate EBs into motor neurons, retinoic acid (final concentration of 0.1 μM) and smoothened agonist (final concentration of 1 μM) were added into the medium for 5 d. Differentiated motor neurons were further purified through fluorescence-activated cell sorting based on the eGFP fluorescence, driven by Hb9 promoter activation during differentiation. Purified eGFP+ motor neurons were plated on a confluent astrocyte layer (35,000 per well) or directly (50,000 per well) on poly-D-lysine-coated eight-well culture slides (BD Biosciences) in motor neuron culture medium (DMEM/F-12, 5% horse serum, 1% Glutamax, 2% B27, 1% N-2 supplement, 1% penicillin/streptomycin, 10 ng/ml GDNF, 10 ng/ml CNTF, and 10 ng/ml BDNF). Half of the motor neuron medium was replaced with fresh medium supplemented with growth factors every day. Fresh medium was replenished with adenosine (final conc.: 300 nM or 1 μM), A1R antagonist DPCPX (final conc.: 50 or 200 nM, Tocris Bioscience), or A2aR antagonist KW6002 (final conc.: 1 or 10 μM, Axon Medchem Reston, VA) for up to 21 d.
Determination of disease onset and end stage
Early disease onset in mice was determined by the loss of 10% forelimb grip strength from its baseline value. Briefly, the forelimb grip strength of an individual mouse was measured once a week starting at postnatal day 60 (P60) (for A2aR+/−SOD1G93A+, A2aR−/− SOD1G93A+, and A2aR+/+SOD1G93A+ mice) or every 4 d starting at P90 (for KW6002 administration), using the grip-strength meter (model 1027 DSM; Columbus Instruments). We first calculated the average of three peak grip-strength values as the baseline for forelimb grip strength for each mouse. We then assessed the 10% decreased value based on the baseline and determine the age at which the grip strength equals a 10% decrease of baseline value. The weight was measured along with the grip strength. Late disease onset was determined by the loss of 10% weight. Disease end stage was determined by a delayed (30 s) righting reflex. We started to analyze the righting reflex at P100. Both the measurement of grip strength and weight was performed in a complete blind manner.
Immunoblot
Freshly dissected (mouse) or frozen (human) spinal cord ventral horns were homogenized in lysis buffer (Tris–HCl 20 mm, NaCl 140 mm, EDTA 1 mm, SDS 0.1%, Triton 1% and glycerol 10%). 50 μg of total cell lysate was loaded into 4–15% gradient SDS-PAGE gels. Separated proteins were transferred onto a PVDF membrane (Bio-Rad) for 1 h. The membrane was blocked with 3% BSA in TBST (Tris buffer saline with 0.1% Tween 20) then incubated with a specific primary antibody overnight at 4 °C. On the following day, the membrane was incubated with HRP-conjugated secondary antibody (1:5000) diluted in TBST. Bands were visualized on CL-XPosure™ film (Thermo Scientific) by ECL Plus chemiluminescent substrate (Thermo Scientific). The following antibodies were used on the immunoblots: anti-A1R (1:300, Abcam), anti-A2aR (1:2500, Millipore), anti-Iba 1 (1:1000, Wako), anti-β-actin (1:3000, Sigma), and anti-GFAP (1:30,000, Dako).
Immunohistochemistry
ESMNs that are co-cultured with WT astrocytes for 3 d were washed three times with PBS and fixed in 4% PFA for 30 min. Cells were then washed three times with PBS and incubated with blocking solution (0.2% Triton X-100, 5% goat serum, and 1% BSA) for 30 min, followed by overnight incubation with the primary antibodies A2aR (1:200, Millipore) at 4 °C. After overnight incubation, cells were washed three times with PBS, and the immunostaining signal was detected with an Alexa Fluor-555-conjugated secondary antibody (1:2000, Jackson ImmunoResearch). Confocal images were captured with a Nikon A1R confocal microscope using NIS-elements software under a 20× lens (Nikon Instruments).
