

SUMMARY
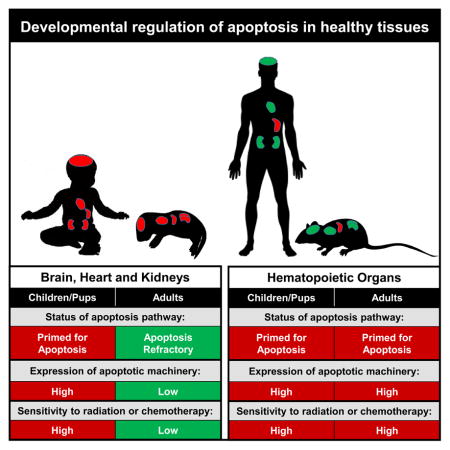
It is not understood why healthy tissues can exhibit varying levels of sensitivity to the same toxic stimuli. Using BH3 Profiling, we find that mitochondria of many adult somatic tissues, including brain, heart and kidneys, are profoundly refractory to pro-apoptotic signaling, leading to cellular resistance to cytotoxic chemotherapies and ionizing radiation. In contrast, mitochondria from these tissues in young mice and humans are primed for apoptosis, predisposing them to undergo cell death in response to genotoxic damage. While expression of the apoptotic protein machinery is nearly absent by adulthood, in young tissues its expression is driven by c-Myc, linking developmental growth to cell death. These differences may explain why pediatric cancer patients have a higher risk of developing treatment-associated toxicities.
Graphical Abstract
To explain how pediatric, compared to adult, cancer patients have a higher risk for treatment-associated toxicities, Sarosiek et al. find that many tissues in children and young mice are primed for apoptosis whereas adult tissues are not due to differences in the expression of apoptotic proteins.
INTRODUCTION
The intrinsic, or mitochondrial, pathway of apoptosis is an evolutionarily-conserved and highly regulated form of cell death that is critical for development and homeostasis of multicellular organisms. The deregulation of apoptosis is associated with many pathologies including cancer (Hanahan and Weinberg, 2000). Apoptosis is triggered when a pro-apoptotic effector protein (BAX or BAK) is activated by an activator BH3-only protein, of which BIM and BID are most potent (Tait and Green, 2013). This results in the oligomerization of BAX or BAK, causing mitochondrial outer membrane permeabilization (MOMP) and consequent release of cytochrome c into the cytosol, where it complexes with APAF-1 to form the apoptosome. This complex activates downstream cysteine proteases, including caspase 3, that dismantle the cell and promote phagocytosis (Galluzzi et al., 2009; Taylor et al., 2008). However, anti-apoptotic proteins in this family (BCL-2, BCL-XL, MCL-1, etc.) can block apoptosis by binding and sequestering monomeric BAX/BAK or BH3-only proteins (Czabotar et al., 2013). In order for apoptosis to occur, anti-apoptotic proteins within the cell must be overwhelmed and BAX and/or BAK activated.
The mitochondrial apoptosis pathway can be activated by a wide variety of cellular stressors including growth factor or nutrient deprivation as well as genotoxic damage from cytotoxic chemotherapies and radiation. In each of these cases, the basal state of the mitochondrial apoptotic pathway can alter the eventual fate of the cell (Sarosiek and Letai, 2016). To directly measure the functional state of the mitochondrial apoptotic pathway in cells, we developed the BH3 Profiling assay, which measures apoptotic priming (proximity of cellular mitochondria to the apoptotic threshold) by delivering titrated doses of distinct pro-apoptotic signals (BH3 peptides) to mitochondria while monitoring MOMP (Ryan and Letai, 2013). In this assay, mitochondria bearing only a small reserve of unbound anti-apoptotic proteins undergo MOMP in response to even relatively low doses of pro-apoptotic peptides and are thus classified as “primed” for apoptosis; primed cells readily die when pro-death signals are generated in response to cellular damage or stress (Ni Chonghaile et al., 2011). In contrast, cells that contain a large reserve of unbound anti-apoptotic proteins are less sensitive to BH3 peptides and are classified as “unprimed;” unprimed cells must experience higher levels of damage or stress to trigger MOMP. Finally, cells that block apoptosis by not expressing sufficient levels of critical components of the cell death machinery (such as BAX and BAK) are classified as “apoptosis refractory.” We have previously shown that patients with primed cancers respond more favorably to chemotherapy than patients with unprimed cancers (Davids et al., 2012; Ni Chonghaile et al., 2011; Vo et al., 2012).
Chemotherapy and radiation treatments have cured cancer in millions of patients (American Cancer Society, 2014), yet the apoptotic cell death that these agents can induce in healthy tissues limits their use. This is especially true in pediatric patients who experience considerably higher levels of treatment-associated toxicity and morbidity from genotoxic agents. For example, brain irradiation is a critical component of the potentially curative treatment for brain tumors. However, radiation can also trigger cell death in healthy neurons in very young patients, resulting in permanent and devastating cognitive deficits with severity being inversely correlated with age (Merchant et al., 2010; Silber et al., 1992). Similarly, children with many types of cancers are commonly treated with anthracyclines including doxorubicin. However, doxorubicin treatment in children can cause thinning of the cardiac ventricular walls, a reduction in ventricular mass and consequent heart failure, with the youngest children again being most at risk (Lipshultz et al., 1995; Trachtenberg et al., 2011). Currently, parents and clinicians must balance the curative potential of these treatments with their potential for causing devastating toxicities. It is unclear why young children are more at risk of developing these toxicities than adults.
Much of the study of apoptosis has been devoted to cancer and hematopoietic tissues, with relatively little study of other healthy somatic tissues. It is currently unknown whether cells that make up distinct tissues have varying levels of apoptotic priming, which could potentially contribute to their different sensitivities to classical apoptosis-inducing agents.
RESULTS
Many adult tissues are apoptosis refractory
We first performed BH3 Profiling on a comprehensive set of adult mouse tissues to detect any potential differences in their apoptotic priming. Cells of the hematopoietic lineage from the periphery (peripheral blood mononuclear cells [PBMCs]), thymus, spleen and bone marrow are the most primed cells in the body among those we studied, as indicated by high mitochondrial depolarization in response to BIM or BID BH3 peptides or full length proteins (Figures 1A–B and S1A). Cells (excluding blood) constituting the large intestine, small intestine, lungs and liver were relatively unprimed, as they required higher doses of BIM or BID BH3 peptides and a longer time period to trigger depolarization. Strikingly, we found adult brain, heart and kidney tissues are far less primed, and nearly completely insensitive to concentrations of BIM and BID BH3 (100 μM) that are sufficient to induce MOMP in nearly every cancer cell line or primary cancer cell we have tested.
Figure 1. Adult brain, heart, and kidney tissues are apoptosis refractory, protected from genotoxic damage.

