

Abstract
Antigen-specific immunotherapy of type 1 diabetes, typically via delivery of a single native β cell antigen, has had little clinical benefit to date. With increasing evidence that diabetogenic T cells react against multiple β cell antigens, including previously unappreciated neo-antigens that can be emulated by mimotopes, a shift from protein- to epitope-based therapy is warranted. To this end, we aimed to achieve efficient co-presentation of multiple major epitopes targeting both CD4+ and CD8+ diabetogenic T cells. We have compared native epitopes versus mimotopes as well as various targeting signals in an effort to optimize recognition by both types of T cells in vitro. Optimal engagement of all T cells was achieved with segregation of CD8 and CD4 epitopes, the latter containing mimotopes and driven by endosome-targeting signals, after delivery into either dendritic or stromal cells. The CD4+ T cell responses elicited by the endogenously delivered epitopes were comparable with high concentrations of soluble peptide and included functional regulatory T cells. This work has important implications for the improvement of antigen-specific therapies using an epitope-based approach to restore tolerance in type 1 diabetes and in a variety of other diseases requiring concomitant targeting of CD4+ and CD8+ T cells.
Keywords: type 1 diabetes, T cells, tolerance, antigen targeting, antigen presentation, epitope, mimotope, diabetogenic, dendritic cell, stromal cell
Graphical Abstract
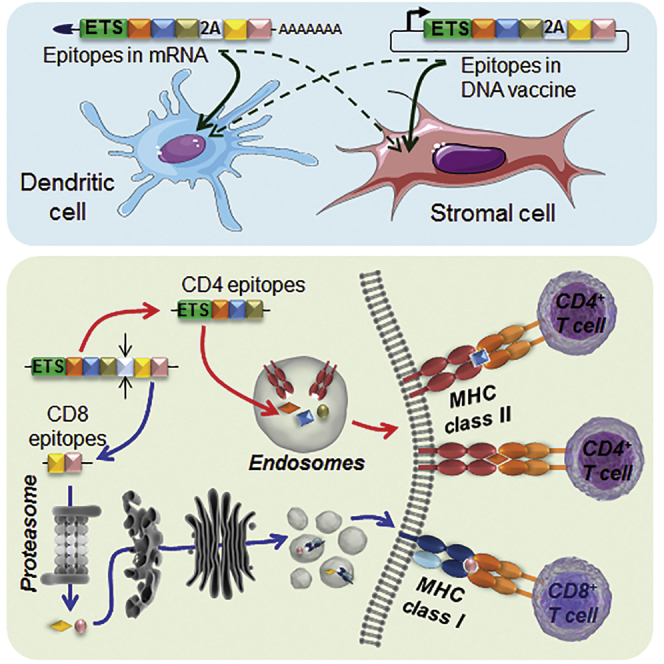
Introduction
In type 1 diabetes (T1D), insulin-producing β cells are progressively and specifically eliminated by an autoimmune attack. A number of self-antigens specific to these β cells are targeted by CD4+ and CD8+ T cell responses as well as by autoantibodies. In non-obese diabetic (NOD) mice, strong evidence points toward a specific insulin epitope (B9–23) as an initial driving antigen,1 with an immune response later diversifying to other insulin epitopes and to other β cell antigens. In humans, there are multiple antigens involved,2, 3 although it is unclear whether there is a common initial antigen because patients are more genetically diverse than NOD mice. Regardless, a large number of overlapping T cell epitopes and autoantibody-targeted antigens have been described in both species.2 Isolation of diabetogenic T cell clones from insulitic lesions has not always led to easy identification of their cognate antigen, with particular T cell clones being poorly responsive to peptides derived from native antigens. Recently, post-translational modifications of antigens4, 5 and generation of hybrid peptides6, 7 have been shown to generate neo-epitopes that constitute more efficient and physiologic antigens for the stimulation of these particular T cell clones, which previously required mimotopes identified from peptide libraries for stimulation.8, 9 Thus, attempts to target diabetogenic T cells for tolerance by simply delivering native protein antigens may be futile, and this may explain the poor efficacy of antigen-specific immunotherapy (ASIT) trials so far.10 In contrast, preclinical evaluation of native peptides versus mimotopes in disease prevention (NOD mice) and humanized mouse models has demonstrated the superior ability of mimotopes to target T cells for tolerance induction, at least in the case of insulin B9–23 peptide.11, 12 Furthermore, tetramer reagents incorporating insulin mimotopes also identify more circulating insulin-reactive T cells than those made with the native insulin epitope.13 These observations strongly support the use of epitope-based strategies for T1D ASIT, whereby epitopes and mimotopes appropriate for specific patients would be combined, integrated, and properly presented for effective engagement of diabetogenic T cells.
Although delivery of epitopes/mimotopes in the form of peptides is a straightforward approach, peptides have drawbacks related to their short half-life, solubility, rapid dilution in vivo, and production costs. Expression of peptides within antigen-presenting cells (APCs) from nucleic acids (exogenous DNA or RNA) generates an antigen reservoir for more sustained presentation, provided there is appropriate subsequent processing of the expressed epitopes. Endogenous expression of CD8 epitopes by a variety of major histocompatibility complex class I (MHC-I)+ cells can effectively mediate deletion of autoreactive CD8+ T cells.14, 15, 16, 17 Moreover, endogenous CD4 epitopes can be re-directed to endosomes or lysosomes18, 19, 20, 21, 22 and may contribute to induction of tolerance.21, 22 Co-expression of multiple CD4 and CD8 epitopes offers the unique possibility of bridging potentially pathogenic T cells and regulatory T cells (Tregs) to enable linked suppression.23, 24 In the present study, we have explored the endogenous delivery of epitopes/mimotopes from multiple β cell antigens into dendritic cells (DCs) and stromal cells (SCs) and determined conditions for the optimal recognition of all expressed epitopes by both CD4+ and CD8+ T cells. With a novel construct design, we integrated, within a single construct, strong native epitopes along with mimotopes not found within native proteins and delivered a sufficient antigen load per cell, allowing, for example, non-professional APCs such as SCs to induce and/or selectively expand Tregs. These constructs can be used, for example, to modify tolerogenic DCs ex vivo or as tolerogenic DNA vaccines in vivo. Their application goes beyond the treatment of T1D to cover other organ-specific autoimmune diseases as well as immunogenic DNA/RNA vaccines to treat infections and malignancies.
Results
Design of Tandem Epitope DNA Constructs
We chose to express five epitopes recognized by T cells that can be isolated from T cell receptor (TCR)-transgenic mice: the overlapping InsB9–23 and InsB15–23 from insulin, ChgA358–371 (also known as WE14) from chromogranin A, IGRP206–214 from islet-specific glucose-6-phosphatase catalytic subunit-related protein, and GAD65286–300 from glutamate decarboxylase (Figure 1; Table S1). These epitopes were included in our constructs as “native epitopes only” (NEO). To make the native epitopes + mimotopes (NEM) constructs, the InsB9–23 and ChgA358–371 epitopes were replaced by the InsB9–23 R22E mimotope25, 26 and 1040-79 mimotope,8 which emulate more disease-relevant MHC-peptide binding conformations or neo-epitopes (Figures 1A and 1B; Table S1). The synthesized DNA sequences for these constructs were cloned into our lentiviral (LV) vector co-expressing GFP27 by themselves or alongside one of four possible targeting signals (TSs) for endosomes or lysosomes. Sequences TFR1–118 (“TFR,” from transferrin receptor),19, 20 Ii1–80 (“Ii short,” from the invariant chain),18 and Ii1–214 (“Ii long,” from the invariant chain),20 corresponding to endosome-targeting signals (ETSs), were placed upstream of the epitopes (Figure 1). The lysosome-targeting signal (“Lamp1,” from lysosomal-associated membrane protein 1) consisted of the LAMP-1166–382 tail placed downstream of the epitopes and a CD161–23 leader sequence upstream.19 In light of our initial data, we subsequently developed a new construct design (“NMS”), whereby CD4 and CD8 epitopes from NEM were rearranged within the construct and separated by a T2A cleavage site, allowing the generation of two polypeptides upon translation (Figure 1C). This construct was produced with TFR or Ii short as ETS. Constructs in LV vectors were transduced into bone marrow-derived DC or SC lines that were then sorted based on an intermediate level of GFP overlap between all groups (Figure S1). These APCs were then co-cultured with one of the five purified T cell clones listed in Table S1.
