

ABSTRACT
Joint injury causes post‐traumatic osteoarthritis (PTOA). About ∼50% of patients rupturing their anterior cruciate ligament (ACL) will develop PTOA within 1–2 decades of the injury, yet the mechanisms responsible for the development of PTOA after joint injury are not well understood. In this study, we examined whole joint gene expression by RNA sequencing (RNAseq) at 1 day, 1‐, 6‐, and 12 weeks post injury, in a non‐invasive tibial compression (TC) overload mouse model of PTOA that mimics ACL rupture in humans. We identified 1446 genes differentially regulated between injured and contralateral joints. This includes known regulators of osteoarthritis such as MMP3, FN1, and COMP, and several new genes including Suco, Sorcs2, and Medag. We also identified 18 long noncoding RNAs that are differentially expressed in the injured joints. By comparing our data to gene expression data generated using the surgical destabilization of the medial meniscus (DMM) PTOA model, we identified several common genes and shared mechanisms. Our study highlights several differences between these two models and suggests that the TC model may be a more rapidly progressing model of PTOA. This study provides the first account of gene expression changes associated with PTOA development and progression in a TC model. © 2016 The Authors. Journal of Orthopaedic Research Published by Wiley Periodicals, Inc. J Orthop Res 35:474–485, 2017.
Keywords: osteoarthritis, RNA sequencing, tibial compression, ACL
Post‐traumatic osteoarthritis (PTOA) is a painful and debilitating disease that is caused by mechanical destabilization of the joint and injury to the articular cartilage; however, the molecular and cellular mechanisms leading to cartilage degeneration due to trauma are not well understood. It has been demonstrated that inflammation,1 abnormal subchondral bone properties2 and loss of response to mechanical load3 all contribute to the development of OA. Many individuals developing OA are asymptomatic until significant joint damage has occurred,4 at which point the only available long‐term treatment options are surgical replacement of the joint and/or pain management.5 Therefore, identifying and characterizing OA biomarkers for detecting and tracking the progression of the disease combined with developing new pharmacologic interventions aimed to minimize cartilage damage triggered by joint injury, are vital scientific endeavors.
In the past decade, using human biopsy and animal OA models, new insights about joint OA pathogenesis were uncovered. To date, several studies have evaluated molecular changes associated with human arthritic joint tissues including: Synovium,6 meniscus,7 cartilage,8, 9 osteophytes,10 and subchondral bone.11 Many of these studies revealed molecular changes associated with late stages of OA but only a few examined earlier molecular events because of clinical limitations. It is difficult to discriminate asymptomatic OA tissues and compare it to age matched healthy controls. Instead, mouse models that mimic human OA have been used with great success to study OA pathogenesis and to identify putative molecular and genetic factors driving the progression of the disease.12, 13
Though OA is commonly diagnosed by visible damage to the articular cartilage, more recent assessments of OA have been migrating to evaluate the entire joint, and perceive the disease as a multi factorial, multi cell‐type phenotype.14, 15 In this study, we used a non‐invasive tibial compression (TC) mouse model that closely mimics traumatic anterior cruciate ligament (ACL) rupture in humans to study molecular mechanisms driving PTOA development and progression.16 We hypothesized that a global gene expression profiling of TC injured knee joints at early (1 day or 1 week post injury) and late (6 or 12 weeks post injury) time points will unveil novel candidate genes and molecular mechanisms responsible for PTOA development and progression, and reveal new insights into OA progression in TC model. Through RNA sequencing analysis (RNAseq) we identified 1446 differentially regulated genes in injured joints, including several long noncoding RNAs (lncRNAs) that have not been previously studied in the context of PTOA. Furthermore, we compared our data with gene expression data generated from the surgical destabilization of the medial meniscus (DMM)17, 18 model of PTOA, to understand the commonalities and differences between these two models.
MATERIALS AND METHODS
Animals and Tibial Compression (TC) Joint Injury
Wildtype C57B/L6 mice underwent injury by applying a TC load (10–12 N) to the right knee of 16 weeks old male mice, as previously described.16 All animal experiments were conducted in accordance with institutional animal care and use committee guidelines at Lawrence Livermore National Laboratory and University of California, Davis.
Histology
Injured and uninjured (contralateral) joints were collected at 1 day, 1‐, 6‐, and 12 weeks (n = 5 per group) post injury. Joints were dissected, fixed in 4% paraformaldehyde, decalcified using 0.5 M EDTA, infiltrated in increasing concentrations of isopropanol, equilibrated into mineral oil, and embedded into paraffin wax. Six micrometer paraffin sections were stained on glass slides using 0.1% Safranin‐O and 0.05% Fast Green using standard procedures (IHC world) and imaged using a Leica DM5000 microscope. Three blind reviewers independently assessed OA severity using a modified OARSI19 scale to examine the medial compartment of injured and uninjured joints (sagittal views) (grade scale 0–0.5 normal; 1–2 mild; 3–4 moderate; 5–6 severe cartilage damage).
Micro‐Computed Tomography (micro‐CT) Analysis and Osteophyte Quantification
Quantification of epiphyseal trabecular bone of the distal femur and osteophyte volume was carried out as previously described.20 In brief, fixed joints were embedded into 1.5% agarose to a desired angle and scanned with micro‐computed tomography (SCANCO micro‐CT 35, Brüttisellen, Switzerland). Analysis was performed to quantify the microstructure of trabecular bone of the distal femoral epiphysis and osteophyte formation surrounding the joint. Joints were imaged under the guidelines for micro‐CT analysis for rodent bone structure (X‐ray tube potential = 55 kVp, intensity = 114 mA, 10 mm isotropic nominal voxel size, integration time = 900 ms).21 Quantification of trabecular bone volume fraction (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), and other microstructural parameters was done using the tools provided by the manufacturer.16 Lastly, osteophyte volume was calculated at terminal time points (6 and 12 weeks post injury), and included all mineralized tissues in the joint space except naturally ossified structures (patella, fabella, and anterior/posterior horns of the menisci). Statistical analysis was performed using a paired t‐test to compare injured and contralateral knees.