TRAP mRNA extraction and QRT-PCR
Ribosomal bound translating mRNAs from spinal cord astrocytes were specifically extracted using immunoprecipitation of BAC ALDH1L1 TRAP transgenic mouse spinal cords, as previously described (Doyle et al., 2008; Higashimori et al., 2013). Briefly, the anti-eGFP antibody (HtzGFP-19C8) was coupled to magnetic beads (Dynabeads, Invitrogen) as described in the kit instructions. Spinal cord supernatant was prepared and mixed with eGFP antibody-coupled beads for immunoprecipitation. Following immunoprecipitation, RNA was isolated using the Trizol reagent and precipitated with isopropanol. The total RNA from the cortex was prepared using the Trizol reagent. RNA was then converted to cDNA using a high archive cDNA synthesis kit (Applied Biosystems, Foster city, CA). The relative abundance of A1R and A2aR transcripts was assessed using SYBR-Green (Life Technologies) and custom made primers (A2aR, F: 5′GCCATCCCATTCGCCATCA3′, R: 5′GCAATAGCCAAGAGGCTGAAGA3′; A1R, F: 5′TGTGCCCGGAAATGTACTGG3′, R: 5′TCTGTGGCCCAATGTTGATAAG3′). β-Actin was used as an endogenous control for the normalization of RNA quantity.
Measurement of forelimb grip strength
The forelimb grip strength was measured as described previously (Kawamata et al., 2014; Yamanaka et al., 2008). Grip strength was assessed using a grip-strength meter (model 1027 DSM; Columbus Instruments). We decide to focus on forelimb grip strength, as it gives consistent readout (Yamanaka et al., 2008), while the hindlimb measurement is subject to high variability. Measurements of grip strength and animal weight usually start at P60. The baseline grip strength was calculated as the average of three peak grip strength values. There is an initial training period for the animals just before the first measurement on the same day. Animals were not trained to perform the grip strength task prior to P60.
Statistical analysis
Student’s t-test or one-way ANOVA, followed by Bonferroni’s test, were mainly used to determine statistical significance. The linear mixed-effects model was used to calculate the significance for all longitudinal forelimb grip strength changes. The Log-Rank analysis was used to determine the significance for Kaplan–Meier curves. P < 0.05 was considered to be statistically significant. Results were presented as mean ± S.E.M., unless otherwise stated.
Results
A2aR is highly enriched in spinal motor neurons and exhibits selectively increased expression in ALS
Previous studies found that A1R is ubiquitously expressed across different CNS regions, while the A2aR is highly enriched in the striatum (Morelli et al., 2010). Although early receptor binding assays suggest that adenosine receptors are expressed in the spinal cord (Choca et al., 1987), the relative expression levels of each adenosine receptor in the spinal cord, especially on motor neurons, remain unclear. We determined the relative expression levels of the A2aR in different CNS regions by immunoblot. The murine A2aR is primarily present (N80% immunoreactivity) in a dimer conformation on our immunoblot (~85 KDa), consistent with previous observation (Bairam et al., 2009). As shown in Fig. 1A, A2aR is highly and selectively enriched in the spinal cord compared to other CNS regions examined. In contrast, A1R expression (primarily in the monomer conformation in immunoblot) in the spinal cord is significantly lower than that in other examined regions (Fig. 1B). We further attempted to determine the relative expression of A1R and A2aR in motor neurons and astroglia in spinal cord sections using immunostaining. Although we tried a few different antibodies with optimized immunostaining procedures, the A2aR immunostaining signals are not specific with these antibodies and procedures. Alternatively, we employed the translational ribosome affinity purification (TRAP) approach (Doyle et al., 2008) to selectively isolate ribosome bound translating mRNA from spinal cord astrocytes of BAC ALDH1L1 TRAP transgenic mice. The expression of eGFP-tagged ribosome subunit allows pull-down of assembled ribosomes and associated mRNAs during active translations in the cell. Selective activation of the ALDH1L1 promoter in astrocytes has been previously characterized (Cahoy et al., 2008; Yang et al., 2011), and we have previously validated the specificity of TRAP-isolated astrocyte mRNAs (Higashimori et al., 2013). As shown in Fig. 1C, we found that the astroglial A1R translating mRNA is significantly higher than total spinal cord A1R mRNA levels, while the astroglial A2aR translating mRNA is significantly lower than spinal A2aR mRNA levels. As equal numbers of neurons and glial cells are generally observed in the mammalian central nervous system (CNS) (Azevedo et al., 2009), our results suggest that A2aR is enriched in non-astroglial cells in the spinal cord, including motor neurons. Although the A2aR antibody gives no specific and clear immunostaining on spinal cord sections, the A2aR immunostaining on cultured ESMNs found clear, specific A2aR immunoreactivity on ESMNs (Fig. 1D), consistent with previous observations that A2aR is found in spinal cord primary motor neurons (Mojsilovic-Petrovic et al., 2006).