Figure 1.

Design of Epitope-Expressing Constructs
(A and B) The NEO group of constructs contains only native CD4 and CD8 epitopes (A). The native InsB9-23 CD4 epitope includes the InsB15–23 CD8 epitope. The NEM group of constructs contains two mimotopes expected to more efficiently stimulate BDC12-4.1 and BDC2.5 T cells (B). Both groups are produced with or without TS among four tested: TFR, Lamp1, Ii short, and Ii long. Note that, for the Lamp1 lysosome TS, the CD161-23 signal precedes the epitopes, whereas LAMP-1166–382 is positioned at the end. (C) The NMS group of constructs contains the same epitopes as NEM, but CD4 and CD8 epitopes are segregated into two polypeptides after translation using the T2A cleavage site. The InsB9–23 mimotope and the InsB15–23 epitope are also segregated. In the expressed polypeptide(s), each epitope is flanked by at least two additional amino acid residues from the protein of origin on each side.
Mimotopes and ETSs Facilitate Engagement of CD4+ T Cells by DCs
Although high concentrations of WE14 have been shown to stimulate BDC2.5 T cells,28 no response was measured when expressed by any NEO constructs (Figure 2; Figures S2A, S2B, S3A, S3B, S4A, and S4B), comparable with control DCs without antigen (data not shown). In contrast, the 1040-79 mimotope (NEM constructs) elicited modest T cell recognition in the absence of TSs and significantly enhanced recognition with all ETSs but not Lamp1 (Figure 2; Figures S3A, S3B, S4A, and S4B). Likewise, the response of BDC12-4.1 T cells to the native InsB9–23 peptide (NEO constructs) was not measurable beyond the background seen with untransduced DCs (data not shown), even with TSs (Figures S2C, S2D, S3C, S3D, S4C, S4D, and S5). Engagement of the R22E mimotope (NEM constructs) was evident with all ETSs but not Lamp1, and no response was measured in absence of ETSs, in contrast to the 1040-79 mimotope. The response of G286 CD4+ T cells to the GAD65286–300 peptide without ETS was the most robust among all CD4+ T cell clones tested but was still greatly improved with the TFR ETS (Figure S6). The similar response measured in NEO and NEM constructs was consistent with the epitope being the same (Figures 1A and 1B).
Figure 2.

Stimulation of CD4+ T Cells from BDC2.5 Mice
(A–C) DCs were lentivirally transduced to express constructs containing no ETS, TFR1–118 ETS, or Ii1–80 ETS. Stimulation was measured by CD25 upregulation and T cell division. Data show representative dot plots of violet cell proliferation dye against CD25 (A) and the mean ± SD of % CD25+ (B) and percent divided (C) from at least three technical replicates (representative of three of four experiments, except NMS/Ii short, two experiments). t test analysis: *p < 0.05, **p < 0.01. Stimulation with latex beads coated with anti-CD3/CD28 gave >90% proliferation (data not shown).
Targeting Signals Hinder the Presentation of CD8 Epitopes Expressed by DCs
The maximal response of IGRP206–214-specific NY8.3 CD8+ T cells was seen with DCs expressing the NEO or NEM construct without any TS, whereas this response was significantly reduced with any of the TS tested (Figure 3; Figures S7 and S8), except with Ii short in one occurrence (Figure S8). Similarly, although G9C8 CD8+ T cells are weak responders to the InsB15–23 epitope, they were more efficiently engaged without TS than with TSs (Figure S9). Because both NEO and NEM constructs carry the same IGRP206–214 epitope (Figures 1A and 1B), the responses were very similar within each TS group (Figure 3; Figures S7 and S8). In contrast, the InsB15–23 epitope was overlapping with the InsB9–23 epitope; thus, it was affected by the R22E mutation in the NEM constructs. Although this mutation improved engagement of the CD4+ T cell clone, it abrogated that of the CD8+ T cell clone (compare NEO and NEM, Figure S9).
Figure 3.

Stimulation of CD8+ T Cells from NY8.3 Mice
(A–D) DCs were lentivirally transduced to express constructs containing no ETS or one of four tested TSs. Stimulation was measured by T cell division. Data show the representative dot plots (A) and mean ± SD (B–D) from three technical replicates (A and B and C and D are from two independent experiments representative of three of five experiments). The NMS/Ii short construct was not yet available when the experiment shown in (A) and (B) was done. t test analysis: *p < 0.05, ***p < 0.005.
Epitope Segregation Achieves Optimal Presentation of Both CD4 and CD8 T Cell Epitopes
When the NMS constructs were used (Figure 1C), the response of CD4+ T cell clones was as good as with the NEM constructs with ETS, if not better (Figure 2; Figures S2 and S5). Using the NMS constructs with ETS, the response of CD8+ T cell clones was rescued in most instances compared with NEM constructs with ETS, to a level similar to NEM constructs without ETS (Figures 3A–3C; Figures S7 and S9C). Thus, optimal presentation of all epitopes was achieved only when epitopes of interest were arranged and produced as two separate polypeptides, one containing CD4 epitopes that is targeted to endosomes and another one containing CD8 epitopes that is expected to remain cytosolic for proteasome processing.
Targeting of Diabetogenic T Cells by Stromal Cells
Given the potential of various types of SCs to present antigens to T cells in a tolerogenic fashion, we asked whether our constructs could be used to allow such SCs to engage T cells. For these studies, we used two different cell lines of fibroblastic lineage: DAPg7 cells, which are derived from I-Ak mice and stably overexpress I-Ag7, and PCRC-5 cells, which are immortalized NOD lymph node SCs expressing Kd but have no detectable basal I-Ag7 expression. All cells transduced to express the constructs were sorted based on GFP expression. DAPg7 cells expressed I-Ag7, PD-L1, and CD40 and no ICOSL or CD86 (Figure S10). Using these cells as APCs, BDC2.5 CD4+ T cells were stimulated only with the mimotope-expressing NEM constructs (Figure 4). However, the response to the mimotope in the absence of TS was lower than what was seen with DCs. As part of these studies, we also started to use CD4+ T cells from BDC12-4.1 mice on a NOD.TCRαKO background, which prevents unwanted pairing between the transgenic Vβ chain and endogenous Vα chains. On this genetic background, the T cell responses were greatly enhanced in amplitude, and some response to mimotope without TS was evident (compare Figure S11 with Figures S2C, S5B and S5C). A significant increase in the stimulation of both CD4+ T cell clones was seen with all ETSs but not Lamp1 (Figures 4D and 4E; Figures S11D and S11E). PCRC-5 cells were characterized as fibroblastic reticular cells, being positive for podoplanin, PDGFRα, and Sca1 and negative for the endothelial marker CD31 and epithelial marker CD326 (Figure S12A). These cells were differentiated fibroblasts (negative for the mesenchymal progenitor maker CD90) positive for Kd and negative for I-Ag7. Some variability was seen in the expression of GFP and Kd between transduced cell lines (Figure S12B), and because the CD8 epitopes were the same between all constructs, we picked constructs between NEO and NEM that were most uniform phenotypically for the studies. Consistent with the observations made with DCs, the presentation of CD8 epitopes, based on the response of NY8.3 T cells, was severely blunted by all TSs unless CD8 epitopes were segregated from ETS/CD4 epitopes (Figure 5). In contrast, G9C8 CD8+ T cells failed to respond with any transduced SC line and only responded to beads coated with anti-CD3/CD28 (data not shown). These data suggest that the differential processing of epitopes from our constructs is equivalent between DCs and SCs and that both types of APCs benefited from the new construct design, allowing efficient engagement of both CD4+ and CD8+ T cells.