RNA Isolation and Sequencing (RNAseq)
Injured and contralateral joints (1 day [n = 5], 1‐ [n = 5], 6‐ [n = 3], and 12‐weeks [n = 3]) were dissected and cut at the base edges of femoral and tibial joint regions with small traces of soft tissues to preserve the intact knee joint. Dissected joints (between 0.25 and 0.3 g total weight) were then cut into small pieces and submerged in RNA Later (Qiagen) and stored at 4°C until processing. RNA Later solution was removed and dissected joints were homogenized in Qiazol lysis solution (Qiagen); RNA was isolated utilizing RNeasy Qiagen kits according to manufacturer's instructions. Isolated RNA (between 1 and 2 µg) was sequenced using an Illumina HiSeq 2000.
RNAseq Data Analysis
RNAseq data quality was checked using “FastQC” (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). Sequence reads were aligned to mouse genome (mm10) using “TopHat.”22 Differential gene expression analysis was conducted using an FPKM (fragments per kilobase of transcript per million mapped reads) based strategy and a count based strategy, to reduce the number of false positive discoveries. In the FPKM based strategy differentially expressed genes (DEGs) were identified using “Cufflinks” and “Cuffdiff.”23 A gene was considered significantly differentially expressed when its false discovery rate (FDR) corrected p‐value was < 0.05 and fold change was greater than 1.5. Subsequently, DEGs were filtered based on their expression values and low expressing genes with FPKM value < 2 were removed. In the count based strategy “featureCounts”24 was used to perform read summarization on reads mapped with “TopHat.” Subsequently, the data was normalized using “voom”25 and DEGs with fold change > 1.5 and p‐value < 0.05 were identified using “limma.”26 Genes identified by both methods as significantly differentially expressed were used to generate a list of high‐confidence DEGs. These high‐confidence DEGs were used for further analyses. Venn diagrams were created using R package “VennDiagram.” Heatmap was generated using heatmap.2 function in R package “gplots.”
Microarray Data Analysis
Previously published microarray data were downloaded from Gene Expression Omnibus (GEO) and the data analysis was conducted using Bioconductor.27 Affymetrix data7, 10, 28 preprocessing and normalization were performed using RMA method.29 Agilent data17 were background corrected with Normexp, normalized within arrays with loess and between arrays with Aquantile.26 Differentially expressed genes were identified using “limma.”26 Genes with p‐value < 0.05, FDR corrected p‐value < 0.1 and fold change > 1.5 were considered significantly differentially expressed.
Functional Annotation
Gene ontology analysis was performed using DAVID30 and enriched gene ontology terms (p‐value < 0.01) were identified. Long noncoding RNA (lncRNA) gene annotations were obtained from GENCODE.31 ToppCluster was used to cluster differentially expressed genes from different time points based on phenotype enrichment32 and Cytoscape was used for cluster visualization.33 Mouse phenotype data was obtained from MGI database (http://www.informatics.jax.org/).
RESULTS
Molecular Changes Associated With PTOA Development
Knee injury was induced by applying a single compressive load (10–12 N) to the right knee (Fig. 1A), as previously described.16, 34 Injured and contralateral joints were examined histologically and by RNAseq at 1 day, 1‐, 6‐, and 12 weeks post injury (Fig. 1B). Contralateral controls revealed minimal pathological changes at all times examined (Fig. 1C). Furthermore, no obvious morphological changes were observed histologically at 1 day post injury (Fig. 1D), suggesting that no articular fracture or damage was initially introduced by the compressive load and knee dislocation. As previously reported, we observed severe cartilage erosion (Fig. 1E and F) with osteophyte formation (Fig. 1G–I) at 6‐ and 12 weeks post injury.16, 34 Significant subchondral bone loss was also observed at 6‐ and 12 weeks post injury (Fig. 1J).
Figure 1.
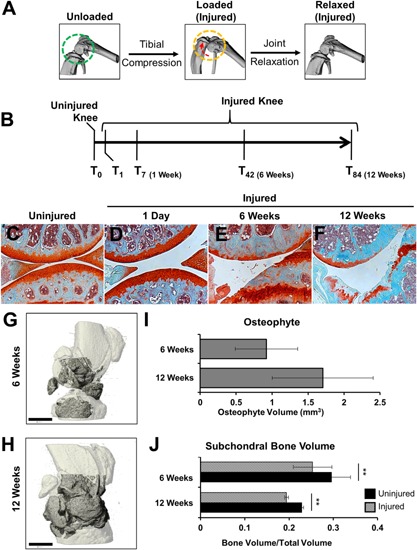
Histological evaluations of tibial compression (TC) OA injury. (A) TC overload leads to joint destabilization though ACL dislocation. The direction of joint displacement is indicated by the red arrow. (B) Time line where mice were injured and joints were collected at 1 day, 1‐, 6‐, and 12 weeks for either histology or RNA sequencing. Histological assessment of uninjured (C) and injured joints at various time points post injury (D–F) by Safranin‐O and Fast Green staining. Micro‐CT highlight regions (dark gray) of osteophyte formation in 6‐ (G) and 12‐ (H) weeks injured joints. (I) Micro‐CT quantification of osteophyte formation in injured joints. (J) Quantification of femoral subchondral trabecular bone formation between injured and uninjured joints. All histological images were presents were taken at 10× magnification. Scale bar is 1 mm; ** p < 0.01.
The FPKM and the count based methods (Fig. 2A) jointly identified a total of 1446 DEGs (Suppl. Table S1), where 599, 644, 511, and 201 DEGs were found at 1 day, 1‐, 6‐, and 12 weeks post injury, respectively (Fig. 2B and C). At 1 day, several genes up/down‐regulated in injured joints in response to injury were also found to be up/down‐regulated in contralateral joints compared to age matched WT controls. However, by 1 week post injury the expression of many of the genes reverted back to WT control levels (Suppl. Fig. S1). The largest overlap among the DEGs was found between 1 day and 1 week post injury, where 272 up‐ and 23 down‐regulated genes were in common (Fig. 2B and C). We also identified 15 genes that were up‐regulated at all time points examined (Table 1). Interestingly, no genes were found to be down‐regulated at all time points. Genes up‐regulated at all time points included extracellular matrix (ECM) components (Fn1,9 collagens 3, 5, and 6,35, 36 Cthrc1, Thbs3), matrix metabolic enzymes (Mmp3 37) and Wnt signaling proteins (Sfrp2 and Wisp2). In addition, it included ECM and cell adhesion proteins (SRPX2 and TNN) and a synovial fibroblast cell surface marker38 (THY1). We also identified 18 lncRNAs including Dnm3os, Rian, H19, and Snhg18 differentially regulated in injured knee joints compared to uninjured controls (Table 2).
Figure 2.