(A) Tissues were isolated from adult mice (>P60) and BH3 Profiled. Mitochondrial potential under each treatment condition was measured every 5 min during the experimental time course and plotted relative to maximum value of negative control (DMSO). Percent depolarization (indicative of MOMP) is calculated for each BH3 peptide treatment relative to DMSO (0%) and FCCP (100%). Representative traces are shown (mean ± SD, ≥ 3 independent experiments [IEs]). (B) Summary BH3 Profiling data across healthy adult mouse tissues. ≥3 IEs. (C) After whole body gamma irradiation, apoptosis was measured via a caspase 3 activity assay. Data were compiled from 5 animals for each treatment across 3 IEs with bars representing means. (D) After whole body irradiation, apoptosis was detected via immunohistochemistry for cleaved caspase 3. Data were compiled from 2 animals for each treatment across 2 IEs with bars representing means. (E) Tissues were collected from adult mice and immunoblotting was performed. Densitometry was performed across immunoblots from 3 IEs and averaged (blue heatmap on right). GAPDH is loading control. Relative molecular weight markers are shown on left. (F) Expression of indicated proteins in adult human tissues was assessed via the Kim et al. (2014) mass spectrometry dataset. (G) Tissues were isolated from adult mice and BH3 Profiled in the presence of recombinant BAX. Representative traces are shown (mean ± SD, ≥3 IEs). See Figure S1 and Tables S1–S2
We previously found that apoptotic priming is a strong determinant of cancer cell fate in response to cytotoxic chemotherapies. We hypothesized that healthy tissue sensitivity to genotoxic agents would be dependent on their degree of priming. To test this hypothesis, we exposed live adult mice to 8 Gray of gamma radiation and found that the degree of apoptosis induced in the tissues corresponded to their level of apoptotic priming (Figure 1C–D, S1B–C).
We next asked whether the profound differences in mitochondrial priming in adult tissues could be due to differences in expression of BCL-2 family proteins. While we might have expected to find reduced expression of pro-apoptotic proteins or increased expression of their anti-apoptotic counterparts, we instead found that both pro- and anti-apoptotic proteins were lacking in apoptosis refractory tissues (Figure 1E). BAX and BAK, two proteins that are required for mitochondrial apoptosis, were nearly undetectable in refractory tissues. These tissues also expressed lower levels of Caspases 3 and 8 to potentially further suppress cell death. We did not observe major differences in the expression of XIAP, a potent inhibitor of caspase activity, across adult tissues.
We next examined the levels of these proteins in human tissues by mining mass-spectrometry-based proteome data (Kim et al., 2014) and found that adult human tissues exhibit a similar pattern of protein expression to that of mice (Figure 1F, Tables S1 and S2), suggesting that mouse and human tissues regulate apoptosis similarly, which is consistent with previous work (Reed et al., 2003).
To test whether the lack of BAX and BAK in apoptosis refractory tissues was preventing activator-induced MOMP, we tested whether addition of recombinant BAX to mitochondria would restore sensitivity to BH3 peptides. While recombinant BAX alone did not induce MOMP, we detected efficient and complete MOMP in adult brain, heart and kidney tissues when administering BAX concurrently with even minute amounts of BIM (Figure 1G). These data indicate that neither BAX nor BAK are expressed at levels sufficient for MOMP in adult brain, heart and kidney tissues. Moreover, in these tissues there is lower expression of nearly all proteins comprising the mitochondrial apoptotic machinery, both pro- and anti-apoptotic, and pre- and post-mitochondrial. This apoptotic resistance presumably protects vital, long-lived cells constituting these largely post-mitotic tissues from aberrant cell death. Operationally, we defined cells as apoptosis refractory when MOMP is not induced by BIM or BID BH3 at even 100 μM, yet efficiently induced by low doses (1 μM) of BIM BH3 in the presence of exogenous BAX. It is important to note that although cells may be designated as apoptosis refractory based on the BH3 Profiling assay, this designation is made at a single time point and in the absence of stress. There may exist conditions under which apoptosis refractory cells upregulate critical pro-apoptotic proteins to acquire apoptotic sensitivity.
Early in life, tissues are primed for apoptosis
Young children frequently experience neuro- and cardiotoxicity when treated with radiation or genotoxic chemotherapies. We therefore hypothesized that the apoptotic pathway may be more active in these tissues in young mammals as compared to adults. To test this, we measured levels of apoptotic priming in newborn mice and found that brain, heart and kidney mitochondria in embryonic and very young mice are extremely primed for apoptosis (Figure 2A). In fact, mitochondria in neonatal brain cells were almost as primed for apoptosis as adult splenocytes. We next sought to delineate the transition between being highly primed and apoptosis refractory in relevant tissues by making serial measurements over time. Each tissue exhibited a distinct temporal program to transition from being primed to being apoptosis refractory with the sharpest decreases occurring shortly after birth (P0–P5) (Figure 2B). In contrast, the spleen maintained a high level of priming throughout development and into adulthood (Figure 2B).
Figure 2. Embryonic and early postnatal brain, heart, kidney and liver tissues are primed for apoptosis and sensitive to genotoxic damage.

(A) Tissues were isolated from postnatal day 0 mice (P0) and BH3 Profiled. Representative traces are shown (mean ± SD, ≥3 IEs). (B) Summary BH3 Profiling data across tissues during embryonic and postnatal development. Each point represents an average of 3 measurements in each mouse tissue across at least 5 IEs. (C) After whole body irradiation or doxorubicin injection, tissues were collected and caspase activity was measured via an enzymatic assay. Each point represents an average of 3 measurements in each mouse tissue across at least 5 IEs. (D) After whole body irradiation, apoptosis was detected via IHC for cleaved caspase 3. Representative images, 2 IEs. Olfactory bulb (OB) and cerebral cortex (CC) are indicated. Scale bars are 200 μm. (E–F) Cleaved caspase 3-positive (E) or TUNEL-positive (F) cells were counted per 40X field in sham-treated or irradiated tissues from young (P0–P2) or adult (P60+) mice processed via IHC. Data are compiled from 2 IEs with bars representing means. See Figure S2.
We next sought to test the functional consequences of these developmental changes in apoptotic priming. We chose to again stress cells with gamma radiation, which applies an equal and reproducible level of genotoxic damage across tissues and models radiation therapy in pediatric cancer patients. Mice at various stages of postnatal development were treated with whole-body gamma irradiation and apoptosis was quantified. We detected extensive caspase 3/7 activation post radiation, at doses of 0.50 – 8 Gy, in brain, heart and kidney tissues in P0–P2 mice, which was measurably reduced each day of postnatal development and finally silenced (insensitivity to 8 Gy) by P13, P15 and P12 in brain, heart and kidney, respectively (Figure 2C and S2A). Splenocytes, which are primed throughout life, consistently activated caspases in response to damage. We utilized immunohistochemical (IHC) staining for cleaved caspase 3 and TUNEL to confirm that the cells undergoing apoptosis in each tissue in response to radiation are not blood cells (Figure 2D–F and S2B–F).
Very young patients suffer increased iatrogenic cardiotoxicity from anthracyclines compared with non-elderly adults. To determine whether the dynamic levels of apoptotic priming during postnatal development may contribute to this difference, we injected mice at various postnatal stages with the anthracycline doxorubicin and measured caspase activation in the heart, again modeling pediatric cancer treatment in humans. In neonatal heart tissue, we detected caspase activation, which decreases with age in a manner similar to that following radiation damage (Figure 2C).