Fig. 1.
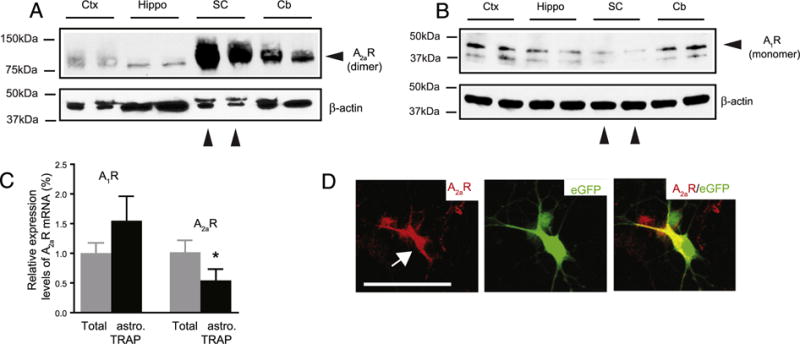
A2aR is highly enriched in spinal motor neuron. Representative immunoblot of A2aR (A) and A1R (B) protein expression in mouse spinal cords. Ctx: cortex; hippo: hippocampus; SC: spinal cord; Cb: cerebellum. C. Relative A1R and A2aR translating mRNA levels in spinal astrocytes and in total spinal cords, N = 3 mice/group, *, P < 0.05 D. Immunostaining of A2aR on eGFP+ ESMNs in culture. Scale bar: 50 μm.
To determine whether A1 and A2aR expression is pathologically altered in ALS, we examined their expression in the spinal cords of SOD1G93A mice. As the end-stage tissues usually show the consequence of many pathological changes that may not reflect the initial pathogenic mechanisms, we focused on the symptomatic onset period (P100–110). Interestingly, A2aR expression in SOD1G93A spinal cords is increased 3 fold compared to that in wild type mice (Figs. 2A–B), while no significant changes of A1R expression were found (Figs. 2C–D). To determine whether the changes of A2aR in SOD1G93A mice is relevant to human ALS pathology, we obtained post-mortem human ALS spinal cord tissues (University of Maryland brain and tissue bank, Baltimore, MD, summarized in Table 1) and examined the expression of A1R and A2aR. The human A2aR is primarily present as a monomer (N90% immunoreactivity) on immunoblots (Fig. 2E). The discrepancy in the dominant conformation of human and mouse A2aR on immunoblot may simply reflect the protein sequence difference of human A2aR from the mouse A2aR. Despite the variability, we observed a clear trend of up-regulation of A2aR in majority of human ALS spinal cords examined (Figs. 2E, G), while the A1R levels are not significantly altered (Fig. 2F). Consistently, up-regulation of A2aR has also been previously observed in lymphocytes of human ALS patients (Vincenzi et al., 2013).
Fig. 2.
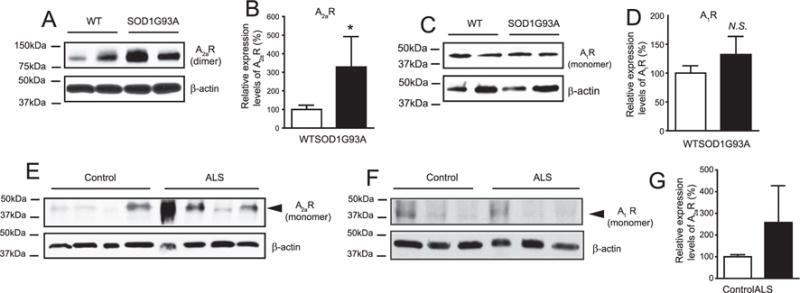
A2aR (but not A1R) is selectively up-regulated in spinal cord of SOD1G93A mice and post-mortem human ALS samples. A representative immunoblot of A2aR (A), A1R (C) and corresponding quantification (A2aR, B and A1R, D) in lumbar cords of WT and SOD1G93A mice (P100–P110); *, P < 0.05 determined by the Student’s t-test, N = 4–6 mice/sample; N.S.: not significant; A representative immunoblot of A2aR (E) and A1R (F) in spinal cords of post-mortem human ALS patients and controls. G. Quantification in spinal cords of post-mortem human ALS patients and controls; *, P < 0.05 determined by the Student’s t-test, N = 6/group.
Table 1.