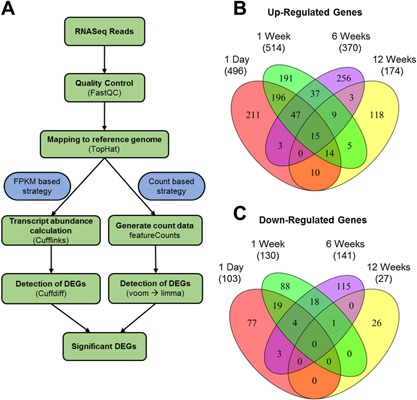
RNA sequencing analysis methodology and differentially expressed transcripts. (A) Flowchart of RNAseq data analysis. All of the differentially regulated genes presented were identified by both the FPKM based method and the count based method as significantly differentially expressed (p < 0.05). Common differentially up (B) and down (C) regulated genes between every time point post TC injury. The total number of genes per category is in brackets beneath each time point.
Table 1.
Transcripts That Were Up‐Regulated in Injured Joints, at All Time Points Examined
| No. | Gene Name | 1 Day | 1 Week | 6 Weeks | 12 Weeks |
|---|---|---|---|---|---|
| 1 | Mmp3 | 4.46 | 5.68 | 11.74 | 6.14 |
| 2 | Col3a1 | 4.10 | 5.74 | 4.05 | 3.93 |
| 3 | Cthrc1 | 3.83 | 5.05 | 1.86 | 2.92 |
| 4 | Sfrp2 | 3.41 | 4.61 | 3.18 | 3.03 |
| 5 | Wisp2 | 3.08 | 2.67 | 1.81 | 2.67 |
| 6 | Tnn | 2.69 | 4.22 | 3.17 | 3.02 |
| 7 | Col5a1 | 2.52 | 3.80 | 2.43 | 2.17 |
| 8 | Col6a3 | 2.24 | 4.39 | 3.14 | 2.39 |
| 9 | Srpx2 | 2.21 | 2.94 | 2.53 | 2.48 |
| 10 | Thy1 | 2.11 | 2.50 | 2.18 | 2.52 |
| 11 | Col6a2 | 2.10 | 4.31 | 2.41 | 2.81 |
| 12 | Col6a1 | 2.07 | 4.17 | 2.29 | 2.44 |
| 13 | Col5a2 | 2.05 | 2.89 | 2.70 | 2.65 |
| 14 | Thbs3 | 1.80 | 3.95 | 4.10 | 2.50 |
| 15 | Fn1 | 1.58 | 3.02 | 4.46 | 2.93 |
Values represent the fold changes between injured and uninjured joints (p < 0.05).
Table 2.
Long Noncoding RNAs Differentially Expressed in Injured Joints
| No. | LncRNA Name | 1 Day | 1 Week | 6 Weeks | 12 Weeks |
|---|---|---|---|---|---|
| 1 | Dnm3os a | 0.672 | 1.324 | ns | ns |
| 2 | Rian | 0.942 | 2.002 | ns | ns |
| 3 | 2810433D01Rik | 1.022 | ns | ns | ns |
| 4 | H19 b | ns | 0.98 | ns | ns |
| 5 | Snhg18 | ns | 0.777 | ns | 1.028 |
| 6 | 2610203C20Rik | ns | 0.892 | ns | ns |
| 7 | Gm11974 | ns | ns | 1.706 | ns |
| 8 | E330020D12Rik | ns | ns | 1.807 | ns |
| 9 | AI504432 | ns | ns | 0.942 | ns |
| 10 | Tmem134 | ns | ns | ns | 0.981 |
| 11 | 2310065F04Rik | −1.044 | ns | ns | ns |
| 12 | C130080G10Rik | −2.921 | ns | ns | ns |
| 13 | 1810044D09Rik | ns | −1.007 | ns | ns |
| 14 | Plet1os | ns | −0.988 | ns | ns |
| 15 | 2610306M01Rik | ns | ns | −0.616 | ns |
| 16 | 0610040B10Rik | ns | ns | −1.647 | ns |
| 17 | BC018473 | ns | ns | −1.04 | ns |
| 18 | 2610035D17Rik | ns | ns | −0.87 | ns |
lncRNAs previously shown to function during skeletal development;
or be dysregulated in OA cartilage.
Values represent the log2 fold changes between injured and uninjured joints (p < 0.05)
Gene Ontology Analysis of Differentially Regulated Genes
For up‐regulated genes, gene ontology categories corresponding to vasculature development, cell adhesion, extracellular matrix organization, extracellular structure organization, and collagen fibril organization were enriched at all time points examined (Table 3). Categories corresponding to regulation of cell proliferation and chondroitin sulfate proteoglycan metabolic process were enriched at all but the 12 week time point. Angiogenesis and hypoxia genes were enriched only at 1 day and 1 week post injury, while categories covering bone and cartilage development and collagen catabolism were enriched only at 1‐ and 6 weeks post injury correlating with the emergence of osteophyte formation and cartilage degradation as observed by histological analysis and micro‐CT (Fig. 1E and F). Wnt signaling was enriched only at 1 week, while genes associated with cell cycle were enriched at 6 weeks post injury. Categories corresponding to immune responses were enriched at 1 day, 6‐ and 12 weeks post injury, while markers of inflammation were enriched at 1 day and 12 weeks post injury. Inflammation related genes up‐regulated at 12 weeks post injury include several complement pathway components such as C1qa, C1qc, Cfb, and Cfp. These data suggest that significant cartilage remodeling occurs shortly after injury, while immune responses oscillate, with an early phase at 1 day and a later phase initiating at 6 weeks post injury.
Table 3.