Figure 4.

Stimulation of BDC2.5 CD4+ T Cells by Transduced DAPg7 Cells
(A–C) Comparison of constructs with mixed epitopes (NEO and NEM) and segregated epitopes (NMS) for CD25 upregulation (A) and T cell division (B), with representative dot plots (C), gated on live CD4+ singlets. (D and E) Comparison of mixed epitope constructs (NEO and NEM) without or with four types of TSs for CD25 upregulation (D) and T cell division (E). Data show the mean ± SD from three biological replicates (three donor transgenic mice) and from one of two similar experiments. Paired t test analysis: *p < 0.05, **p < 0.01, ***p < 0.005.
Figure 5.

Stimulation of NY8.3 CD8+ T Cells by Transduced PCRC-5 Cells
(A–C) Comparison of constructs with mixed epitopes (NEO/NEM, gray bars) and segregated epitopes (NMS, striped bars) for CD25 upregulation (A) and T cell division (B), with representative dot plots (C), gated on live CD8+ singlets. (D and E) Comparison of mixed epitope constructs without or with four types of TSs for CD25 upregulation (D) and T cell division (E). Data show the mean ± SD from three technical replicates and are representative of three experiments, one with round-bottom wells and two with flat-bottom wells. t test analysis: *p < 0.05, **p < 0.01, ***p < 0.005.
Comparison between Endogenous and Exogenous Peptide Delivery to Stromal Cells
Because epitopes are typically delivered in vitro to APCs in the form of soluble peptides, we sought to determine how the response to endogenously produced peptides compares with that obtained with various concentrations of exogenous peptide. The profile of the response of three T cell clones (BDC2.5, BDC12-4.1/TCRαKO, and NY8.3) to peptide-pulsed SCs was the same whether T cell division or CD25 upregulation was measured (Figure S13). BDC2.5 and NY8.3 T cells were confirmed to be the most responsive (they are also the most pathogenic in vivo), whereas BDC12-4.1 T cells exhibited a lower affinity to antigen, even with the mimotope peptide. CD25 expression and T cell division in response to exogenous peptides were highly correlated (Figure 6). The CD4+ T cell responses to transduced DAPg7 cells fall on the same trend line as soluble peptide: the best constructs (NMS with TFR or Ii short) gave a response equivalent to 10 μM soluble InsB9–23 R22E mimotope and to 0.1–1 μM soluble 1040-79 mimotope, whereas the response to the weakest constructs (NEM without ETS) was equivalent to 10- to 100-fold lower exogenous peptide concentrations (Figures 6A, 6B, and 7C). Thus, these constructs proved to be very efficient for SCs, eliciting CD4+ T cell responses otherwise achieved with very high concentrations of soluble peptide. The CD8+ T cell response to transduced PCRC-5 cells, surprisingly, did not follow the trend line of the exogenous peptide (Figure 6C): these T cells responded by CD25 upregulation with minimal proliferation. The extent of CD25 expression was comparable with that achieved with 1–10 nM IGRP206–214 peptide.
Figure 6.

Comparison between Endogenously Expressed Epitopes and Exogenous Peptides for the Stimulation of Diabetogenic T Cells by Stromal cells
The percentages of CD25+ and divided cells (gated on CD4+ or CD8+ T cells) are plotted against each other for various tandem epitope constructs and exogenous peptide concentrations. (A) Stimulation of CD4+ T cells from BDC12-4.1 (TCRαKO) mice by DAPg7 cells transduced with the indicated construct or pulsed with InsB9–23 R22E mimotope. (B) Stimulation of CD4+ T cells from BDC2.5 mice by DAPg7 cells transduced with indicated construct or pulsed with the 1040-79 mimotope. (C) Stimulation of CD8+ T cells from NY8.3 mice by PCRC-5 cells transduced with the indicated construct or pulsed with IGRP206–214 peptide. Responses to concentrations above 90 nM are not shown (saturation). Each dot is a biological (A and B) or technical (C) replicate from a representative experiment (from three experiments). A polynomial trend line with coefficient of correlation (R2) for the soluble peptide titration is indicated on each graph (a linear trend line also gives a R2 > 0.9).
Figure 7.

Comparison between Dendritic Cells and DAPg7 Stromal Cells for the Stimulation of CD4+ T Cells from BDC2.5.Foxp3/GFP Mice with Exogenous and Endogenous Peptides
(A–D) The percentages of CD25+ versus divided cells (A and C) and CD25+ versus Foxp3/GFP+ cells (B and D) are shown for a range of soluble 1040-79 mimotope peptide concentrations (indicated on each graph) and the NMS/TFR construct. Each dot is a biological replicate (n = 3). A linear or polynomial trend line with coefficient of correlation (R2) for the soluble peptide titration is indicated on each graph. (E and F) Representative plots showing proliferation (VCPD dilution) and Foxp3/GFP and Lag-3 expression. All data are gated on live singlet CD4+ T cells. DCs (A, B, and E, left) were pulsed with peptide or electroporated with NMS/TFR mRNA (0.4 or 2 μg/106 cells). For DAPg7 cells (C, D, and F, right), we used either control cells pulsed with the same peptide dilutions as DCs or the line transduced with NMS/TFR. Data are from an experiment using flat-bottom wells, with similar data obtained in an experiment using round-bottom wells (data not shown).
Comparison between Endogenous and Exogenous Peptide Delivery to Dendritic Cells
We repeated the analysis of BDC12-4.1 CD4+ T cell responses to DCs transduced with different constructs, this time using T cells from BDC12-4.1.TCRαKO mice. We also compared these responses with one of our NMS constructs that was produced as mRNA for electroporation (a technique more amenable to clinical translation than viral vectors29) and with serial dilutions of exogenous peptide. The response of these T cells was more robust than their counterpart isolated from NOD wild-type mice, with some response now seen with NEM/no ETS (compare Figure S14 with Figures S5B and S5C). Moreover, the T cell response to the mRNA version was comparable with its LV counterpart under the transfection conditions used (Figure S14). The LV and mRNA versions of NMS/TFR were comparable in the induction of CD25 (equivalent to 0.1–1 μM soluble InsB9–23 R22E mimotope) but induced more proliferation than any of the exogenous peptide concentrations (Figure S15A). The response of BDC2.5 CD4+ T cells to both LV and mRNA versions of NMS/TFR was equivalent to 10–100 nM soluble 1040-79 mimotope, with CD25 expression and T cell division highly proportional (Figure 7A; Figure S15B). Finally, the responses of NY8.3 and G9C8 CD8+ T cells to peptide-pulsed DCs had a very different profile than with peptide-pulsed SCs. At the lowest concentrations (∼10–100 pM), the T cells proliferate vigorously with minimum CD25 upregulation, whereas, at higher concentrations (from 1 nM to 1 mM), proliferation decreases as CD25 increases (Figures S15C and S15D). In contrast, the response of NY8.3 CD8+ T cells to endogenous epitopes showed a linear correlation between CD25 and proliferation (Figure S15C). The reduced proliferation of CD8+ T cells at high concentrations of exogenous peptide was not due to a deficit of secreted interleukin-2 (IL-2) (Figures S15E and S15F). However, the response to the IGRP206–214 epitope after mRNA delivery was weaker than with the LV construct (Figure S15C). Furthermore, endogenous epitopes induced substantially less proliferation of CD8+ T cells than soluble peptides at comparable CD25 levels (Figures S15C and S15D), which is reminiscent of what was seen with PCRC-5 cells as APCs (Figure 6C).