Finally, we sought to determine whether the changes in apoptotic priming were dependent on an in vivo milieu or whether they could be driven by cell-autonomous mechanisms. To test this, we isolated hippocampal neurons from embryonic day 19 (E19) rat brains and measured apoptotic priming and chemosensitivity of these cells at regular intervals as they matured in vitro (Figure 3A–C). These non-proliferating (Figure 3D) neurons underwent the same developmentally-defined transition from high apoptotic priming to apoptotic resistance that was evident in vivo. Their sensitivity to the classical apoptosis inducers staurosporine and doxorubicin followed a similar pattern (Figure 3E).
Figure 3. Apoptotic priming and chemosensitivity decrease as primary rat hippocampal neurons mature in vitro.

(A) Primary neurons were isolated from E19 rat embryos and allowed to mature in vitro, with representative images shown at indicated time points (days in vitro). Note that the same cells (those inside box on low magnification) are shown across the 4 time points in the higher magnification window. Scale bars are 200 μm. (B) Primary neurons were BH3 Profiled at indicated days in vitro. Representative traces are shown (mean ± SD, 2 IEs). (C) Summary of BH3 Profiling data of primary neurons at indicated time points. (D) Neuron nuclei were counted at indicated time points. (E) Chemosensitivity of rat hippocampal neurons in vitro. Each point represents an average of 3 measurements at each time point across 2 IEs.
Age-related apoptotic priming is regulated via BAX and BAK
The contrast in apoptotic priming in vital tissues between young and adult mice prompted us to explore the age-related expression of BCL-2 family proteins. Because we had previously shown that exogenous BAX supplementation is sufficient to reverse mitochondrial resistance to pro-apoptotic signals, we focused on the developmental regulation of BAX and BAK. In the spleen, we found the critical effectors BAX and BAK and potent activator BH3-only proteins BIM and BID to be consistently expressed during the entire lifespan of the animals (Figure 4A). Major anti-apoptotic proteins were also expressed in the spleen consistently throughout life, although a downregulation of BCL-XL and MCL-1 and upregulation of BCL-2 with age was noted. Expression of caspase 3 and APAF-1 was unchanged over time. These results were consistent with BH3 profiling data showing high levels of priming throughout life.
Figure 4. Key components of the apoptotic machinery are lost during postnatal development.

(A) Healthy tissues were collected from mice at indicated ages and immunoblotted. (B) Expression of indicated proteins in fetal and adult human tissues assessed via proteome data (Kim et al., 2014). See Figure S3.
In contrast to the spleen, we found the expression of BCL-2 family members in the mouse brain during postnatal development to be dynamic. BAX was highly expressed in the mouse brain at P0–P5, but continually reduced into adulthood (Figure 4A). BAK was reduced in a similar manner yet expression levels were lower overall (relative to adult spleen). BCL-XL, as well as caspases 8 and 9 were consistently expressed while BIM, BID, MCL-1 and BCL-2 were also reduced during postnatal brain development. Finally, both caspase 3 and APAF-1 were strongly downregulated during postnatal development, further contributing to the suppression of apoptosis in adult brain tissue.
Similar results to the brain were also observed in heart, kidney and liver with BAX and BAK being rapidly downregulated following birth (Figure 4A). We confirmed this downregulation occurs in the non-blood cells that comprise these tissues via IHC (Figure S3). We also found that adult liver tissue expresses low, yet detectable, levels of BAK (Figure S3B), which is consistent with the higher sensitivity of adult hepatocytes to BID over BIM (Figure 1B) due to their activation preferences (Sarosiek et al., 2013). It is notable that anti-apoptotic proteins tended to be downregulated in these tissues as well, further demonstrating that adult tissues are not protected from apoptosis by high expression of anti-apoptotic proteins. Instead, there appears to be a wholesale dismantling of the apoptotic machinery to render these cells apoptosis refractory, preserving their survival. The expression levels of VDAC, which can facilitate cytochrome c release from mitochondria during MOMP, and IAP proteins (XIAP, CIAP1) also remained largely unchanged across all tissues.
We also found BAX and BAK, along with other key components of the apoptotic machinery, to be strongly downregulated in human adult brain, heart and liver tissues relative to fetal tissues (Figure 4B and Tables S1–S2).
Healthy tissues differ in utilizing BAX versus BAK to undergo apoptosis
BAX and BAK have non-overlapping roles in regulating apoptosis and their activity can be modulated selectively (Sarosiek et al., 2013; Shamas-Din et al., 2014). Our results showed that BAX and BAK are downregulated in tissue-specific manners as they transition from being apoptotically primed to refractory. We therefore utilized mouse models to determine the distinct contributions of BAX and BAK to the activation of apoptosis in healthy tissues. Neonatal brain tissue in WT mice was efficiently depolarized by the BIM (~80%) and, to a lesser extent, BID (40%) peptides (Figures 5A–B and S4). In Bax−/− neonates, however, we found the responses to BIM and BID significantly reduced, which was not evident in Bak1−/− mice. We therefore hypothesized that loss of Bax would protect brain tissue from radiation-induced apoptosis while loss of Bak1 would not. In agreement with the BH3 Profiling data, we found that loss of Bax prevented nearly all caspase 3 activation post radiation while loss of Bak1 had no effect (Figure 5C). Thus, in the early postnatal brain, BAX and not BAK is the dominant effector that is engaged to trigger apoptosis.
Figure 5. BAX and BAK dependence in early postnatal mouse tissues.

(A) Tissues were collected from P0 mice of indicated genotypes and BH3 Profiled. Representative traces are shown (mean ± SD, ≥ 3 IEs). (B) Summary BH3 Profiling data from P0–P2 mouse tissues. Each point represents an average of 3 measurements in each tissue across 4 IEs. Bars represent means (C) Summary caspase 3 activity data from P0–P2 mouse tissues after whole body irradiation. Each point represents an average of 3 measurements in each tissue across 11 IEs. Bars represent means. See Figure S4.
We then found that each tissue exhibits its own pattern of effector dependence. Splenocytes can execute apoptosis via BAX or BAK and although loss of BAX is somewhat protective, both must be lost in order to prevent all radiation-induced apoptosis (Figure 5C). In the neonatal heart, loss of either BAX or BAK reduced responses to peptides and radiation, indicating that both effectors are present, yet at limited levels, consistent with our immunoblotting results (Figure 4A). Cells within the neonatal kidney also contain both BAX and BAK, and loss of either reduced peptide responses. However, loss of BAX meaningfully reduced caspase activation after irradiation while loss of BAK did not. Finally, we found that neonatal hepatocytes contained both BAX and BAK and required the loss of both in order to significantly reduce apoptosis post radiation. In all cases, the knockout of Bax preferentially dulled responses to the BIM BH3 peptide while loss of Bak1 preferentially dulled responses to the BID BH3 peptide, which is in agreement with reported specificity of activator/effector interactions (Sarosiek et al., 2013). These results may enable the prevention of damage-induced apoptosis in neonatal tissues by inhibiting BAX alone (brain) or both BAX and BAK (heart, kidney, liver, spleen).
Higher apoptotic priming in young mice contributes to doxorubicin-induced cardiotoxicity
Young hearts are considerably more sensitive to doxorubicin than non-elderly adult hearts. Clinically, the degree of cardiotoxicity observed correlates with age, with the youngest children being most at risk for developing symptoms including decreased ejection fraction (EF), thinning of ventricular walls, and an overall reduction in heart size (ventricular mass), sometimes referred to as “Grinch syndrome” (Lipshultz et al., 2014). We hypothesized that the differences in apoptotic priming in cardiomyocytes from young versus adult hearts may contribute to the heightened risk of developing cardiotoxicity in young patients.