Geneo Ontology Enriched Categories for Differentially Expressed Genes, at All Time Points Examined
| 1 Day | 1 Week | 6 Weeks | 12 Weeks | ||||||
|---|---|---|---|---|---|---|---|---|---|
| GO ID | GO Category | No. Genes | p‐Value | No. Genes | p‐Value | No. Genes | p‐Value | No. Genes | p‐Value |
| Up‐regulated genes | |||||||||
| GO:0001944 | Vasculature development | 29 | 5.08E‐11 | 25 | 2.59E‐08 | 14 | 6.20E‐04 | 8 | 0.00671 |
| GO:0007155 | Cell adhesion | 41 | 4.26E‐09 | 63 | 3.16E‐23 | 41 | 6.48E‐14 | 17 | 3.33E‐05 |
| GO:0030198 | Extracellular matrix organization | 17 | 5.39E‐09 | 25 | 4.35E‐17 | 17 | 3.13E‐11 | 8 | 3.04E‐05 |
| GO:0030199 | Collagen fibril organization | 7 | 1.06E‐05 | 7 | 1.04E‐05 | 9 | 1.66E‐09 | 5 | 3.08E‐05 |
| GO:0043062 | Extracellular structure organization | 18 | 2.64E‐07 | 29 | 7.22E‐17 | 19 | 1.87E‐10 | 8 | 3.50E‐04 |
| GO:0042127 | Regulation of cell proliferation | 25 | 0.00604 | 26 | 0.00291 | 23 | 2.98E‐04 | ns | ns |
| GO:0050654 | Chondroitin sulfate proteoglycan metabolic process | 4 | 0.00728 | 5 | 5.96E‐04 | 5 | 1.58E‐04 | ns | ns |
| GO:0009611 | Response to wounding | 27 | 9.91E‐07 | 18 | 0.00789 | ns | ns | 12 | 2.49E‐04 |
| GO:0001525 | Angiogenesis | 14 | 3.66E‐05 | 12 | 6.04E‐04 | ns | ns | ns | ns |
| GO:0001666 | Response to hypoxia | 7 | 0.00581 | 8 | 0.00119 | ns | ns | ns | ns |
| GO:0006954 | Inflammatory response | 23 | 7.64E‐08 | ns | ns | ns | ns | 10 | 1.70E‐04 |
| GO:0006955 | Immune response | 33 | 4.93E‐07 | ns | ns | 18 | 0.00523 | 16 | 1.68E‐05 |
| GO:0060348 | Bone development | ns | ns | 15 | 1.83E‐06 | 14 | 2.01E‐07 | ns | ns |
| GO:0051216 | Cartilage development | ns | ns | 10 | 1.61E‐04 | 11 | 1.30E‐06 | ns | ns |
| GO:0001501 | Skeletal system development | ns | ns | 30 | 2.19E‐10 | 22 | 4.84E‐08 | ns | ns |
| GO:0001503 | Ossification | ns | ns | 14 | 2.94E‐06 | 14 | 5.57E‐08 | ns | ns |
| GO:0030574 | Collagen catabolic process | ns | ns | 4 | 0.00860 | 5 | 2.04E‐04 | ns | ns |
| GO:0001558 | Regulation of cell growth | ns | ns | 13 | 3.69E‐06 | ns | ns | 5 | 0.00890 |
| GO:0006508 | Proteolysis | ns | ns | ns | ns | ns | ns | 19 | 0.00414 |
| GO:0006956 | Complement activation | ns | ns | ns | ns | ns | ns | 4 | 0.00388 |
| GO:0016477 | Cell migration | ns | ns | 18 | 1.47E‐04 | ns | ns | 8 | 0.00540 |
| GO:0002062 | Chondrocyte differentiation | ns | ns | ns | ns | 6 | 2.94E‐05 | ns | ns |
| GO:0002063 | Chondrocyte development | ns | ns | ns | ns | 3 | 0.00844 | ns | ns |
| GO:0002694 | Regulation of leukocyte activation | ns | ns | ns | ns | 11 | 4.79E‐04 | ns | ns |
| GO:0006029 | Proteoglycan metabolic process | ns | ns | 6 | 0.00139 | 8 | 1.63E‐06 | ns | ns |
| GO:0006260 | DNA replication | ns | ns | ns | ns | 10 | 0.00174 | ns | ns |
| GO:0006790 | Sulfur metabolic process | ns | ns | ns | ns | 7 | 0.00703 | ns | ns |
| GO:0007017 | Microtubule‐based process | ns | ns | ns | ns | 12 | 0.00157 | ns | ns |
| GO:0007049 | Cell cycle | ns | ns | ns | ns | 29 | 5.47E‐06 | ns | ns |
| GO:0008630 | DNA damage response, signal transduction resulting in induction of apoptosis | ns | ns | ns | ns | 4 | 0.00785 | ns | ns |
| GO:0009100 | Glycoprotein metabolic process | ns | ns | ns | ns | 11 | 3.31E‐04 | ns | ns |
| GO:0030203 | Glycosaminoglycan metabolic process | ns | ns | ns | ns | 6 | 7.19E‐04 | ns | ns |
| GO:0031214 | Biomineral formation | ns | ns | ns | ns | 7 | 3.38E‐05 | ns | ns |
| GO:0006928 | Cell motion | ns | ns | 22 | 4.97E‐04 | ns | ns | ns | ns |
| GO:0016055 | Wnt receptor signaling pathway | ns | ns | 12 | 4.98E‐04 | ns | ns | ns | ns |
| GO:0048870 | Cell motility | ns | ns | 18 | 0.00103 | ns | ns | ns | ns |
| GO:0051674 | Localization of cell | ns | ns | 18 | 0.00103 | ns | ns | ns | ns |
| GO:0000904 | Cell morphogenesis involved in differentiation | ns | ns | 15 | 0.00111 | ns | ns | ns | ns |
| GO:0007169 | Transmembrane receptor protein tyrosine kinase signaling pathway | ns | ns | 13 | 0.00386 | ns | ns | ns | ns |
| GO:0007167 | Enzyme linked receptor protein signaling pathway | ns | ns | 16 | 0.00453 | ns | ns | ns | ns |
| GO:0001649 | Osteoblast differentiation | ns | ns | 6 | 0.00672 | ns | ns | ns | ns |
| GO:0048771 | Tissue remodeling | ns | ns | 6 | 0.00672 | ns | ns | ns | ns |
| GO:0006897 | Endocytosis | ns | ns | 12 | 0.00911 | ns | ns | ns | ns |
| GO:0030036 | Actin cytoskeleton organization | ns | ns | 11 | 0.00987 | ns | ns | ns | ns |
| GO:0006935 | Chemotaxis | 11 | 4.81E‐04 | ns | ns | ns | ns | ns | ns |
| GO:0010876 | Lipid localization | 10 | 0.00565 | ns | ns | ns | ns | ns | ns |
| GO:0022604 | Regulation of cell morphogenesis | 9 | 0.00341 | ns | ns | ns | ns | ns | ns |
| GO:0030029 | Actin filament‐based process | 13 | 0.00193 | ns | ns | ns | ns | ns | ns |
| GO:0045597 | Positive regulation of cell differentiation | 13 | 0.00184 | ns | ns | ns | ns | ns | ns |
| Down‐regulated genes | |||||||||