Human ALS and control spinal cord tissues examined in the current study. All human control and ALS spinal cord tissues were obtained from the University of Maryland Brain & Tissue Bank (UMB, Baltimore, MD).
| UMB# | Age (year) | Gender | Post mortem interval (hour) | Region | Tissue type |
|---|---|---|---|---|---|
| 5444 | 79 | M | 10 | Cervical spinal cord | Frozen (control) |
| 5511 | 80 | F | 10 | Cervical spinal cord | Frozen (control) |
| 5657 | 82 | M | 22 | Cervical spinal cord | Frozen (control) |
| 5352 | 81 | M | 17 | Cervical spinal cord | Frozen (control) |
| 5458 | 64 | M | 13 | Cervical spinal cord | Frozen (control) |
| 5452 | 67 | M | 23 | Cervical spinal cord | Frozen (control) |
| 4837 | 78 | F | 12 | Cervical spinal cord | Frozen (ALS) |
| 5717 | 82 | M | Unknown | Cervical spinal cord | Frozen (ALS) |
| 5740 | 86 | F | 11 | Cervical spinal cord | Frozen (ALS) |
| 5774 | 77 | F | 28 | Cervical spinal cord | Frozen (ALS) |
| 1314 | 66 | M | 13 | Cervical spinal cord | Frozen (ALS) |
| 5724 | 65 | M | 25 | Cervical spinal cord | Frozen (ALS) |
A2aR-mediated adenosine signaling induces motor neuron cell death
We have previously observed elevated release of ATP from SOD1G93A astrocyte cultures (Kawamata et al., 2014). As extracellular ATP is often rapidly hydrolyzed into adenosine, we decided to investigate whether adenosine signaling is involved in SOD1G93A+ astrocyte-mediated motor neuron cell death in vitro. Daily treatment with an A2aR selective antagonist KW6002 (1–10 μM) significantly (P < 0.05) and consistently increases survival of motor neurons (from 24% ± 3% to 41% ± 2%) co-cultured with SOD1G93A+ astrocytes (Figs. 3A, C). Additionally, ESMNs in co-cultures treated with KW6002 have more observable neurites (revealed by βIII-tubulin staining) and larger cell bodies (Figs. 3A–B), further indicating that these motor neurons maintain healthier structural integrity. The treatment of a selective A1R antagonist 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), however, has no observable effect on the survival of cultured motor neurons (Fig. 3C). Together with the modest expression of A1R in spinal cord motor neurons (Figs. 1B–C), these results suggest that A1R is unlikely to be involved in SOD1G93A+ astrocyte-induced motor neuron death in vitro.
Fig. 3.
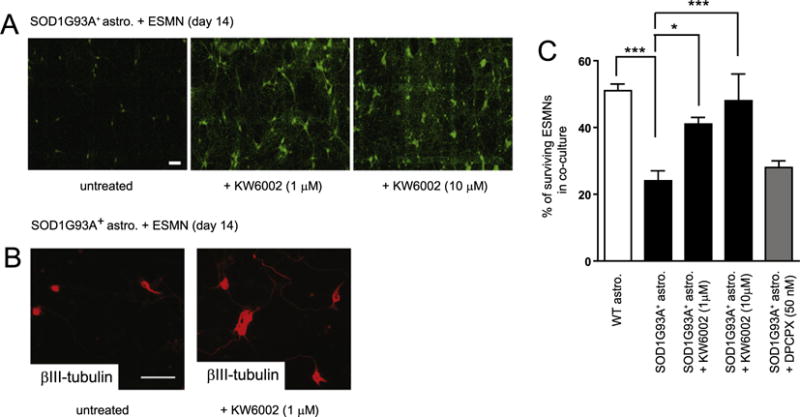
Direct treatment of A2aR antagonist significantly protects ESMNs from SOD1G93A+ astrocyte-induced cell death. A. A representative image of ESMNs following A2aR antagonist KW6002 treatment in ESMN and SOD1G93A+ astrocyte co-cultures. B. Immunostaining of βIII-tubulin in astrocyte and ESMN co-cultures following KW6002 treatment. C. The quantification of ESMNs following A2aR antagonist KW6002 treatment in ESMN and SOD1G93A+ astrocyte co-cultures. ***, P < 0.001, *, P < 0.05 from one-way ANOVA test and Bonferroni post-test; astro.: astrocytes. N = 264–418 neurons from three independent experiments.