We developed a mouse model of doxorubicin-induced cardiotoxicity by injecting mice with 3 doses of doxorubicin (5 mg/kg) over the course of one week, starting at days P5/6 (primed for apoptosis), P11/12 (unprimed), or P60–80 (apoptosis refractory) (Figure 6A). Our dosing schedule is reduced in intensity (3 doses instead of 5) from one previously used to model chronic doxorubicin cardiotoxicity in adult mice (Zhang et al., 2012), which, when tested in young mice induced prohibitively high levels of cardiotoxicity as evidenced by arrhythmias and death (data not shown). Using echocardiograms at day 14, we found significant thinning of the interventricular septal (IVS;d) and posterior ventricular walls (LVPW;d) in M-mode echocardiogram tracings at diastole in animals that began receiving injections at P5/6, but not those that began as adults (Figure 6B–D, Movies S1–S2). Moreover, animals treated at a young age exhibited a profound decrease in EF and left ventricular mass while adults did not. Masson’s Trichrome staining (MTS) of hearts after treatment showed focal areas of altered myocyte architecture with signs of early injury (loss of cross striations, disorganization of myofilaments resulting in abnormal staining, and hypochromatic nuclei) in mice treated starting at P5/6 but not adults (Figure S5A).
Figure 6. Mouse model of pediatric doxorubicin cardiotoxicity.

(A) Schematic representation of the experiment. WT mice of different ages were injected intraperitoneally with 3 doses of doxorubicin at 5 mg/kg on days 0, 4 and 7. Echocardiograms to assess heart function were performed on day 14. (B) Representative M-mode echocardiogram tracings are shown from WT mice treated at P5 or P60+. (C) Representative parasternal long-axis views at the level of the papillary muscle are shown from WT mice treated at P5. (D) Summary echocardiogram data. Each point represents an average of 2 measurements in each animal across ≥ 5 IEs. Bars represent means. (E) P5–P6 mice of indicated genotypes were treated as in (A). Bars represent means. See Figure S5 and Movies S1–S2.
We next asked if suppression of the intrinsic apoptotic pathway could reverse these effects of doxorubicin. Using knockout mice to model pharmacologic inhibition, we found that loss of both Bax and Bak1 was required to consistently reverse doxorubicin-induced damage in neonatal mouse hearts (Figure 6E). In addition, knockout of Bax and Bak1 in P5/6 mice treated with doxorubicin showed dramatically reduced, although not completely lost, evidence of early injury in MTS histology (Figure S5B). Therefore, young heart tissue expresses both BAX and BAK at sufficient levels to activate apoptosis and thus inhibition of both of these effectors is needed to mitigate doxorubicin-induced thinning of ventricular walls and reduction in ventricular mass. In addition, there may also be a non-apoptotic component of doxorubicin-induced cardiotoxicity since the BAX/BAK double knockout mice still exhibited some signs of early injury based on MTS histology, although symptoms were greatly reduced. Taken together, our results show that hearts in young mice exhibit more severe clinically-relevant symptoms of doxorubicin cardiotoxicity than adults, potentially due to the increased BAX and BAK expression in apoptotically primed young hearts (Figure 2B) that renders them hypersensitive to doxorubicin (Figure 2C).
Apoptotic priming is modulated by c-Myc
The concerted loss of several genes responsible for regulating apoptosis suggested that a master developmental program or transcription factor may be regulating multiple members of this pathway. One such potential modulator is c-Myc (hereafter Myc), a transcription factor that drives cellular growth and proliferation in normal as well as cancerous cells (Dang et al., 2006). Several links between Myc and apoptosis have been reported, including the sensitization of Myc over-expressing cells to a variety of pro-apoptotic stimuli (Bissonnette et al., 1992; Egle et al., 2004; Evan et al., 1992; Murphy et al., 2008) and its direct regulation of BAX expression (Mitchell et al., 2000). We thus hypothesized that tissues early in development express Myc at levels that are sufficient to drive the expression of BAX and potentially other apoptosis-related genes, making them primed for apoptosis, and that loss of BAX expression in adulthood is related to loss of Myc.
Consistent with our hypothesis, we found that Myc is expressed at higher levels in young brain, heart and kidney tissues as compared to adult (Figure 4A). The human proteome database did not contain sufficient data for Myc expression. If Myc was driving expression of pro-apoptotic genes, we would expect Myc-expressing cells to be more primed. To test, we utilized flow cytometry-based BH3 Profiling, which allows for single-cell measurements of priming concurrently with evaluation of extracellular or intracellular factors. We measured priming and nuclear Myc expression (indicative of activation) in cells from P0 tissues and found that Myc-positive brain, kidney and liver cells were, as expected, more primed than Myc negative cells (Figure 7A). We did not detect any Myc-positive cells within the respective adult tissues, which we again found to be dramatically less primed than neonatal tissues. In heart tissues, the extended processing necessary for this analysis resulted in prohibitively high levels of cytochrome c loss in even untreated cells, thus preventing analysis in that tissue.
Figure 7. Apoptotic priming is modulated by Myc and priming is dynamically regulated in human brain tissue.

(A) Flow cytometry-based BH3 Profiling was performed on indicated tissues collected from neonatal or adult mice and loss of cytochrome c (indicative of apoptosis) was measured after treatment with negative control (DMSO), positive control alamethicin (AlaM), or BH3 peptides. For neonatal tissues, each point represents a flow cytometry-based measurement of either Myc negative or positive cells (as determined by co-staining for nuclear Myc) from a single tissue sample. For adult tissues, each point represents a flow cytometry-based measurement of Myc negative cells. All cells analyzed by flow cytometry were CD45 negative to exclude blood cells from analysis. Data shown are for either 3 (neonatal) or 2 (adult) IEs. (B) Fluorescence-based BH3 Profiling was performed on brain tissue (neocortex) of litter-matched, neonatal Myc WT or heterozygous mice. Each point represents analysis of a single animal across 2 IEs. (C) MycER mice were treated with vehicle (oil) or tamoxifen (tam) to activate Myc for 3 days and then irradiated (8 Gy). TUNEL+ cells were counted per 20X field. (D) Flow cytometry-based BH3 Profiling was performed on tissues from (C) and each point represents the loss of cytochrome c in cells that are nuclear Myc-positive vs -negative from each tamoxifen-treated animal across 2 IEs. (E) After injection with vehicle or concanavalin A, liver tissue was collected from mice at indicated time points and Myc positivity was measured in hepatocytes via flow cytometry. Each point represents one animal across 2 IEs. (F) 96 hours after injection with concanavalin A, liver tissue was collected and flow cytometry-based BH3 Profiling was performed on hepatocytes while monitoring nuclear Myc expression. Each point represents one animal across 3 IEs. (G) ChIP-qPCR was performed to measure Myc occupancy on promoters for positive control (Mybbp1a) and negative control (Untr6) genes, along with genes of interest. Values shown are binding events detected per μg chromatin. In (A–G), bars represent means. (H) Representative BH3 Profile traces (mean ± SD) from healthy human brain tissue obtained during resection of seizure foci. (I) Summary of fluorescence-based BH3 Profiling data from human brain tissues. Each point represents an average of 3 measurements in each human brain specimen across at least 5 IEs. BAX mRNA expression across human brain regions were assessed at indicated ages. (J) Immunoblotting for BAX across healthy human brain specimens tested in (H–I). Numbers indicate months post conception. Heatmap represents expression of BAX normalized to GAPDH levels. See Table S3.