| GO:0005996 | Monosaccharide metabolic process | 8 | 5.51E‐05 | 9 | 2.11E‐05 | 6 | 0.00834 | ||
| GO:0046164 | Alcohol catabolic process | 4 | 0.00455 | 4 | 0.00731 | 4 | 0.00886 | ||
| GO:0006096 | Glycolysis | ns | ns | 4 | 2.43E‐03 | 4 | 2.96E‐03 | ||
| GO:0006091 | Generation of precursor metabolites and energy | ns | ns | 10 | 2.95E‐05 | 16 | 1.28E‐10 | ||
| GO:0014706 | Striated muscle tissue development | ns | ns | 6 | 0.00107 | 5 | 0.00965 | ||
| GO:0045668 | Negative regulation of osteoblast differentiation | 3 | 0.00167 | ns | ns | ns | ns | ||
| GO:0006071 | Glycerol metabolic process | 3 | 6.17E‐03 | ns | ns | ns | ns | ||
| GO:0006094 | Gluconeogenesis | 4 | 1.40E‐04 | ns | ns | ns | ns | ||
| GO:0006641 | Triglyceride metabolic process | 4 | 8.28E‐04 | ns | ns | ns | ns | ||
| GO:0030278 | Regulation of ossification | 4 | 0.00255 | ns | ns | ns | ns | ||
| GO:0016051 | Carbohydrate biosynthetic process | 4 | 8.97E‐03 | ns | ns | ns | ns | ||
| GO:0016052 | Carbohydrate catabolic process | 5 | 7.97E‐04 | ns | ns | ns | ns | ||
| GO:0008610 | Lipid biosynthetic process | 8 | 6.43E‐04 | ns | ns | ns | ns | ||
| GO:0007517 | Muscle organ development | ns | ns | 9 | 1.17E‐05 | ns | ns | ||
| GO:0003012 | Muscle system process | ns | ns | 6 | 5.40E‐05 | ns | ns | ||
| GO:0009408 | Response to heat | ns | ns | 4 | 9.59E‐04 | ns | ns | ||
| GO:0046034 | ATP metabolic process | ns | ns | ns | ns | 8 | 1.96E‐06 | ||
| GO:0006119 | Oxidative phosphorylation | ns | ns | ns | ns | 6 | 3.16E‐05 | ||
| GO:0022900 | Electron transport chain | ns | ns | ns | ns | 7 | 9.14E‐05 | ||
| GO:0015992 | Proton transport | ns | ns | ns | ns | 5 | 3.66E‐04 | ||
For down‐regulated genes monosaccharide metabolic process and alcohol catabolic process were enriched at all but the 12 weeks post injury time point. Categories corresponding to glycolysis, generation of precursor metabolites and energy, and striated muscle tissue development were enriched at 1 week and 6 weeks post injury. Genes associated with negative regulation of osteoblast differentiation, and carbohydrate and lipid biosynthetic processes were enriched at 1 day, while muscle organ development, muscle system process, and response to heat were enriched at 1 week post injury. Categories corresponding to ATP metabolic process, oxidative phosphorylation, and electron transport chain were enriched at 6 weeks post injury. We found several components of mitochondrial electron transport chain including Ndufa1, Ndufa2, Ndufb3, and Uqcr10 to be down‐regulated at 6 weeks post injury suggesting aberrant mitochondrial activity. The 12 week time point did not reveal enrichment for any ontology categories.
Phenotype Enrichment Analysis
We performed a comparative phenotype enrichment analysis using ToppCluster32 and identified enriched mouse phenotypes associated with the DEGs. For up‐regulated genes, inflammatory/immune response related phenotypes and muscle phenotypes were found to be enriched at 1 day post injury, whereas both 1‐ and 6 weeks data showed enrichment for cartilage and bone phenotypes. Three categories: Arthritis, abnormal cutaneous collagen fibril morphology, and abnormal tendon morphology were enriched at all time points, except 12 weeks post injury. We identified 26 genes with arthritis phenotypes up‐regulated at 1 day, 1‐ and/or 6 weeks (Fig. 3).
Figure 3.
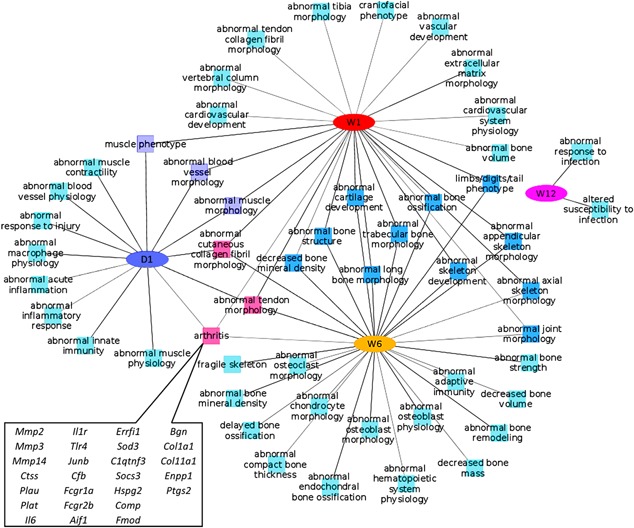
Enriched phenotypes associated with genes up‐regulated at 1 day (D1), 1‐ (W1), 6‐ (W6), and 12 weeks (W12) post injury. Up‐regulated genes associated with Arthritis phenotypes are highlighted at the bottom, left side.
While 1 day, 1‐ and 6 weeks data shared a significant overlap, the 12 weeks post injury data did not have any overlap with any other time points, and showed enrichment for only two categories: Abnormal response to infection and increased susceptibility to infection, suggesting that the severe cartilage damage observed histologically at this point (Fig. 1F) may render the organism susceptible to secondary health consequences due to viral or bacterial exposure (Fig. 3). Genes down regulated at 1‐ and 6 weeks showed enrichment for muscle phenotypes; by contrast, 1 day and 12 weeks genes did not show enrichment for any mouse phenotypes. Genes associated with abnormal glycogen and triglyceride levels were also found to be enriched at 1 week post injury. These data suggest that at early stages post injury, inflammation, and vasculature are in play with the attempt to repair and remodel both cartilage and bone. However at later stages post injury, the enrichment in abnormal bone and cartilage morphology categories are consistent with a damaged joint that has accrued significant morphological changes in bone and cartilage such as articular cartilage erosion and osteophyte formation, which are consistent with the histological and micro‐CT evaluations (Fig. 1E–J).