To further determine whether adenosine signaling is directly involved in motor neuron cell death, ESMNs and wild type astrocyte co-cultures were treated with adenosine (300 nM and 1 μM), modestly higher than the physiological adenosine levels (100–200 nM), on a daily basis. The percentage of surviving motor neurons was calculated, as previously described (Kawamata et al., 2014). Surprisingly, direct treatment with adenosine is sufficient to induce motor neuron cell death at both 300 nM and 1 μM (Figs. 4A–B). Although A2aR is enriched in spinal cord neurons, it is still possible that adenosine-induced motor neuron death may result from the activation of astroglial A2aR. To rule out the involvement of astroglial A2aR, we then treat the ESMN culture alone with adenosine and found that direct treatment of adenosine can still induce motor neuron cell death (Figs. 4C–D), clearly indicating that adenosine-induced motor neuron cell death is likely to be directly mediated by A2aR on motor neurons.
Fig. 4.
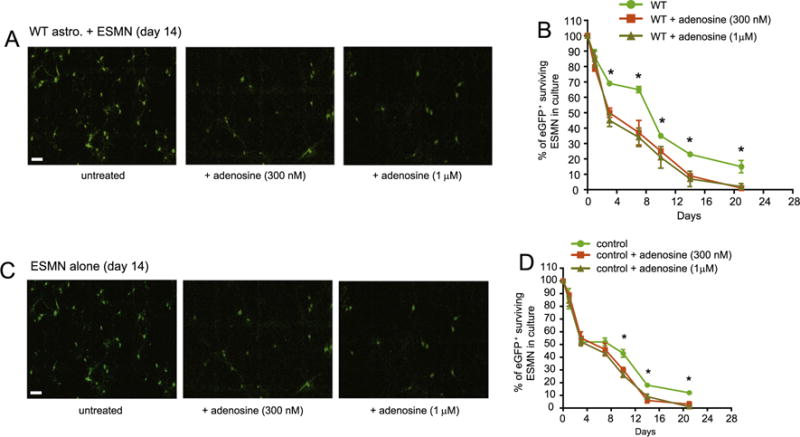
Direct treatment of adenosine is sufficient to induce ESMN cell death in cultures Representative images (A) and quantification (B) of ESMNs following adenosine treatment in ESMNs co-cultured with wild type astrocytes. N = 161–354 neurons from three independent experiments. Representative images (C) and quantification (D) of ESMNs following adenosine treatment in ESMN cultures alone. *, P < 0.05 from one-way ANOVA test and Bonferroni post-test, N = 121–254 neurons from three independent experiments.
In vivo pharmacological inhibition and partial genetic ablation of A2aR significantly delays disease progression in SOD1G93A mice
Encouraged by our in vitro results, we next determined whether pharmacological inhibition of A2aR is sufficient to alter motor behaviors and the overall survival of SOD1G93A mice. We administered sterile KW6002 (3 mg/kg, permeable to the blood–brain barrier) or PBS (sham control) by daily intraperitoneal injection (i.p.) to SOD1G93A mice starting at P90–95. We chose KW6002 because it is permeable to the blood–brain barrier and has the longest in vivo half-time (3–4 h) compared to other A2aR antagonists (Xu et al., 2005). The dose is determined based on previously published in vivo experiments with KW6002 in rodents (Aoyama et al., 2000). As shown in Fig. 5A, daily injection of KW6002 significantly (P = 0.01) and consistently delayed the progressive decrease of forelimb grip strength in SOD1G93A mice. KW6002 injections also significantly delayed late disease onset (Fig. 5B, from 121 ± 1.7 d to 129 ± 1.8 d), and extended the overall survival (from 137 ± 2.4 to 144 ± 1.9) of SOD1G93A mice (Figs. 5C–D). We did not observe significant sex difference in the overall survival in SOD1G93A mice following KW6002 administration.
Fig. 5.
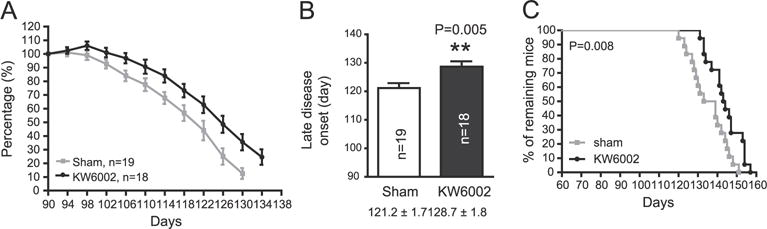
Daily in vivo administration of A2aR antagonist KW6002 significantly delays disease progression in SOD1G93A mice A. Longitudinal forelimb grip strength change of SOD1G93A mice that received KW6002 injections. P = 0.013 with the linear mixed-effects model; B. KW6002 injections significantly delay the late onset of disease in SOD1G93A mice; P = 0.005 with the unpaired Student’s t-test; C. Survival of individual experimental SOD1G93A mice following KW6002 injections. P = 0.008 with the Log-Rank (Mantel–Cox) test for the survival curve; *, P < 0.05; **, P < 0.01; N = 18–19 mice/group.