Different Propensity of Dendritic and Stromal Cells to Induce Foxp3 in T Cells
Given that one of the major goals of ASITs is to generate Treg populations in response to delivered epitopes, we used CD4+ T cells from BDC2.5.Foxp3/GFP reporter mice to evaluate whether the type of antigen delivery (endogenous versus exogenous), the antigen dose, and the type of APCs have any influence on the upregulation of Foxp3 after T cell engagement. Because the CD4+ T cell responses appeared to be equivalent in DCs transduced with LV and DCs electroporated with mRNA (1 μg/106 cells), we only used electroporated DCs thereafter. We compared two doses of antigen mRNA (0.4 and 2 μg/106 cells) that were completed to a total of 20 μg mRNA/106 cells with GFP mRNA for normalization. The response of BDC2.5.Foxp3/GFP, based on CD25 expression and cell division, was identical to that of BDC2.5 mice (compare Figure 7A with Figure S15B and Figure 7C with Figure 6B). However, the CD25 depletion step performed previously as part of CD4+ T cell enrichment only removed Foxp3hi cells, whereas some CD25− Foxp3lo cells remained present (Figure S16). T cells stimulated with DCs experienced a small (less than 2-fold) but significant (p < 0.02) increase in Foxp3+ cells at low peptide concentrations (0.1–10 nM) but not at higher concentrations (Figure 7B; Figure S17A). In contrast, those stimulated with DAPg7 cells saw a 5-fold increase in the proportion of Foxp3+ cells (p < 0.04) at the highest peptide concentrations (0.1–1 μM) (Figure 7D; Figure S17A). When delivered endogenously (mRNA or LV), this CD4 epitope induced the highest percentage of Foxp3+ cells in both types of APCs with the most significant increase (p < 0.003) (Figures 7B and 7D). DCs induced fewer Foxp3+ cells that nonetheless expressed higher levels of Foxp3 (compared with DAPg7 cells as APCs, p < 0.03) (Figures 7E and 7F). It was not clear whether these Foxp3+ T cells were de novo-induced or the result of a preferential expansion of the few Foxp3+ CD25− T cells present at the beginning of the culture. Our data suggest that endogenous epitope delivery to DCs and SCs led to at least as many Foxp3+ cells that would be obtained with variable doses of soluble peptide, although these Foxp3+ cells may differ phenotypically and functionally depending on the type of APC involved.
MHC-II+ Stromal Cells Induce Lag-3hi IL-10-Secreting CD4+ T cells
Because the induction of Foxp3 was relatively modest, we explored whether CD4+ T cells engaged by stromal cells were tolerized in other ways and whether they had any suppressive ability. Lag-3 has been identified as a functional marker in subsets of Tregs.30 We found that Lag-3 expression mirrored CD25 expression and cell division in percentage (Figures S17B–S17E), but T cells stimulated by DAPg7 SCs expressed higher levels of Lag-3 relative to CD25 than those stimulated by DCs (Figures 7E and 7F; Figures S17E and S17F). Furthermore, CD4+ T cells cultured with antigen-expressing DAPg7 cells secreted IL-10 (Figure 8A), whereas the same CD4+ T cells cultured with anti-CD3/CD28 beads in the presence of transforming growth factor β (TGF-β) and IL-2 expressed higher levels of Foxp3 (Figure 8B) but no Lag-3 (data not shown) and no IL-10 (Figure 8A). We next stimulated carboxyfluorescein succinimidyl ester (CFSE)-labeled polyclonal CD25-depleted Thy1.1+ NOD T cells (containing CD4+ and CD8+ T cells) with anti-CD3/CD28-coated beads in the presence of polyclonal NOD CD25-depleted Thy1.2+ CD4+ T cells or sorted populations from BDC2.5 Thy1.2+ CD4+ T cells previously stimulated with DAPg7 cells with or without NMS/Ii short LV to assess the suppressive potential of IL-10+ Lag-3hi cells at different target/regulator ratios. In these secondary cultures, we observed greater IL-10 levels and greater suppression with BDC2.5 T cells that had divided at least once, irrespective of Foxp3 levels, whereas those that did not divide contributed less IL-10 and were less suppressive (Figures 8C–8F). Thus, delivery of our constructs to SCs that are MHC-II+ enable efficient antigen presentation that results in tolerance.
Figure 8.

DAPg7 Stromal Cells Induce Suppressive Tregs that Secrete IL-10
(A) Production of IL-10 by BDC2.5 CD4+ T cells cultured for 3 days with anti-CD3/CD28-coated beads with or without TGF-β (1 ng/mL)/IL-2 (10 ng/mL) or with DAPg7 cells with or without NMS/TFR or NMS/Ii short LV constructs. (B) Proliferation and Foxp3/GFP expression of the same cells after 3 days and sorting gates for the suppression assay (the same color for each sorted population was used throughout the figure). (C) Production of IL-10 3 days after secondary culture of polyclonal NOD Thy1.1+ T cells with sorted BDC2.5 Thy1.2+ T cells from the primary culture in the presence of anti-CD3/CD28-coated beads. (D) Proliferation of CFSE-labeled Thy1.1+ T cells. Shown are representative proliferation histograms gated on CD4+ or CD8+ T cells. (E and F) Proliferation of Thy1.1+ CD4+ (E) and CD8+ (F) T cells in the presence of polyclonal CD25-depleted Thy1.2+ CD4+ T cells (purple bars) or sorted populations of BDC2.5 T cells (the bar color corresponds to the gates shown in B). Two Thy1.1 to Thy1.2 T cell ratios were used, 1:1 and 9:1, containing 50% and 10% putative suppressor cells, respectively. Data show the mean ± SD and are representative of two independent experiments, each with three technical replicates. t test analysis: *p < 0.05, **p < 0.01.