Comparison With Gene Expression Changes Induced by DMM Surgery
DMM is a widely used and validated animal model for studying PTOA.13 Gene expression profiling have been recently conducted on DMM injured mouse knee joints, and significant transcriptional changes were reported at 2‐, 4‐, and 16 weeks time points for whole‐joint derived RNA28 and at 1‐, 2‐, and 6 weeks time points for micro‐dissected articular cartilage derived RNA.17 We compared TC gene expression data to DMM model17, 28 to determine the commonalities and differences between TC induced and DMM induced PTOA, at the molecular level. 485 (whole joint; 472 up‐ and 13 down‐regulated) and 189 (micro‐dissected cartilage; 168 up‐ and 21 down‐regulated) of the DMM differentially expressed transcripts overlapped with our TC data, at least at one time point examined. A full list of these overlapping genes is provided in Supplementary Table S1. For significantly up‐regulated genes in whole joint RNA, 1 week post‐TC and 4 weeks post‐DMM injury had the largest overlap, with 382 shared (74.32% of TC genes) differentially expressed genes, while only 228 transcripts overlapped between the 1 week post‐TC and 2 weeks post‐DMM (whole joint). When micro‐dissected DMM cartilage was compared to our data, 1 week post‐TC and 2 weeks post DMM (cartilage) had the largest overlap, with 139 shared genes (27% of TC genes) (Table 4). Only 67 transcripts overlapped between the 1‐week post‐TC and 1‐week post‐DMM (cartilage). The 6 weeks TC data shared only 32 genes with the 6 weeks cartilage data but had a 125 gene overlap with the 4 weeks DMM whole joint data. Very few genes were found to overlap among the differentially down‐regulated transcripts or between our 12 weeks and the whole joint DMM 16 week data (Table 4).
Table 4.
Gene Expression Similarities Between TC (Tibial Compression) and DMM (Destabilization of Medial Meniscus) at Various Time Points Post Injury
| Time Point | TC | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 Day (496) | 1 Week (514) | 6 Weeks (370) | 12 Weeks (174) | |||||
| No. Genes | % Genes | No. Genes | % Genes | No. Genes | % Genes | No. Genes | % Genes | |
| DMM up‐regulated transcripts | ||||||||
| 1 week (C) | n/c | 67 | 13.04 | n/c | n/c | |||
| 2 weeks (C) | n/c | 139 | 27.04 | n/c | n/c | |||
| 2 weeks (WJ) | 168 | 33.87 | 228 | 44.36 | 68 | 18.38 | 26 | 14.94 |
| 4 weeks (WJ) | 262 | 52.82 | 382 | 74.32 | 125 | 33.78 | 36 | 20.69 |
| 6 weeks (C) | n/c | n/c | 32 | 8.65 | n/c | |||
| 16 weeks (WJ) | 21 | 4.23 | 41 | 7.98 | 28 | 7.57 | 14 | 8.05 |
| 1 Day (103) | 1 Week (130) | 6 Weeks (141) | 12 Weeks (27) | |||||
|---|---|---|---|---|---|---|---|---|
| No. Genes | % Genes | No. Genes | % Genes | No. Genes | % Genes | No. Genes | % Genes | |
| DMM down‐regulated transcripts | ||||||||
| 1 week (C) | n/c | 4 | 3.08 | n/c | n/c | |||
| 2 weeks (C) | n/c | 17 | 13.08 | n/c | n/c | |||
| 2 weeks (WJ) | 5 | 4.85 | 6 | 4.62 | 1 | 0.71 | 0 | 0.00 |
| 4 weeks (WJ) | 9 | 8.74 | 6 | 4.62 | 1 | 0.71 | 0 | 0.00 |
| 6 weeks (C) | n/c | n/c | 4 | 2.84 | n/c | |||
| 16 weeks (WJ) | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 |
C, cartilage (Bateman et al.)17; WJ, whole joint (Loeser et al.)18; n/c comparison was not conducted.
Number of overlapping genes are presented as the net number of genes up‐ (a) or down‐ (b) regulated in both the TC and DMM datasets, as well as percentage of the entire gene expression data set described for the TC time points.
TC model uniquely identified 582 up‐, 295 down‐, and 15‐mixed (up or down at different time points) regulated genes (Suppl. Table S1). This includes several immune/inflammatory response related genes such as Ccr2, C1qa, C1qb, C1qc, and Cfb; bone development related genes such as Spp1, Ctgf, Dmp1, Gabbr1, and Pth1r; cell adhesion genes such as Emilin1, Gpnmb, Itga5, and Stab1 and energy metabolism genes such as Ndufa1, Ndufa2, Ndufb3, Uqcr10, and Sdhb.
DISCUSSION
The TC induced ACL rupture model of PTOA is a new animal model that has not yet been widely explored to study mechanisms of joint OA development. This study provides the first account of whole genome expression profiles to obtain new insights into the temporal progression of the disease in the TC PTOA model. Despite being a noninvasive procedure, and exhibiting no obvious morphological or structural damages to tissues other than the ACL (Fig. 1C), we observed the largest transcriptional changes at early time points, with 599 and 644 transcripts being differentially expressed, at 1 day and 1 week post injury, respectively. Also, most of the differentially expressed genes were up‐regulated, with less than 30% of differentially expressed genes being significantly down‐regulated, at any time point examined.
Consistent with reports by Gardiner et al.9 where they examined transcriptional changes in micro‐dissected cartilage derived from DMM injured joints, we also identified Tnn (tenasin N) and Fn1 (fibronectin 1) as two transcripts consistently up‐regulated at all time points examined (Table 1). Also, similar to previous reports on DMM gene expression time‐course experiments,9, 28 the TC model of PTOA also undergoes a decline in transcriptional changes with time, where by 12 weeks post injury, only 201 transcripts are differentially expressed, suggesting that the joint adapts to the injury, with time, to reach a new molecular homeostasis, despite the enormous articular cartilage loss evident at this time point (Fig. 1F).