Although KW6002 is permeable to brain–blood barrier and has been characterized as a potent and selective A2aR antagonist, in vivo drug delivery could be variable and inefficient which may compromise the dissection of the effect of KW6002 in SOD1G93A mice. To further determine the effect of A2aR inhibition on motor phenotypes and the survival of SOD1G93A mice, we genetically ablated A2aR in SOD1G93A mice by generating A2aR+/−SOD1G93A and A2aR−/− SOD1G93A mice. The A2aR−/− mice themselves have no observable motor phenotypes. Interestingly, the complete ablation of A2aR expression has no observable effect on forelimb grip strength and overall survival in A2aR−/−SOD1G93A mice (Figs. 6A and E). Given the widespread expression of A2aR in the body, it is likely that the complete loss of A2aR significantly affects non-CNS physiology. Previous studies have found that the A2aR−/− mice become hypoalgesic and develop high blood pressure with increased heart rate (Ledent et al., 1997). In contrast, 50% reduced A2aR expression in SOD1G93A mice significantly delays the decrease of forelimb grip strength in A2aR+/− SOD1G93A mice (Fig. 6B). A2aR+/− SOD1G93A mice also live significantly longer than either A2aR+/+SOD1G93A or A2aR−/−SOD1G93A (P = 0.04) mice (Figs. 6C–D), suggesting that the partial suppression of A2aR indeed is neuroprotective and delays disease progression in the SOD1 mouse model of ALS.
Fig. 6.
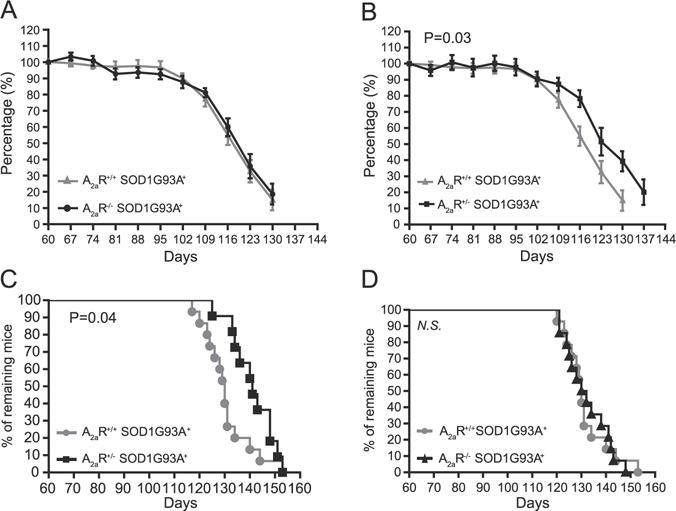
Partial genetic ablation of A2aR significantly delays disease progression in SOD1G93A mice. Longitudinal forelimb grip strength change between A2aR+/+ SOD1G93A+ and A2aR−/−SOD1G93A+ (A, P = 0.77), A2aR+/+ SOD1G93A+ and A2aR+/− SOD1G93A+ (B, P = 0.03). Survival curves of A2aR+/+SOD1G93A+, A2aR+/− SOD1G93A+ mice (C, P = 0.04, Log-rank test) and A2aR+/+ SOD1G93A+, A2aR−/−SOD1G93A+ mice (D, N.S.: not significant); N = 11–15 mice/group.