Expression of Myc was associated with higher apoptotic priming, but this relationship may not be causal, prompting us to test whether Myc expression was necessary to maintain high priming in neonatal tissues. We therefore measured priming in the cerebral cortex of either WT or Myc+/− mice (Hofmann et al., 2015). As expected, the loss of one allele of Myc resulted in reduced sensitivity to BIM as compared to WT littermates (Figure 7B).
Next, we sought to determine whether activating Myc in adult tissues would be sufficient to reactivate apoptosis in refractory cells. We first utilized the well-characterized Gt(ROSA)26Sortm1.1(MYC/ERT2)GEV (hereafter MycER) mouse model that was developed to study the physiological effects of acute Myc activation in adult tissues (Murphy et al., 2008). These mice express a cDNA encoding human c-Myc fused at its C terminus to the hormone-binding domain of 4-hydroxytamoxifen (4-OHT)-responsive mutant murine estrogen receptor, enabling us to activate Myc systemically with injections of tamoxifen. Activation of Myc in normally apoptosis refractory brain and kidney tissue, or unprimed liver tissue increased sensitivity to genotoxic damage (Figure 7C). Notably, we found that Myc-positive cells within these tissues were more primed for apoptosis than their Myc-negative counterparts as measured via flow cytometry-based BH3 Profiling (Figure 7D). Our data suggests that Myc activation may reenable the apoptotic pathway in apoptosis refractory tissues.
After acute liver damage, hepatocytes are able to re-enter the cell cycle and proliferate in order to replace dead cells (Tzung et al., 1997). Based on our previous data, we hypothesized that hepatocytes from adult liver tissue that are actively expressing Myc and proliferating would become more primed for apoptosis. We tested this by inducing immune-mediated liver damage via injection of Concanavalin A (ConA), which triggers active liver regeneration and hepatocyte proliferation (Trautwein et al., 1998). After injection with ConA, we observed a profound increase in Myc-positive cells within the liver (~0% Myc-positive in untreated animals) (Figure 7E), as hepatocytes proliferated to replace dead cells. We directly compared the level of priming in Myc-positive versus Myc-negative hepatocytes and found the former to be significantly more sensitive to BIM and BID BH3 peptides (Figure 7F). Thus, using both genetic and physiologic models, we found that activation of Myc increases mitochondrial priming.
Because modulation of Myc was sufficient to alter priming in the expected manner, we next sought to test whether Myc directly drives the expression of apoptosis-regulating genes in neonatal tissues. Myc activates the transcription of its target genes by binding specific E-box (Enhancer box) elements in gene promoters (Mitchell et al., 2000). We therefore used chromatin immunoprecipitation followed by quantitative polymerase chain reaction (ChIP-qPCR) to test for Myc occupancy in the E-boxes present in the promoters of various BCL-2 family member genes. In agreement with our hypothesis that Myc may be modulating BAX expression in neonatal tissues, we detected the Bax promoter being bound by Myc in neonatal brain, kidney, liver and spleen tissues (Figure 7G). Furthermore, Myc binding to the Bax promoter was lost in the adult brain and kidney but not the spleen, consistent with the spleen maintaining a high level of priming throughout life (note that adult heart and liver tissues were not tested). We also detected E-boxes in the promoters of pro-apoptotic Bcl2l11 (Bim), which is consistent with previous reports (Campone et al., 2011; Muthalagu et al., 2014), and Bid, and confirmed that Myc bound to these promoters. Taken together, these data indicate that Myc drives an apoptotically primed state by directly activating transcription of pro-apoptotic genes Bax, Bim, and Bid.
Human brain regulates apoptosis during development similarly to mouse
The dynamic regulation of apoptosis in murine tissues dramatically affected responses to genotoxic damage and prompted us to ask whether the same temporal regulation could be directly observed in human tissues. One source of non-malignant, viable human brain tissue is from pediatric and adult patients that undergo removal of seizure foci to control otherwise intractable seizures caused by epilepsy, trauma or other pathology. Seizure foci are mapped in the human brain and resected, inevitably along with some ostensibly healthy brain tissue (Bittigau et al., 2003). We obtained fresh, healthy brain tissue from over 20 such patients from 4 months to 21 years of age for BH3 Profiling analysis (Table S3). As with mice, we found human brain tissue from young patients to be significantly more sensitive to BIM and BID BH3 peptides than those from adults (Figure 7H–I). There existed a consistent downregulation of apoptotic priming during postnatal human brain development, predisposing the youngest children to radiation- and chemotherapy-induced neurotoxicity. In our study, there was a period of higher heterogeneity in apoptotic priming among patients between 2 and 6 years old, after which, the brain transitions to full apoptotic resistance. Finally, we mined mRNA expression data in the human brain during prenatal and postnatal development (Miller et al., 2014) and found that BAX expression is dramatically reduced during development, starting even prenatally (Figure 7I), which we confirmed at the protein level in our samples (Figure 7J). The highest mRNA expression of BAX was found in fetal brain, suggesting that the highest levels of apoptotic priming, perhaps similar to those observed in the youngest (P0–P2) mice, would be found in fetuses and pre-term infants.
DISCUSSION
The study of apoptosis has been traditionally dominated by the study of cancer cells and lymphocytes, the ubiquitous presence of functioning machinery of the mitochondrial apoptotic pathway in which has led to a general acceptance that this pathway is present in all cells. We were surprised to find that heart, kidney, and brain in adult mice lacked the proteins that regulate the mitochondrial apoptotic pathway. It appears that the mitochondrial apoptotic machinery is largely absent in these adult tissues, which we designate as being “apoptosis refractory.” This apoptosis refractory state is achieved as part of a regulated postnatal developmental program in both mice and humans and is apparently reversible, as stress can restore apoptotic sensitivity to cells previously apoptosis refractory. A common feature of embryonic, immediately postnatal, and regenerating tissues is a greater degree of proliferation. We believe that in all of these tissues, enhanced priming is the price that cells pay for the capacity to proliferate. Cancer cells often share many features of embryonal cells, including lack of differentiation, enhanced proliferation, and stem-like function (Daley, 2009). The relatively primed nature of cancer cells may be thought of as re-establishment of yet another embryonic program in cancer cells.
Previous studies have shown that ectopically expressed Myc can foster pro-apoptotic signaling (Bissonnette et al., 1992; Evan et al., 1992; Murphy et al., 2008). We demonstrate here that endogenous, physiological Myc regulates apoptosis sensitivity across a range of tissues during mammalian development. Myc therefore provides one important mechanistic link between developmental proliferation, expression of pro-apoptotic genes, and apoptotic priming. However, we suspect that there are additional modulators of properties of this significance. In addition, our findings raise the question of whether other programmed cell death pathways including extrinsic (cell death receptor mediated) apoptosis, necroptosis (Zhou and Yuan, 2014), and ferroptosis (Dixon et al., 2012) are also dynamically regulated during postnatal development.