In addition, our model captures many of the transcriptional changes previously reported for DMM damaged cartilage, both reported for whole joints and micro‐dissected cartilage. We identified several genes including Mmp3, Errfi1, and C1qtnf3 with known arthritis phenotype as differentially regulated in TC data and both DMM datasets. C1qtnf3 has previously been implicated in autoimmune arthritis39; however, further studies are required to understand its role in OA. We also identified several regulators of Wnt signaling including Sfrp1, Sfrp2, Sfrp4, Dkk2, and Dkk3 and TGFβ signaling including Ltbp1, Fbn1, and Fbn2 commonly changed in both the TC and DMM models. Our 1 week data has the largest overlap with the 4 weeks post injury DMM28 whole joint data, where 74% of our up‐regulated transcripts were also found transcriptionally elevated in the DMM injured joints. We also found significant overlap between our 1 week post injury data and DMM injury pure cartilage data, where 67 up‐regulated genes (13%) overlapped with the 1 week and 139 up‐regulated genes (27%) overlapped with 2 weeks post DMM injury (Table 4). These data provides confirmation that the TC model recapitulates a large proportion of the gene expression reported for the DMM model, both at the whole joint and at the pure cartilage level. However these data also shows that these molecular changes happened at a faster rate in the TC model, suggesting that the TC injury may be a rapidly progressing PTOA model than DMM.
We also compared our data to previously published expression data of different human OA tissues including synovium,6 osteophyte,10 tibial plateau,11 articular cartilage,8, 40 and meniscus7 and found that majority of genes commonly differentially regulated between both human OA and TC induced mouse OA correspond to osteophyte and subchondral bone changes (Suppl. Table S2), consistent with osteophyte formation being a major hallmark of PTOA in humans and recapitulated in the TC mouse model examined here (Fig. 1G and H).
Interestingly, few genes up‐regulated in the TC PTOA time course are found to be significantly up‐regulated in the patient‐derived OA cartilage. In particular one gene, Col6A2 is up‐regulated in TC joints, at all times points examined, but significantly up‐regulated in human OA articular cartilage only. Mutations in this protein are primarily associated with myopathies,41 but may also have an important role in ECM remodeling in PTOA. Serpines are peptidase inhibitors and Serpine2 has been shown to up‐regulated in cultured chondrocytes in response to IL‐1α to inhibit MMP13 expression. We find Serpine1 to be significantly up‐regulated at 1 day and 1 week post injury, and similarly this molecule is up‐regulated in OA articular cartilage, suggesting that the upregulation of this peptidase may represent a regulatory mechanism for repressing the expression of cartilage catabolic enzymes.
Several genes identified in our study including Fndc1, Fads3, Edil3, C1qtnf6, Il1r1, Ptgs2, Rel, Tlr4, and Aldh1a2 were previously identified by genome wide association studies (GWAS) as potential OA, rheumatoid arthritis (RA), or ankylosing spondylitis (AS) susceptibility loci. These genes commonly altered in both PTOA and other degenerative joint diseases may highlight molecular components of potentially shared mechanisms that contribute to joint degeneration.
Our study also allowed us to examine transcriptional changes in several lncRNAs. We identified 18 lncRNAs differentially transcribed at least at one time point, 10 that were up‐ and 8 that were down‐regulated (Table 2). Two of these lncRNAs, Dnm3os, and H19 have been previously described in the context of skeletal development42 or OA,43 but all other lncRNAs identified herein have yet to be studied in the context of PTOA development. Furthermore, since we do not observe broad activation or repression across all time points examined, but rather see groups of lncRNAs transcriptionally changed at single time points, we speculated these noncoding RNAs may have unique functions to modulate the transcription of “function specific” cohorts of genes. Also, it would be interesting to determine whether some of these lncRNAs are modulated by the immune system, since initial changes in inflammation may be able to trigger large cascades of gene expression by repressing or activating these regulatory RNAs.
A list of possible genes that remain to be explored as candidate biomarkers or local joint therapeutics includes Ssc5d, a gene primarily expressed in monocytes/macrophages and T‐lymphocytes44 and Cemip (KIAA1199), a gene involved in hyaluronan (HA) metabolism.45 SSC5D, a soluble scavenger protein, was previously identified to be elevated in synovial fluid of OA patients.46 HA plays an important role in maintaining the integrity of articular cartilage by providing lubrication between the femoral and tibial surfaces. Cemip (KIAA1199) is a gene that enhances HA catabolism in the synovium45 and improves growth and angiogenesis of synovial tissue.47 However its role in OA developing joints has not yet been explored and inhibitors of Cemip may potentially prevent cartilage degradation post injury.
Our study also identified several novel genes including Suco, Sorcs2, and Medag. Suco (Opt) encodes a widely expressed rough endoplasmic reticulum‐localized integral membrane protein. Mice lacking Suco develop acute onset of skeletal defects including impaired bone formation and spontaneous fractures.48 Suco is > 2‐fold up‐regulated at 6 weeks post injury suggesting a role in aberrant bone remodeling and/or osteophyte formation. Sorcs2 is a member of vacuolar protein sorting 10 family proteins (VPS10Ps) with a potential role in protein trafficking and cell signaling.48 Recent GWAS study identified Sorcs2 as a candidate loci associated with cranial cruciate ligament rupture in Newfoundland dogs,49 however this gene has not been previously studied in the context of osteoarthritis. Medag, a gene we found up‐regulated at 1 day, 1‐ and 6 weeks has been shown to play a role in adipose tissue development50; however, its role in osteoarthritis has yet to be explored.
There are a few potential limitations to our study. First, we utilized the contralateral joint as controls instead of age matched sham injured joints. Because of this, it is difficult to distinguish between changes mediate by the injury that had systemic effects on both joints. In addition, there may also be changes occurring in the contralateral as a result of altered loading (more use of one joint) and changes in the injured joint/leg due to reduced mobility, disuse and pain. Second, because we are sequencing whole joints instead of individual tissues of the joint, to tease out where the gene expressions are coming from presents a challenge. However, because OA is considered a “joint disease,” comprehensive intact joint analysis may allow us to identified changes in tissues that may not normally be assumed to contribute to cartilage degradation or remodeling, such as muscle. Because of these caveats, we may lose some genes that are differentially expressed in a small area, or genes that are normally expressed broadly, but are affected regionally. These challenges may be overcome by examining candidate proteins for their tissue specific expression.
Our study uniquely introduces the gene expression changes associated with a new, noninvasive model of PTOA.34 This model closely mimics human ACL injuries resulted from a single high impact damage that leads to PTOA development. Our data provides evidence that a significant number of changes in the TC model correlate with both whole joint derived RNA and micro‐dissected cartilage derived RNA from the DMM model. Our study also highlights the differences between two models at the molecular level where our earlier time points (1 week) revealed more similarities with the later DMM time points (4 weeks), suggesting that TC model may be a more rapidly progressing PTOA model. Therefore, the TC model may be more suitable for investigating rapidly progressing joint failure and events occurring at earlier stages of traumatic injury. The pathways and candidate genes presented herein represent additional opportunities for investigating new potential therapeutic targets and susceptibility loci for PTOA.