Discussion
In this study, we showed that A2aR is highly enriched in spinal (motor) neurons and its expression is selectively and significantly increased in spinal cords with ALS conditions (SOD1G93A mice and post-mortem human ALS samples). In addition, we found that direct adenosine treatment is sufficient to induce ESMN cell death in culture. Subsequent pharmacological inhibition and 50% reduction of A2aR significantly protect ESMN from SOD1G93A+ astrocyte-induced cell death and delay the disease progression of SOD1G93A mice. Our results suggest that A2aR-mediated adenosine signaling induces the selective spinal motor neuron degeneration observed in the SOD1G93A mouse model of ALS. These results provide interesting evidence about a novel toxic effect of adenosine on motor neurons. An enhanced A2aR signaling in the neuromuscular junctions at the pre-symptomatic (but not at the symptomatic) stages of SOD1G93A mice has also been observed (Nascimento et al., 2014). Although our observation indicates that the A2aR expression is altered at the symptomatic stage, the specific disease stages from which the A2aR signaling is altered in SOD1G93A mice likely resulted from different regions examined in our study and the previous study (Nascimento et al., 2014). Although chronic administration of caffeine, a non-specific antagonist of all adenosine receptors, significantly shortens overall survival of SOD1G93A mice without obvious effect on the body weight and motor phenotypes (Potenza et al., 2013), this likely resulted from the pan-inhibition of all adenosine receptors, especially the neuroprotective A1R.
Extracellular adenosine is usually derived from the hydrolysis of ATP and released through exocytosis or channels from multiple cell types in the CNS (Blutstein and Haydon, 2013). In pathological conditions, extracellular adenosine levels are often significantly elevated (Yoshida et al., 1999), as neurons lose their structural integrity during degeneration and glia become reactive in response to neuronal degeneration. We have observed increased release of ATP from cultured SOD1G93A+ astrocytes (Kawamata et al., 2014). How adenosine-induced A2aR activation is toxic to spinal motor neurons remains unclear. As A2aR activation is excitatory to spinal motor neurons (Miyazaki et al., 2008) and excitotoxicity is considered one of the primary mechanisms for spinal motor neuron degeneration (Cleveland and Rothstein, 2001), it is likely that elevated adenosine extensively stimulates pathologically increased A2aR and subsequently contributes to motor neuron excitotoxicity. In addition, pharmacological inhibition of A2aR-mediated transactivation of TrkB protects primary motor neurons from excitotoxic insults in cultures (Mojsilovic-Petrovic et al., 2006). In vivo conditional deletion of TrkB from mature motor neurons also attenuates mutant SOD1 toxicity (Zhai et al., 2011). Thus, the suppression of A2aR activation may also attenuate pathological transactivation of TrkB and protect motor neurons from degeneration. The specific downstream mechanisms of A2aR activation and the involvement of alternative ATP-mediated purinergic signaling in motor neuron degeneration in ALS remain to be investigated in the future.
Acknowledgments
This work is partially supported by The Packard Center for ALS Research (YY). We thank Dr. Kevin Eggan for providing mouse ES cells. We thank Tufts Center for Neuroscience Research (CNR) for providing valuable core facilities.
Footnotes
All authors have no conflict of interest.
References
- Aoyama S, Kase H, Borrelli E. Rescue of locomotor impairment in dopamine D2 receptor-deficient mice by an adenosine A2A receptor antagonist. J Neurosci. 2000;20:5848–5852. doi: 10.1523/JNEUROSCI.20-15-05848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armentero MT, Pinna A, Ferre S, Lanciego JL, Muller CE, Franco R. Past, present and future of A(2A) adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol Ther. 2011;132:280–299. doi: 10.1016/j.pharmthera.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob Filho W, Lent R, Herculano-Houzel S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009;513:532–541. doi: 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
- Bairam A, Joseph V, Lajeunesse Y, Kinkead R. Altered expression of adenosine A1 and A2A receptors in the carotid body and nucleus tractus solitarius of adult male and female rats following neonatal caffeine treatment. Brain Res. 2009;1287:74–83. doi: 10.1016/j.brainres.2009.06.064. [DOI] [PubMed] [Google Scholar]
- Blutstein T, Haydon PG. The importance of astrocyte-derived purines in the modulation of sleep. Glia. 2013;61:129–139. doi: 10.1002/glia.22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Boison D, Chen JF, Fredholm BB. Adenosine signaling and function in glial cells. Cell Death Differ. 2010;17:1071–1082. doi: 10.1038/cdd.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Brundege JM, Dunwiddie TV. Role of adenosine as a modulator of synaptic activity in the central nervous system. Adv Pharmacol. 1997;39:353–391. doi: 10.1016/s1054-3589(08)60076-9. [DOI] [PubMed] [Google Scholar]