Discussion
Tolerogenic APCs need to engage autoreactive T cells through presentation of self-antigen-derived epitopes to mediate deletion, anergy, or Treg programming. One of the prerequisites for the success of ASITs is the efficient delivery of relevant antigens to these APCs, of which there are two types that are known to perform some or all of the above functions. The first group of target APCs is DCs, which are efficient at acquiring exogenous protein antigens and presenting peptides onto MHC-II and, to a more limited extent, MHC-I, depending on their cross-presentation capabilities. These cells can potentially become immunogenic under inflammatory conditions, and researchers have explored the possibility of generating stably tolerogenic DCs in vitro for cell therapy to treat autoimmunity, with a few initial clinical studies recently completed.31, 32 The ability of these cells to specifically engage autoreactive T cells for tolerance induction may rely on the presentation of appropriate epitopes.32 SCs constitute the second group of target APCs. Because they lack the extensive endocytic activity of DCs, their antigen presentation relies on endogenous expression, which is essentially limited to MHC-I. Subsets of SCs also express MHC-II and can process endogenous peptides onto MHC-II primarily through autophagy (as epitomized by medullary thymic epithelial cells33). SCs can also acquire MHC-II/peptide complexes from DCs.34 Although these cells are less amenable for cell therapy, lacking the motility and migratory properties of DCs, they tend to be more easily transfected than DCs using a variety of non-viral vectors35 and may, therefore, constitute better targets for delivery strategies such as tolerogenic DNA vaccines.36
In clinical trials for T1D ASIT, delivery of antigens in the form of proteins has been by far the most common approach.10 However, the disappointing results of such trials have led us to question whether the use of a single antigen is sufficient to restore tolerance to all targeted antigens. In theory, a regulatory response to one epitope can help promote a regulatory response to other epitopes (infectious tolerance) as long as these epitopes are linked; that is, presented by the same APC.37 Although linkage is obvious for epitopes derived from the same antigen, the chance that APCs simultaneously present epitopes from different β cell antigens is low when a single antigen is provided and still not guaranteed when different antigens are co-administered. Hybrid proteins made of epitope-containing regions of several autoantigens can be produced and appear to be more efficient than soluble peptides at promoting tolerance.38 Recombinant proteins for ASIT cost more to produce than DNA and may not undergo certain post-translational modifications during production. Many epitopes recognized by autoreactive T cells turn out to be modified or hybrid peptides,3, 4, 6, 7, 39 which further suggests that conventional ASIT strategies are suboptimal and limited. Thus, incorporation of hybrid peptides or mimotopes emulating post-translationally modified peptides identified in human patients will be required to more efficiently engage and tolerize diabetogenic T cells. Finally, protein antigens are not acquired by SCs, thereby excluding an important group of tolerogenic APCs as players. Nucleic acid-based systems, on the other hand, allow endogenous delivery of antigens, most efficiently in SCs. DNA vaccines expressing proinsulin lead to delayed loss of C-peptide and reduced frequency of proinsulin-specific CD8+ T cells in human patients,36 which is consistent with good MHC-I presentation of endogenous peptide. However, the effect on CD4+ T cells is more limited unless the antigen is targeted for secretion. Endogenous epitopes and mimotopes can also be targeted to endosomes or lysosomes18, 19, 20, 21 to allow tolerization of specific diabetogenic CD4+ T cells directly by the transfected APC.21
We proposed that endogenous delivery of multiple epitopes would have several advantages: targeting more types of APCs as opposed to the protein version of multi-epitope constructs,38 easy generation of expression constructs as DNA or RNA that incorporate non-native sequences to target neo-epitopes, and targeting epitopes from different antigens to favor intermolecular linkage and more effective immunoregulation, all with the convenience of a single construct. Although TSs have been known for some time to enhance the presentation of endogenous CD4 epitopes, it was not clear whether there was a need to optimize the concomitant presentation of endogenous CD4 and CD8 epitopes. We tested endogenous delivery to DCs and SCs of polypeptides containing five epitopes targeted in NOD mice. Presentation of all expressed epitopes was confirmed based on stimulation of their respective specific T cell clone, indicating that all peptides were processed correctly for presentation.
For the CD4+ T cell clones that were tested against both native peptide and mimotope, we only saw stimulation with the mimotope. This is consistent with the pronounced immunological and clinical differences seen between native InsB9–23 and its mimotope using peptide vaccination in animal models.11, 12 Likewise, the naturally processed ChgA358–371 (WE14) epitope is poorly immunogenic on its own, but, when fused with insulin-derived peptide to form a neo-epitope resembling the previously identified mimotope, it strongly stimulates BDC2.5 T cells.7 ETS from the transferrin receptor and the invariant chain significantly enhanced the engagement of CD4+ T cell clones, as measured by T cell proliferation and CD25 upregulation, the latter being the most sensitive response. The short invariant chain ETS was as good, if not better, than the longer form; however, the shorter form is preferred because smaller constructs are easier to package and provide more epitope copies per total amount of DNA/RNA. It was also as good, if not better, than the TFR ETS. In contrast, the lysosome-targeting Lamp1 TS did not enhance presentation of our CD4 epitopes. It is possible that the polypeptide(s) expressed by our constructs is/are differentially processed in the lysosomes because of different protease content.40 Indeed, antigens targeted to endosomes and lysosomes can be skewed toward a different peptide repertoire.19 A study using the TS of another lysosome protein (lysosomal integral membrane protein II) to target another BDC2.5 mimotope found an increased response of BDC2.5 CD4+ T cells.21 Different mimotopes, TSs, and contexts (in vitro versus in vivo) may account for this difference.
CD8+ T cell responses were also suboptimal when TSs were used in conjunction with mixed CD4/CD8 epitopes, suggesting that CD8 epitopes were being diverted away from proteasome processing. In a few instances, although not consistently, the short invariant chain ETS gave a CD8+ T cell response comparable with the no TS group, with epitope segregation. Interestingly, although the Lamp1 TS did not improve CD4+ T cell engagement, it was able to hinder engagement of CD8+ T cells, further suggesting that the lack of an effect by the Lamp1 TS may be a processing rather than a targeting issue. G9C8 CD8+ T cells are very-low-affinity T cells requiring high concentrations of InsB15–23 peptide for stimulation. These T cells responded when the most efficient constructs were delivered to DCs, but not SCs, and only to the non-mutated version of the epitope.41 The low affinity of these T cells may be sufficiently compensated by high-avidity presentation by DCs (high MHC-II levels), whereas SCs may be unable to stimulate because of lower levels of MHC-II. Although CD4+ T cells responded in the same way to exogenous and endogenous epitopes (relative CD25 expression and proliferation), we observed that endogenous epitopes tend to induce less proliferation of CD8+ T cells despite similar engagement, based on CD25 expression. It is possible that MHC/peptide complexes in this case are better “stabilized” by constant direct binding of exogenous peptides and that CD25 upregulation and proliferation require a different duration of engagement controlled by MHC complex density and renewal rate.