Very young pediatric cancer patients are subject to severe side effects of radiation and chemotherapy from which older children and adults are relatively spared. We show that the apoptotic responses of heart and brain tissues depend on the developmentally regulated priming of the mitochondria. It is possible that this regulation is a major contributor to the hypersensitivity of very young tissues to genotoxic agents. We find this correlation holds in the brains of very young humans. Moreover, we find we can reverse this phenotype by un-priming mitochondria of young tissues by genetic means, potentially modeling pharmacologic inhibition.
Earlier work in this field reported the seemingly contrasting findings that individual BCL-2 family members were modulated with time in select tissues(Kole et al., 2013; Polster et al., 2003a) (Kole et al., 2013; Polster et al., 2003; Shi et al., 2012; Soane et al., 2008) and that BAX and BAK were highly and broadly expressed in somatic tissues including the brain (Brustovetsky et al., 2003; Krajewski et al., 1994). Our work aims to clarify how apoptosis is regulated in healthy tissues and also highlight the challenges in inferring phenotypic changes due to changes in levels of individual BCL-2 family proteins. The large number of proteins within this family and their nuanced pro- and anti-apoptotic effects, which can be further modulated by post-translational modifications, requires the use of a functional test of apoptotic priming to measure the net integration of these signals.
It is important to note that BH3 Profiling does not assess the state of post-MOMP regulators of apoptosis such as IAP proteins or caspases, which are also known to affect cell fate (Holly et al., 1999; Martin, 2002). In addition, the abilities of cells to die via the extrinsic apoptotic pathway (Fulda and Debatin, 2006) or inflammation-associated pathways (Martin et al., 2012) in response to genotoxic damage are not directly probed by BH3 Profiling. Finally, we acknowledge that even in a primed cell, whether apoptotic machinery is actually engaged following genotoxic damage depends on factors such as DNA damage responses and p53 competence (Kandioler-Eckersberger et al., 2000; Loewer et al., 2010; Rich et al., 2000). Combining BH3 Profiling with tests of the expression or adequacy/activity of these other factors/pathways, as we have attempted to do to some extent here, would provide the most complete understanding of the pretreatment state of the cell or tissue and how that impacts cell fate in response to damage or stress.
While our results show that many healthy tissues are apoptosis refractory in the adult, we do not mean to rule out the possibility that various chronic injuries or disease states might restore apoptotic sensitivity in these tissues. In fact, increased levels of neuronal BAX and/or BAK have been reported in patients with Alzheimer’s disease (MacGibbon et al., 1997; Shimohama, 2000), which would be expected to contribute to the neuronal death that characterizes this pathology. BAX expression has also been observed to increase in different liver pathologies, including hepatitis and cirrhosis (Jr et al., 2000; Liang et al., 2007) and is increased following Mcl1 deletion in mouse cardiomyocytes, potentially contributing to the apoptosis reported in that model (Wang et al., 2013). Indeed, we found that chemical liver injury and the resulting regenerative proliferation increased apoptotic priming in adult mice. Such findings suggest that the program we observe is reversible, as we observed with the increased priming post activation of Myc in the regenerating liver or MycER mouse model. More study is needed to understand how disease states may impact apoptotic priming.
Our analysis shows that virtually all cells within neonatal brain, heart and kidney are primed for apoptosis while in adults virtually all cells within these tissues are apoptosis refractory. We did not measure priming levels or changes in priming among different types of cells within each tissue. It remains possible, however, that apoptotic priming of cell subsets are differentially regulated in development. Future work will focus on mapping how individual cell types within organs regulate apoptosis.
Our findings suggest that there may potentially be ways to improve the therapeutic index of cancer treatments through the use of agents that selectively modulate BCL-2 family member function. For example, because of the developing brain’s dependence on BAX and not BAK for apoptosis, clinicians could utilize a BAX inhibitor or RNAi-based knockdown of BAX while administering brain irradiation for the treatment or prophylaxis of acute lymphocytic leukemia (ALL) with CNS involvement. This strategy would potentially prevent apoptosis in neurons, which express only BAX, while still allow ALL cells, which express both BAX and BAK (Haferlach et al., 2010), to undergo apoptosis via BAK. It may also be possible to modulate developmental programs or Myc levels via recently developed strategies (Delmore et al., 2011) to induce transient apoptotic resistance in healthy tissues prior to administering radiation or chemotherapy. Beyond cancer, our data have implications for medical conditions such as head trauma in pediatric patients, which induces neuronal death via apoptotic and excitotoxic pathways, while in adults only the excitotoxic pathway is engaged (Pohl et al., 1999). A BAX (or BAX/BAK) inhibitor could prevent the apoptotic component of this neuronal death, potentially rescuing some of the ill effects of these injuries in young children. Although no inhibitors of BAX and/or BAK have reached the clinic, efforts are currently underway to develop such agents (Hetz et al., 2005; Lessene, 2015).
Our findings may also impact the development of anti-cancer agents targeting cell death. Efforts to develop small molecule direct activators of BAX for cancer therapy have begun (Gavathiotis et al., 2012). The BIM BH3 peptide is itself a BAX activator. Our finding that the mitochondria of most adult somatic tissues are relatively insensitive to BIM BH3 makes the direct activation of BAX for cancer therapy more attractive. However, based on our findings these same tissues in pediatric patients could prove to be exquisitely sensitive, and thus efforts to utilize such agents in very young children should be accompanied by consideration of the apoptotic sensitivity of their normal tissues.
EXPERIMENTAL PROCEDURES
Animals: Mice were housed and bred in accordance with the policies and regulations set forth by the Dana-Farber Cancer Institute’s Institutional Animal Care and Use Committee (IACUC). All animal experiments were approved by IACUC under DFCI protocols 11-008, 12-049 and UK Home Office licenses 70-7950 and 70-8645.
Human brain specimens: All specimens were collected under IRB-approved tissue collection protocol #09-02-0043 at Boston Children’s Hospital and transferred to DFCI via Office for Human Research Studies exemption 13–545. Patients underwent surgery for removal of seizure foci at Boston Children’s Hospital (Table S3) and resected tissues were first delivered to a neuropathologist for evaluation. If available, a de-identified sample (0.2–0.5g) of brain tissue was provided to study investigators in PBS on ice and was immediately processed for BH3 Profiling. Part of the sample (0.1g) was excluded for cryopreservation and was subsequently prepared for immunoblotting as outlined above. The remaining brain tissue was dissociated by repeated pipetting until a single cell suspension was achieved and BH3 Profiled with JC1.
Fluorescence-based BH3 Profiling: Briefly, BH3 peptides or recombinant proteins in T-EB buffer were deposited into each well in a black 384-well plate, 1 treatment per well, in triplicate for each independent experiment. Single cells isolated from indicated tissues were resuspended in T-EB buffer and mixed 1:1 with a dye solution containing digitonin and JC-1 in T-EB. Cells were kept at room temperature for 5 min to allow for cell permeabilization and dye equilibration. Cells were then added to each treatment well in the 384-well plate and fluorescence at 590 nm was measured every 5 min at 32°C for a total of 120 min. Relative mitochondrial depolarization was defined as the magnitude of mitochondrial potential loss resulting from BH3 peptide treatments as compared to negative control DMSO and positive control FCCP.