- Burnstock G. The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology. 1997;36:1127–1139. doi: 10.1016/s0028-3908(97)00125-1. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choca JI, Proudfit HK, Green RD. Identification of A1 and A2 adenosine receptors in the rat spinal cord. J Pharmacol Exp Ther. 1987;242:905–910. [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimori H, Morel L, Huth J, Lindemann L, Dulla C, Taylor A, Freeman M, Yang Y. Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum Mol Genet. 2013;22:2041–2054. doi: 10.1093/hmg/ddt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligoden-drocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Ng SK, Diaz N, Burstein S, Morel L, Osgood A, Sider B, Higashimori H, Haydon PG, Manfredi G, Yang Y. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J Neurosci. 2014;34:2331–2348. doi: 10.1523/JNEUROSCI.2689-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini S, Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem. 2001;79:463–484. doi: 10.1046/j.1471-4159.2001.00607.x. [DOI] [PubMed] [Google Scholar]
- Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- Lupica CR, Proctor WR, Dunwiddie TV. Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: analysis of unitary EPSP variance measured by whole-cell recording. J Neurosci. 1992;12:3753–3764. doi: 10.1523/JNEUROSCI.12-10-03753.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki N, Nakatsuka T, Takeda D, Nohda K, Inoue K, Yoshida M. Adenosine modulates excitatory synaptic transmission and suppresses neuronal death induced by ischaemia in rat spinal motoneurones. Pflugers Arch. 2008;457:441–451. doi: 10.1007/s00424-008-0542-1. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Jeong GB, Crocker A, Arneja A, David S, Russell DS, Kalb RG. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J Neurosci. 2006;26:9250–9263. doi: 10.1523/JNEUROSCI.1856-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli M, Carta AR, Kachroo A, Schwarzschild MA. Pathophysiological roles for purines: adenosine, caffeine and urate. Prog Brain Res. 2010;183:183–208. doi: 10.1016/S0079-6123(10)83010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HW, Bruner RC, Choi DS. Adenosine signaling in striatal circuits and alcohol use disorders. Mol Cell. 2013;36:195–202. doi: 10.1007/s10059-013-0192-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento F, Pousinha PA, Correia AM, Gomes R, Sebastiao AM, Ribeiro JA. Adenosine A2A receptors activation facilitates neuromuscular transmission in the pre-symptomatic phase of the SOD1(G93A) ALS mice, but not in the symptomatic phase. PLoS One. 2014;9:e104081. doi: 10.1371/journal.pone.0104081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Potenza RL, Armida M, Ferrante A, Pezzola A, Matteucci A, Puopolo M, Popoli P. Effects of chronic caffeine intake in a mouse model of amyotrophic lateral sclerosis. J Neurosci Res. 2013;91:585–592. doi: 10.1002/jnr.23185. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sawynok J. Adenosine receptor activation and nociception. Eur J Pharmacol. 1998;347:1–11. doi: 10.1016/s0014-2999(97)01605-1. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Jackson MB. Adenosine-activated potassium conductance in cultured striatal neurons. Proc Natl Acad Sci U S A. 1985;82:4857–4861. doi: 10.1073/pnas.82.14.4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenzi F, Corciulo C, Targa M, Casetta I, Gentile M, Granieri E, Borea PA, Popoli P, Varani K. A2A adenosine receptors are up-regulated in lymphocytes from amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:406–413. doi: 10.3109/21678421.2013.793358. [DOI] [PubMed] [Google Scholar]
- Xu K, Bastia E, Schwarzschild M. Therapeutic potential of adenosine A(2A) receptor antagonists in Parkinson’s disease. Pharmacol Ther. 2005;105:267–310. doi: 10.1016/j.pharmthera.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, Rothstein JD. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia. 2011;59:200–207. doi: 10.1002/glia.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Une F, Utatsu Y, Nomoto M, Furukawa Y, Maruyama Y, Machigashira N, Matsuzaki T, Osame M. Adenosine and neopterin levels in cerebrospinal fluid of patients with neurological disorders. Intern Med. 1999;38:133–139. doi: 10.2169/internalmedicine.38.133. [DOI] [PubMed] [Google Scholar]
- Zhai J, Zhou W, Li J, Hayworth CR, Zhang L, Misawa H, Klein R, Scherer SS, Balice-Gordon RJ, Kalb RG. The in vivo contribution of motor neuron TrkB receptors to mutant SOD1 motor neuron disease. Hum Mol Genet. 2011;20:4116–4131. doi: 10.1093/hmg/ddr335. [DOI] [PMC free article] [PubMed] [Google Scholar]