Responses to soluble peptides in vitro are not a good reflection of in vivo responses because dissociated peptides may be more readily replaced by direct binding of other identical peptides in static culture. In contrast, endogenously delivered epitopes provide a more sustained presentation both in vitro and in vivo via continuous replenishment of MHC/peptide complexes on the cell surface. Constructs expressing multiple epitopes endogenously not only ensure antigen linkage within APCs but may also increase the chance of productive encounters with specific T cells when presentation is more durable. Injected soluble peptides (and multi-epitope proteins) are expected to disperse in vivo and target a large number of APCs with a diluted and limited load of antigen per APC. In contrast, plasmid DNA delivered “naked” or using vectors will target fewer APCs because of limited transfection efficiency but, at the same time, will ensure that a higher amount of antigen is presented per cell. Our data suggest that a high antigen load was required to achieve a high proportion of Foxp3+ cells with SCs. Dosing of 5 μg/mouse/day of soluble insulin mimotope11, 12 would reach a maximal theoretical blood concentration of 2 μM (based on 1.5 mL of blood in an adult mouse), which would allow DCs, but not SCs, to effectively engage specific T cells. MHC-II expression in lymph node SCs is usually low and/or inducible but plays a role in the maintenance of tolerance and Treg numbers.42 PD-L1, expressed by some of these cells, has been implicated in CD8+ T cell tolerance43 and may also contribute to Treg induction.44, 45 Because DAPg7 cells express PD-L1 and low levels of MHC-II, data from these cells may be extrapolated to some lymph node SC subsets. At comparable levels of CD25 induction, DAPg7 SCs induced higher Lag-3 expression in T cells than DCs with endogenous antigen and highest concentrations of soluble peptide. This is likely attributable to differences in the type and level of costimulatory and coinhibitory ligands on the surface of these APCs. After culture with antigen-presenting DAPg7 SCs, CD4+ T cells secreted IL-10 and acquired suppressive functions. Bone marrow-derived DCs were relatively inefficient at inducing Foxp3 by simply presenting the mRNA-derived epitopes to T cells. However, because the amount of mRNA needed to obtain substantial T cell stimulation is small relative to the total amount of mRNA that can be loaded into DCs (≤10%), it is possible to modulate the tolerogenic properties of these DCs with complementary mRNA to overexpress tolerogenic products. Given the increasing clinical use of mRNA-modified DCs in cancer immunotherapy,29 this approach is applicable to cell-based ASIT.46
These extensive in vitro studies, using multiple CD4+ and CD8+ T cell specificities and two types of APCs, demonstrate the potential of tandem epitope constructs to express select epitopes (or mimotopes) from multiple antigens targeted in T1D. While conducting an extensive comparison of targeting signals for the MHC-II pathway, we determined that optimal presentation of all epitopes generally requires CD4 and CD8 epitopes to be segregated so that only CD4 epitopes are driven by these targeting signals. Likewise, overlapping CD4 and CD8 epitopes, as exemplified by InsB9–23 and InsB15–23, should be duplicated and segregated. Weaker epitopes may also be incorporated in higher copy numbers within the construct to further improve recognition. Endogenous delivery of these constructs is applicable to tolerogenic DC therapy and DNA vaccines, all approaches that have proven safe in patients with T1D and other autoimmune diseases31, 32, 36 but whose efficacy is not yet established. In particular, DNA vaccines are more likely to implicate SCs than approaches using exogenous protein antigens because SCs have poorer endocytic activity but are more efficiently transfected by DNA compared with DCs. Follow-up in vivo studies, as currently undertaken in our lab, will need to demonstrate the clinical benefit of delivering CD4/CD8 epitopes from multiple antigens as opposed to a single antigen47 or a single CD4 mimotope.21
Materials and Methods
Mice
Unless otherwise noted, all mice were purchased from The Jackson Laboratory and bred in our barrier facility. Male or female NOD mice (Jax #001976) were used as bone marrow donors for the generation of DCs. The TCR-transgenic mice used for most experiments were BDC2.5 (Jax #004460), BDC12-4.1 (Jax #006303 and #006304), and NY8.3 (Jax #005868). NOD.Foxp3/GFP mice (Jax #025097) were crossed with BDC2.5 mice to produce BDC2.5.Foxp3/GFP mice. G9C8 (TCRαKO) spleens and mice were provided by Dr. Susan Wong (University of Cardiff).48 NOD.TCRαKO mice were derived from G9C8 (TCRαKO) mice and crossed with BDC12-4.1 mice to generate BDC12-4.1.TCRαKO mice. Spleens from G286 mice were shipped to us by Dr. Kristin Tarbell (NIH National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK]).49 NOD.Thy1.1 congenic mice (Jax #004483) were used as donors for suppression assays. Mice were used at 8–16 weeks of age in all experiments. All studies were approved by Columbia University’s Institutional Animal Care and Use Committee.
Tandem Epitope DNA Constructs, Lentiviral Vectors, and mRNA
Epitope and targeting signal-containing constructs (Figure 1) were codon-optimized and synthesized by GeneArt (Thermo Fisher Scientific) and then sub-cloned into the pHR LV system downstream of a cytomegalovirus (CMV) promoter and upstream of internal ribosomal entry site (IRES)-GFP.27 LV particles were produced by calcium/phosphate-based transfection of 293T cells, followed by 100–150× concentration of supernatant collected after 48–60 hr, and titrated as described previously.27 An mRNA version of the NMS/TFR construct was designed and produced by TriLink Technologies as codon-optimized, anti-reverse cap analog (ARCA)-capped mRNA fully substituted with 5-methylcytosine and pseudouridine. These modifications enhance protein production and minimize the immunogenicity of mRNA,50 which is more appropriate for the purpose of tolerance induction.
Antigen-Presenting Cells
DAPg7 fibroblastic cells (I-Ag7 H2-Kk)51 were donated by Dr. Elisabeth Mellins (Stanford University). PCRC-5 cells are NOD lymph node SCs immortalized in our lab by overexpression of human papilloma virus E6/E7 proteins. These cells are H2-Kd+ and I-Ag7− fibroblastic reticular cells. DCs were generated in vitro from the bone marrow of NOD mice depleted of T cells, B cells, and granulocytes after 6–7 days of culture in the presence of GM-CSF and IL-4 (Peprotech, 10 ng/mL).52 DAPg7 and PCRC-5 cells were transduced with the different LV particles, sorted based on similar GFP levels between transduced lines. DCs were transduced overnight with LV particles at MOI 15–20 on days 4–5 of culture, washed on days 5–6 of culture, and harvested on days 6–7 for sorting. Because of variable transduction efficiencies and expression levels, DCs were sorted based on intermediate GFP MFI to normalize expression between all groups. Cell sorting was performed on BD Aria2 or Influx sorters. In some cases, DCs were also electroporated with tandem epitope-expressing mRNA using a GenePulser electroporator (Bio-Rad). DCs (5 × 106 cells) were electroporated in a 4-mm cuvette with up to 20 μg mRNA using a square wave pulse of 10 ms at 325 V (optimized conditions).
Responding T Cells
Spleen and pooled lymph nodes (inguinal, brachial, axillary, cervical, pancreatic, and mesenteric) were isolated by negative selection from donor TCR transgenic mice. CD4+ CD25− T cells were purified from BDC2.5, BDC12-4.1, and G286 mice using the EasySep Mouse CD4+ T Cell Isolation Kit (STEMCELL Technologies) supplemented with biotinylated anti-CD25. CD8+ T cells were purified from NY8.3 and G9C8 mice using the EasySep Mouse CD8+ T Cell Isolation Kit. The T cell purity was confirmed by flow cytometry and was 94%–99% for strains on a NOD background (see Figure S16A for an example) and 75%–85% for strains on a NOD.TCRαKO background. The percentage of CD25+ cells among isolated CD4+ was reduced to less than 1% after depletion (see Figure S16B for an example). Prior to culture, purified T cells were labeled with a green (CFSE) or violet cell proliferation dye (VCPD) (eBioscience).
T Cell Stimulation and Suppression Assays
5 × 104 T cells were co-cultured with 0.5–1 × 104 transduced DAPg7 cells, 2 × 103 transduced PCRC-5 cells, or 2 × 104 transduced DCs in technical or biological replicates for 3 days (round-bottom wells for DCs and flat-bottom wells for SCs unless otherwise noted). GFP-transduced DAPg7 cells, PCRC-5 cells, and DCs were used as negative controls. GFP-transduced DAPg7 and PCRC-5 pulsed with cognate peptides, untransduced DCs pulsed with cognate peptides, and anti-CD3/anti-CD28-coated latex beads were used as positive controls. Some culture supernatant was collected for ELISA, and then T cells were analyzed by flow cytometry for CD4 and/or CD8, GFP, CD25, and/or Lag-3 expression (all antibodies and ELISA kits were from BioLegend) and for CFSE and/or VCPD levels. For suppression assays, a total of 5 × 104 T cells comprising Thy1.1+ target T cells (CFSE-labeled) and Thy1.2+ T cells tested for suppressor function at a ratio ranging from 1:1 to 9:1 were co-cultured with 2 × 104 anti-CD3/anti-CD28-coated latex beads for 3 days. Culture supernatant was collected for ELISA, and then T cells were analyzed by flow cytometry for CD4, CD8, and CFSE.