Flow cytometry-based BH3 Profiling: Briefly, BH3 peptides in T-EB buffer with digitonin were deposited into each well in a 384-well plate. Single cells were resuspended in T-EB buffer and added to each treatment well and incubated for 60 min at 28°C. Peptide exposure was terminated with formaldehyde and cells were stained overnight with an antibody to cytochrome c conjugated to AF647. Cytochrome c positivity was measured on a BD Biosciences LSR II flow cytometer.
Additional experimental procedures can be found in the Extended Experimental Procedures.
Supplementary Material
SIGNIFICANCE.
Pediatric cancer patients treated with ionizing radiation or cytotoxic chemotherapy have a high risk of developing devastating toxicities including cognitive decline and chronic heart failure, limiting the use of potentially curative therapies. These treatments, which rely on the preferential induction of an apoptotic cell death in cancer cells over healthy tissues, are comparatively well tolerated in adults, yet the molecular basis for this difference in sensitivity is unknown. Herein, we make the discovery that apoptosis is dynamically regulated during postnatal development in healthy tissues, altering cell fate in response to genotoxic damage induced by anti-cancer therapies. Importantly, we use mouse models to show that these pathways may be modulated to potentially prevent treatment-associated toxicities.
HIGHLIGHTS.
Cells within healthy adult brain, heart and kidneys are apoptosis refractory.
Early in life, brain, heart and kidney cells are primed for apoptosis.
Dynamic regulation of apoptosis affects cell fate in response to genotoxic damage.
The apoptosis pathway is modulated by c-Myc, linking cellular growth with death.
Acknowledgments
We thank J. Sims, P. Sorger, X. Chi, L. Walensky, D. Andrews, C. Unitt, T. Bowman, O. Pozdnyakova and S. White for providing assistance and B. Mar, J. Montero, G. Joshi and L. Boise for critical review of our manuscript. We gratefully acknowledge funding from the American Cancer Society Postdoctoral Fellowship 121360-PF-11-256-01-TBG (K.A.S.), Alex’s Lemonade Stand Foundation for Childhood Cancers Young Investigator Award (K.A.S), as well as NIH grants K99CA188679 (K.A.S) and RO1CA129974 (A.L). A.L. was a Leukemia and Lymphoma Society Scholar. The authors have no conflict of interest to declare.
Footnotes
AUTHOR CONTRIBUTIONS
K.A.S., C.F., N.M., P.D.B., W.C., S.K.M., A.C., S.F., G.G-M., J.M.R. and J.D. conducted the experiments. K.A.S. and A.L. conceptualized the study and wrote the manuscript. K.A.S., C.C., M.G., J.R.M, B.J., R.L., D.W., J.S., D.J.M., D.R.C., S.R., J.M. and A.L. assisted with the acquisition and analysis of the data. All authors revised the manuscript and approved its content.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- American Cancer Society. Cancer Treatment & Survivorship Facts and Figures 2014–2015. Atlanta Am Cancer Soc. 2014:44. [Google Scholar]
- Bissonnette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359:552–554. doi: 10.1038/359552a0. [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Ikonomidou C. Antiepileptic drugs and apoptosis in the developing brain. Ann N Y Acad Sci. 2003;993:103–114. doi: 10.1111/j.1749-6632.2003.tb07517.x. discussion 123–124. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM, Antonssont B, Jemmerson R. Two pathways for tBID-induced cytochrome c release from rat brain mitochondria: BAK- versus BAX-dependence. J Neurochem. 2003;84:196–207. doi: 10.1046/j.1471-4159.2003.01545.x. [DOI] [PubMed] [Google Scholar]
- Campone M, Noël B, Couriaud C, Grau M, Guillemin Y, Gautier F, Gouraud W, Charbonnel C, Campion L, Jézéquel P, et al. c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol Cancer. 2011;10:110. doi: 10.1186/1476-4598-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2013;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Daley GQ. Common Themes of Dedifferentiation in Somatic Cell Reprogramming and Cancer Common Themes of Dedifferentiation in Somatic Cell Reprogramming and Cancer. Cold Spring Harb Symp Quant Biol. 2009;LXXIII doi: 10.1101/sqb.2008.73.041. [DOI] [PubMed] [Google Scholar]
- Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Davids MS, Deng J, Wiestner A, Lannutti BJ, Wang L, Wu CJ, Wilson WH, Brown JR, Letai A. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012 doi: 10.1182/blood-2012-02-414060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert S, Littlewood TD, Land H, Brooks M, Waters CM, Penn L, Hancock DC. Induction of Apoptosis by c-myc Protein in Fibroblasts. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Aaronson Sa, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Bellairs Ja, Leshchiner ES, Walensky LD. Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol. 2012:1–7. doi: 10.1038/nchembio.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Kohlmann A, Wieczorek L, Basso G, Te Kronnie G, Béné MC, De Vos J, Hernández JM, Hofmann WK, Mills KI, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the international microarray innovations in leukemia study group. J Clin Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg R. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hetz C, Vitte P-A, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou J-C, et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280:42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- Hofmann JW, Zhao X, De Cecco M, Peterson AL, Pagliaroli L, Manivannan J, Hubbard GB, Ikeno Y, Zhang Y, Feng B, et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell. 2015;160:477–488. doi: 10.1016/j.cell.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holly T, Drincic A, Byun Y. Caspase Inhibition Reduces Myocyte Cell Death Induced by Myocardial Ischemia and Reperfusion In Vivo. J Mol …. 1999;1715:1709–1715. doi: 10.1006/jmcc.1999.1006. [DOI] [PubMed] [Google Scholar]
- E, Galuszkova D, Ehrmann J, Ek BV, Murray PG, Kolao Z. Apoptosis-related proteins, BCL-2, BAX, FAS, FAS-L and PCNA in liver biopsies of patients with chronic Hepatitis B virus infection. 2000;6 doi: 10.1007/BF03032363. [DOI] [PubMed] [Google Scholar]
- Kandioler-Eckersberger D, Ludwig C, Rudas M, Kappel S, Janschek E, Wenzel C, Schlagbauer-Wadl H, Mittlböck M, Gnant M, Steger G, et al. TP53 mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin Cancer Res. 2000;6:50–56. [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole aJ, Annis RP, Deshmukh M. Mature neurons: equipped for survival. Cell Death Dis. 2013;4:e689. doi: 10.1038/cddis.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S, Krajewska M, Shabaik a, Miyashita T, Wang HG, Reed JC. Immunohistochemical determination of in vivo distribution of Bax, a dominant inhibitor of Bcl-2. Am J Pathol. 1994;145:1323–1336. [PMC free article] [PubMed] [Google Scholar]
- Lessene G. Targeting cell death pathways with small molecules: playing with life and death at the cellular level to treat diseases. Future Med Chem. 2015 doi: 10.4155/fmc.14.169. [DOI] [PubMed] [Google Scholar]