Author Contributions
S.R.D., J.P.F., and R.J.C. performed the experiments and analyzed the data. C.X., J.H.S., and R.F.F. assisted with the experiments. R.J.C. designed the experiments and wrote the paper. All authors reviewed, edited, and approved the paper.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported by JDRF Transition Award 10-2010-790 and NIH P30 DK063608 Pilot and Feasibility Grant (to R.J.C.) and NIH T32 DK007271 (to J.H.S.). Furthermore, these studies made ample use of the resources of the Columbia Center for Translational Immunology (CCTI) and Diabetes and Research Center (DRC) Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050 and S10OD020056 and DRC Grant P30 DK063608. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank Susan Wong (Division of Infection and Immunity, Cardiff University, UK) for providing G9C8 spleens and mice and for critically reviewing our manuscript. The authors also thank Kristin Tarbell (NIH/NIDDK) for providing spleens from G286 mice, Siu-Hong Ho and Pavel Tishchenko from the Flow Core for assistance with cell sorting, as well as Cory Berkland (University of Kansas) and Donna Farber (CCTI) for feedback on the manuscript.
Footnotes
Supplemental Information includes seventeen figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.omtm.2016.12.002.
Supplemental Information
References
- 1.Nakayama M., Abiru N., Moriyama H., Babaya N., Liu E., Miao D., Yu L., Wegmann D.R., Hutton J.C., Elliott J.F., Eisenbarth G.S. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Lorenzo T.P., Peakman M., Roep B.O. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin. Exp. Immunol. 2007;148:1–16. doi: 10.1111/j.1365-2249.2006.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babon J.A., DeNicola M.E., Blodgett D.M., Crèvecoeur I., Buttrick T.S., Maehr R., Bottino R., Naji A., Kaddis J., Elyaman W. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat. Med. 2016;22:1482–1487. doi: 10.1038/nm.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delong T., Baker R.L., He J., Barbour G., Bradley B., Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes. 2012;61:3239–3246. doi: 10.2337/db12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGinty J.W., Marré M.L., Bajzik V., Piganelli J.D., James E.A. T cell epitopes and post-translationally modified epitopes in type 1 diabetes. Curr. Diab. Rep. 2015;15:90. doi: 10.1007/s11892-015-0657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin N., Wang Y., Crawford F., White J., Marrack P., Dai S., Kappler J.W. N-terminal additions to the WE14 peptide of chromogranin A create strong autoantigen agonists in type 1 diabetes. Proc. Natl. Acad. Sci. USA. 2015;112:13318–13323. doi: 10.1073/pnas.1517862112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delong T., Wiles T.A., Baker R.L., Bradley B., Barbour G., Reisdorph R., Armstrong M., Powell R.L., Reisdorph N., Kumar N. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. 2016;351:711–714. doi: 10.1126/science.aad2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Judkowski V., Pinilla C., Schroder K., Tucker L., Sarvetnick N., Wilson D.B. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 2001;166:908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 9.You S., Chen C., Lee W.H., Wu C.H., Judkowski V., Pinilla C., Wilson D.B., Liu C.P. Detection and characterization of T cells specific for BDC2.5 T cell-stimulating peptides. J. Immunol. 2003;170:4011–4020. doi: 10.4049/jimmunol.170.8.4011. [DOI] [PubMed] [Google Scholar]
- 10.Coppieters K.T., Harrison L.C., von Herrath M.G. Trials in type 1 diabetes: Antigen-specific therapies. Clin. Immunol. 2013;149:345–355. doi: 10.1016/j.clim.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniel C., Weigmann B., Bronson R., von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J. Exp. Med. 2011;208:1501–1510. doi: 10.1084/jem.20110574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serr I., Fürst R.W., Achenbach P., Scherm M.G., Gökmen F., Haupt F., Sedlmeier E.M., Knopff A., Shultz L., Willis R.A. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nat. Commun. 2016;7:10991. doi: 10.1038/ncomms10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakayama M., McDaniel K., Fitzgerald-Miller L., Kiekhaefer C., Snell-Bergeon J.K., Davidson H.W., Rewers M., Yu L., Gottlieb P., Kappler J.W., Michels A. Regulatory vs. inflammatory cytokine T-cell responses to mutated insulin peptides in healthy and type 1 diabetic subjects. Proc. Natl. Acad. Sci. USA. 2015;112:4429–4434. doi: 10.1073/pnas.1502967112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J.W., Epardaud M., Sun J., Becker J.E., Cheng A.C., Yonekura A.R., Heath J.K., Turley S.J. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat. Immunol. 2007;8:181–190. doi: 10.1038/ni1427. [DOI] [PubMed] [Google Scholar]
- 15.Cohen J.N., Guidi C.J., Tewalt E.F., Qiao H., Rouhani S.J., Ruddell A., Farr A.G., Tung K.S., Engelhard V.H. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J. Exp. Med. 2010;207:681–688. doi: 10.1084/jem.20092465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fletcher A.L., Lukacs-Kornek V., Reynoso E.D., Pinner S.E., Bellemare-Pelletier A., Curry M.S., Collier A.R., Boyd R.L., Turley S.J. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J. Exp. Med. 2010;207:689–697. doi: 10.1084/jem.20092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardner J.M., Devoss J.J., Friedman R.S., Wong D.J., Tan Y.X., Zhou X., Johannes K.P., Su M.A., Chang H.Y., Krummel M.F., Anderson M.S. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science. 2008;321:843–847. doi: 10.1126/science.1159407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanderson S., Frauwirth K., Shastri N. Expression of endogenous peptide-major histocompatibility complex class II complexes derived from invariant chain-antigen fusion proteins. Proc. Natl. Acad. Sci. USA. 1995;92:7217–7221. doi: 10.1073/pnas.92.16.7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandes D.M., Vidard L., Rock K.L. Characterization of MHC class II-presented peptides generated from an antigen targeted to different endocytic compartments. Eur. J. Immunol. 2000;30:2333–2343. doi: 10.1002/1521-4141(2000)30:8<2333::AID-IMMU2333>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 20.Diebold S.S., Cotten M., Koch N., Zenke M. MHC class II presentation of endogenously expressed antigens by transfected dendritic cells. Gene Ther. 2001;8:487–493. doi: 10.1038/sj.gt.3301433. [DOI] [PubMed] [Google Scholar]
- 21.Rivas E.I., Driver J.P., Garabatos N., Presa M., Mora C., Rodriguez F., Serreze D.V., Stratmann T. Targeting of a T cell agonist peptide to lysosomes by DNA vaccination induces tolerance in the nonobese diabetic mouse. J. Immunol. 2011;186:4078–4087. doi: 10.4049/jimmunol.0902395. [DOI] [PubMed] [Google Scholar]
- 22.Gardner J.M., Metzger T.C., McMahon E.J., Au-Yeung B.B., Krawisz A.K., Lu W., Price J.D., Johannes K.P., Satpathy A.T., Murphy K.M. Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4+ T cells. Immunity. 2013;39:560–572. doi: 10.1016/j.immuni.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleijwegt F.S., Laban S., Duinkerken G., Joosten A.M., Koeleman B.P., Nikolic T., Roep B.O. Transfer of regulatory properties from tolerogenic to proinflammatory dendritic cells via induced autoreactive regulatory T cells. J. Immunol. 2011;187:6357–6364. doi: 10.4049/jimmunol.1101638. [DOI] [PubMed] [Google Scholar]
- 24.Kleijwegt F.S., Roep B.O. Infectious tolerance as candidate therapy for type 1 diabetes: transfer of immunoregulatory properties from human regulatory T cells to other T cells and proinflammatory dendritic cells. Crit. Rev. Immunol. 2013;33:415–434. doi: 10.1615/critrevimmunol.2013006782. [DOI] [PubMed] [Google Scholar]
- 25.Stadinski B.D., Zhang L., Crawford F., Marrack P., Eisenbarth G.S., Kappler J.W. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc. Natl. Acad. Sci. USA. 2010;107:10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crawford F., Stadinski B., Jin N., Michels A., Nakayama M., Pratt P., Marrack P., Eisenbarth G., Kappler J.W. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc. Natl. Acad. Sci. USA. 2011;108:16729–16734. doi: 10.1073/pnas.1113954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Creusot R.J., Yaghoubi S.S., Kodama K., Dang D.N., Dang V.H., Breckpot K., Thielemans K., Gambhir S.S., Fathman C.G. Tissue-targeted therapy of autoimmune diabetes using dendritic cells transduced to express IL-4 in NOD mice. Clin. Immunol. 2008;127:176–187. doi: 10.1016/j.clim.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stadinski B.D., Delong T., Reisdorph N., Reisdorph R., Powell R.L., Armstrong M., Piganelli J.D., Barbour G., Bradley B., Crawford F. Chromogranin A is an autoantigen in type 1 diabetes. Nat. Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benteyn D., Heirman C., Bonehill A., Thielemans K., Breckpot K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines. 2015;14:161–176. doi: 10.1586/14760584.2014.957684. [DOI] [PubMed] [Google Scholar]
- 30.Huang C.T., Workman C.J., Flies D., Pan X., Marson A.L., Zhou G., Hipkiss E.L., Ravi S., Kowalski J., Levitsky H.I. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Giannoukakis N., Phillips B., Finegold D., Harnaha J., Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34:2026–2032. doi: 10.2337/dc11-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benham H., Nel H.J., Law S.C., Mehdi A.M., Street S., Ramnoruth N., Pahau H., Lee B.T., Ng J., Brunck M.E. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci. Transl. Med. 2015;7:290ra87. doi: 10.1126/scitranslmed.aaa9301. [DOI] [PubMed] [Google Scholar]
- 33.Aichinger M., Wu C., Nedjic J., Klein L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J. Exp. Med. 2013;210:287–300. doi: 10.1084/jem.20122149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubrot J., Duraes F.V., Potin L., Capotosti F., Brighouse D., Suter T., LeibundGut-Landmann S., Garbi N., Reith W., Swartz M.A., Hugues S. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4+ T cell tolerance. J. Exp. Med. 2014;211:1153–1166. doi: 10.1084/jem.20132000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 36.Roep B.O., Solvason N., Gottlieb P.A., Abreu J.R., Harrison L.C., Eisenbarth G.S., Yu L., Leviten M., Hagopian W.A., Buse J.B., BHT-3021 Investigators Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8+ T cells in type 1 diabetes. Sci. Transl. Med. 2013;5:191ra82. doi: 10.1126/scitranslmed.3006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cobbold S.P., Adams E., Nolan K.F., Regateiro F.S., Waldmann H. Connecting the mechanisms of T-cell regulation: dendritic cells as the missing link. Immunol. Rev. 2010;236:203–218. doi: 10.1111/j.1600-065X.2010.00913.x. [DOI] [PubMed] [Google Scholar]
- 38.Kaushansky N., Kerlero de Rosbo N., Zilkha-Falb R., Yosef-Hemo R., Cohen L., Ben-Nun A. ‘Multi-epitope-targeted’ immune-specific therapy for a multiple sclerosis-like disease via engineered multi-epitope protein is superior to peptides. PLoS ONE. 2011;6:e27860. doi: 10.1371/journal.pone.0027860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marré M.L., Profozich J.L., Coneybeer J.T., Geng X., Bertera S., Ford M.J., Trucco M., Piganelli J.D. Inherent ER stress in pancreatic islet β cells causes self-recognition by autoreactive T cells in type 1 diabetes. J. Autoimmun. 2016;72:33–46. doi: 10.1016/j.jaut.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watts C. The endosome-lysosome pathway and information generation in the immune system. Biochim. Biophys. Acta. 2012;1824:14–21. doi: 10.1016/j.bbapap.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrich de Marquesini L.G., Moustakas A.K., Thomas I.J., Wen L., Papadopoulos G.K., Wong F.S. Functional inhibition related to structure of a highly potent insulin-specific CD8 T cell clone using altered peptide ligands. Eur. J. Immunol. 2008;38:240–249. doi: 10.1002/eji.200737762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baptista A.P., Roozendaal R., Reijmers R.M., Koning J.J., Unger W.W., Greuter M., Keuning E.D., Molenaar R., Goverse G., Sneeboer M.M. Lymph node stromal cells constrain immunity via MHC class II self-antigen presentation. eLife. 2014;3:3. doi: 10.7554/eLife.04433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tewalt E.F., Cohen J.N., Rouhani S.J., Guidi C.J., Qiao H., Fahl S.P., Conaway M.R., Bender T.P., Tung K.S., Vella A.T. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood. 2012;120:4772–4782. doi: 10.1182/blood-2012-04-427013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L., Pino-Lagos K., de Vries V.C., Guleria I., Sayegh M.H., Noelle R.J. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc. Natl. Acad. Sci. USA. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Francisco L.M., Salinas V.H., Brown K.E., Vanguri V.K., Freeman G.J., Kuchroo V.K., Sharpe A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creusot R.J., Giannoukakis N., Trucco M., Clare-Salzler M.J., Fathman C.G. It’s time to bring dendritic cell therapy to type 1 diabetes. Diabetes. 2014;63:20–30. doi: 10.2337/db13-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solvason N., Lou Y.P., Peters W., Evans E., Martinez J., Ramirez U., Ocampo A., Yun R., Ahmad S., Liu E. Improved efficacy of a tolerizing DNA vaccine for reversal of hyperglycemia through enhancement of gene expression and localization to intracellular sites. J. Immunol. 2008;181:8298–8307. doi: 10.4049/jimmunol.181.12.8298. [DOI] [PubMed] [Google Scholar]
- 48.Wong F.S., Siew L.K., Scott G., Thomas I.J., Chapman S., Viret C., Wen L. Activation of insulin-reactive CD8 T-cells for development of autoimmune diabetes. Diabetes. 2009;58:1156–1164. doi: 10.2337/db08-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tarbell K.V., Lee M., Ranheim E., Chao C.C., Sanna M., Kim S.K., Dickie P., Teyton L., Davis M., McDevitt H. CD4(+) T cells from glutamic acid decarboxylase (GAD)65-specific T cell receptor transgenic mice are not diabetogenic and can delay diabetes transfer. J. Exp. Med. 2002;196:481–492. doi: 10.1084/jem.20011845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karikó K., Muramatsu H., Welsh F.A., Ludwig J., Kato H., Akira S., Weissman D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008;16:1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nabavieh A., Chou H., Volokhov I., Lee J.E., Purdy L.E., Elliott J.F., Singh B., Madrenas J. Development of an I-Ag7-expressing antigen-presenting cell line: intrinsic molecular defect in compact I-Ag7 dimer generation. J. Autoimmun. 1998;11:63–71. doi: 10.1006/jaut.1997.0176. [DOI] [PubMed] [Google Scholar]
- 52.Creusot R.J., Chang P., Healey D.G., Tcherepanova I.Y., Nicolette C.A., Fathman C.G. A short pulse of IL-4 delivered by DCs electroporated with modified mRNA can both prevent and treat autoimmune diabetes in NOD mice. Mol. Ther. 2010;18:2112–2120. doi: 10.1038/mt.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.