- Liang X, Liu Y, Zhang Q, Gao L, Han L, Ma C, Zhang L, Chen YH, Sun W. Hepatitis B virus sensitizes hepatocytes to TRAIL-induced apoptosis through Bax. J Immunol. 2007;178:503–510. doi: 10.4049/jimmunol.178.1.503. [DOI] [PubMed] [Google Scholar]
- Lipshultz SE, Lipsitz SR, Mone SM, Goorin aM, Sallan SE, Sanders SP, Orav EJ, Gelber RD, Colan SD. Female sex and drug dose as risk factors for late cardiotoxic effects of doxorubicin therapy for childhood cancer. N Engl J Med. 1995;332:1738–1743. doi: 10.1056/NEJM199506293322602. [DOI] [PubMed] [Google Scholar]
- Lipshultz SE, Scully RE, Stevenson KE, Franco VI, Neuberg DS, Colan SD, Silverman LB, Moslehi JJ, Cheng S, Sallan S. Hearts too small for body size after doxorubicin for childhood ALL: Grinch syndrome. J Clin Oncol. 2014;32(suppl) abstr 10021. [Google Scholar]
- Loewer A, Batchelor E, Gaglia G, Lahav G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell. 2010;142:89–100. doi: 10.1016/j.cell.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGibbon G, Lawlor P, Sirimanne E, Walton M, Connor B, Young D, Williams C, Gluckman P, Faull RL, Hughes P, et al. Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimer’s disease hippocampus. Brain Res. 1997;750:223–234. doi: 10.1016/s0006-8993(96)01351-0. [DOI] [PubMed] [Google Scholar]
- Martin SJ. Destabilizing influences in apoptosis: Sowing the seeds of IAP destruction. Cell. 2002;109:793–796. doi: 10.1016/s0092-8674(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Henry CM, Cullen SP. A Perspective on Mammalian Caspases as Positive and Negative Regulators of Inflammation. Mol Cell. 2012;46:387–397. doi: 10.1016/j.molcel.2012.04.026. [DOI] [PubMed] [Google Scholar]
- Merchant TE, Pollack IF, Loeffler JS. Brain tumors across the age spectrum: biology, therapy, and late effects. Semin Radiat Oncol. 2010;20:58–66. doi: 10.1016/j.semradonc.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Ja, Ding SL, Sunkin SM, Smith Ka, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KO, Ricci MS, Miyashita T, Dicker DT, Jin Z, Reed JC, El-Deiry WS. Bax is a transcriptional target and mediator of c-Myc-induced apoptosis. Cancer Res. 2000;60:6318–6325. [PubMed] [Google Scholar]
- Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui Da, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–457. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthalagu N, Junttila MR, Wiese KE, Wolf E, Morton J, Bauer B, Evan GI, Eilers M, Murphy DJ. Report BIM Is the Primary Mediator of MYC-Induced Apoptosis in Multiple Solid Tissues. CellReports. 2014:1–7. doi: 10.1016/j.celrep.2014.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Chonghaile T, Sarosiek Ka, Vo TT, Ryan Ja, Tammareddi A, Moore VDG, Deng J, Anderson KC, Richardson P, Tai YT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl D, Bittigau P, Ishimaru MJ, Stadthaus D, Hübner C, Olney JW, Turski L, Ikonomidou C. N-Methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci U S A. 1999;96:2508–2513. doi: 10.1073/pnas.96.5.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster BM, Basañez G, Young M, Suzuki M, Fiskum G. Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J Neurosci. 2003a;23:2735–2743. doi: 10.1523/JNEUROSCI.23-07-02735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster BM, Robertson CL, Bucci CJ, Suzuki M, Fiskum G. Postnatal brain development and neural cell differentiation modulate mitochondrial Bax and BH3 peptide-induced cytochrome c release. Cell Death Differ. 2003b;10:365–370. doi: 10.1038/sj.cdd.4401158. [DOI] [PubMed] [Google Scholar]
- Reed JC, Doctor K, Rojas A, Zapata JM, Stehlik C, Fiorentino L, Damiano J, Roth W, Matsuzawa S, Newman R, et al. Comparative Analysis of Apoptosis and Inflammation Genes of Mice and Humans Comparative Analysis of Apoptosis and Inflammation Genes of Mice and Humans. 2003:1376–1388. doi: 10.1101/gr.1053803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich T, Allen RL, Wyllie aH. Defying death after DNA damage. Nature. 2000;407:777–783. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods. 2013 doi: 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarosiek KA, Letai A. Directly targeting the mitochondrial pathway of apoptosis for cancer therapy with BH3 mimetics: recent successes, current challenges and future promise. FEBS J. 2016 doi: 10.1111/febs.13714. n/a – n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell. 2013;51:751–765. doi: 10.1016/j.molcel.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamas-Din A, Binder S, Chi X, Leber B, Andrews DW, Fradin C. Distinct lipid effects on tBid and Bim activation of membrane permeabilization by pro-apoptotic Bax. Submitted. 2014;505:1–25. doi: 10.1042/BJ20141291. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhang L, Zhang Y-W, Surma M, Mark Payne R, Wei L. Downregulation of doxorubicin-induced myocardial apoptosis accompanies postnatal heart maturation. Am J Physiol Heart Circ Physiol. 2012;302:H1603–H1613. doi: 10.1152/ajpheart.00844.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S. Apoptosis in Alzheimer’s disease — an update. 2000;5:9–16. doi: 10.1023/a:1009625323388. [DOI] [PubMed] [Google Scholar]
- Silber JH, Radcliffe J, Peckham V, Perilongo G, Kishnani P, Fridman M, Goldwein JW, Meadows aT. Whole-brain irradiation and decline in intelligence: the influence of dose and age on IQ score. J Clin Oncol. 1992;10:1390–1396. doi: 10.1200/JCO.1992.10.9.1390. [DOI] [PubMed] [Google Scholar]
- Soane L, Siegel ZT, Schuh Ra, Fiskum G. Postnatal developmental regulation of Bcl-2 family proteins in brain mitochondria. J Neurosci Res. 2008;86:1267–1276. doi: 10.1002/jnr.21584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Trachtenberg BH, Landy DC, Franco VI, Henkel JM, Pearson EJ, Miller TL, Lipshultz SE. Anthracycline-associated cardiotoxicity in survivors of childhood cancer. Pediatr Cardiol. 2011;32:342–353. doi: 10.1007/s00246-010-9878-3. [DOI] [PubMed] [Google Scholar]
- Trautwein C, Rakemann T, Malek NP, Plümpe J, Tiegs G, Manns MP. Concanavalin A-induced liver injury triggers hepatocyte proliferation. J Clin Invest. 1998;101:1960–1969. doi: 10.1172/JCI504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzung SP, Fausto N, Hockenbery DM. Expression of Bcl-2 family during liver regeneration and identification of Bcl-x as a delayed early response gene. Am J Pathol. 1997;150:1985–1995. [PMC free article] [PubMed] [Google Scholar]
- Vo T-T, Ryan J, Carrasco R, Neuberg D, Ross DJ, Stone R, DeAngelo DJ, Frattini MG, Letai A. Relative Mitochondrial Priming of Malignant Myeloblasts and Normal HSCs Determines Chemotherapeutic Success in AML. Cell. 2012 doi: 10.1016/j.cell.2012.08.038. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27:1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF, Yeh ETH. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- Zhou W, Yuan J. SnapShot: Necroptosis. Cell. 2014;158:464–464. e1. doi: 10.1016/j.cell.2014.06.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.