AUTHORS' CONTRIBUTIONS
G.G.L. designed research; J.C.C., D.K.M., S.H., and B.A.C. performed research; A.S., J.C.C., B.A.C., and G.G.L. data analysis; S.H. and A.N.E. carried out sequencing; and J.C.C., A.S., and G.G.L. wrote the paper.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information.
Supporting Information.
ACKNOWLEDGMENTS
AS, JCC, BAC, and GGL were supported by DOD grant OR130220. BAC was supported by NIH/NIAMS grant AR062603T. This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE‐AC52‐07NA27344.
Jiun C. Chang and Aimy Sebastian contributed equally to the work.
REFERENCES
- 1. Joosten LA, Smeets RL, Koenders MI, et al. 2004. Interleukin‐18 promotes joint inflammation and induces interleukin‐1‐driven cartilage destruction. Am J Pathol 165:959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Osch GJ, van der Kraan PM, Blankevoort L, et al. 1996. Relation of ligament damage with site specific cartilage loss and osteophyte formation in collagenase induced osteoarthritis in mice. J Rheumatol 23:1227–1232. [PubMed] [Google Scholar]
- 3. Glasson SS, Askew R, Sheppard B, et al. 2005. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434:644–648. [DOI] [PubMed] [Google Scholar]
- 4. Glasson SS, Blanchet TJ, Morris EA. 2007. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15:1061–1069. [DOI] [PubMed] [Google Scholar]
- 5. Ma HL, Blanchet TJ, Peluso D, et al. 2007. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage 15:695–700. [DOI] [PubMed] [Google Scholar]
- 6. Lambert C, Dubuc JE, Montell E, et al. 2014. Gene expression pattern of cells from inflamed and normal areas of osteoarthritis synovial membrane. Arthritis Rheumatol 66:960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun Y, Mauerhan DR, Honeycutt PR, et al. 2010. Analysis of meniscal degeneration and meniscal gene expression. BMC Musculoskelet Disord 11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramos YF, den Hollander W, Bovee JV, et al. 2014. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PLoS ONE 9:e103056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gardiner MD, Vincent TL, Driscoll C, et al. 2015. Transcriptional analysis of micro‐dissected articular cartilage in post‐traumatic murine osteoarthritis. Osteoarthritis Cartilage 23:616–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klinger P, Beyer C, Ekici AB, et al. 2013. The transient chondrocyte phenotype in human osteophytic cartilage: a role of pigment epithelium‐derived factor? Cartilage 4:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chou CH, Wu CC, Song IW, et al. 2013. Genome‐wide expression profiles of subchondral bone in osteoarthritis. Arthritis Res Ther 15:R190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fang H, Beier F. 2014. Mouse models of osteoarthritis: modelling risk factors and assessing outcomes. Nat Rev Rheumatolo 10:413–421. [DOI] [PubMed] [Google Scholar]
- 13. Little CB, Hunter DJ. 2013. Post‐traumatic osteoarthritis: from mouse models to clinical trials. Nat Rev Rheumatolo 9:485–497. [DOI] [PubMed] [Google Scholar]
- 14. Kapoor M, Martel‐Pelletier J, Lajeunesse D, et al. 2011. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatolo 7:33–42. [DOI] [PubMed] [Google Scholar]
- 15. Wieland HA, Michaelis M, Kirschbaum BJ, et al. 2005. Osteoarthritis—an untreatable disease? Nat Rev Drug Discov 4:331–344. [DOI] [PubMed] [Google Scholar]
- 16. Christiansen BA, Anderson MJ, Lee CA, et al. 2012. Musculoskeletal changes following non‐invasive knee injury using a novel mouse model of post‐traumatic osteoarthritis. Osteoarthritis Cartilage 20:773–782. [DOI] [PubMed] [Google Scholar]
- 17. Bateman JF, Rowley L, Belluoccio D, et al. 2013. Transcriptomics of wild‐type mice and mice lacking ADAMTS‐5 activity identifies genes involved in osteoarthritis initiation and cartilage destruction. Arthritis Rheum 65:1547–1560. [DOI] [PubMed] [Google Scholar]
- 18. Loeser RF, Olex AL, McNulty MA, et al. 2012. Microarray analysis reveals age‐related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum 64:705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Glasson SS, Chambers MG, Van Den Berg WB, et al. 2010. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18:S17–S23. [DOI] [PubMed] [Google Scholar]
- 20. Lockwood KA, Chu BT, Anderson MJ, et al. 2014. Comparison of loading rate‐dependent injury modes in a murine model of post‐traumatic osteoarthritis. J Orthop Res 32:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bouxsein ML, Boyd SK, Christiansen BA, et al. 2010. Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J Bone Miner Res 25:1468–1486. [DOI] [PubMed] [Google Scholar]
- 22. Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trapnell C, Williams BA, Pertea G, et al. 2010. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liao Y, Smyth GK, Shi W. 2014. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. [DOI] [PubMed] [Google Scholar]
- 25. Law CW, Chen Y, Shi W, et al. 2014. Voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- 27. Gentleman RC, Carey VJ, Bates DM, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loeser RF, Olex AL, McNulty MA, et al. 2013. Disease progression and phasic changes in gene expression in a mouse model of osteoarthritis. PLoS ONE 8:e54633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Irizarry RA, Hobbs B, Collin F, et al. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. [DOI] [PubMed] [Google Scholar]
- 30. Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. [DOI] [PubMed] [Google Scholar]
- 31. Harrow J, Frankish A, Gonzalez JM, et al. 2012. GENCODE: the reference human genome annotation for The ENCODE project. Genome Res 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaimal V, Bardes EE, Tabar SC, et al. 2010. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network‐based dissection of biological systems. Nucleic Acids Res 38:W96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shannon P, Markiel A, Ozier O, et al. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Christiansen BA, Guilak F, Lockwood KA, et al. 2015. Non‐invasive mouse models of post‐traumatic osteoarthritis. Osteoarthritis Cartilage 23:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiao Q, Wei L, Chen C, et al. 2016. Cartilage oligomeric matrix protein and hyaluronic acid are sensitive serum biomarkers for early cartilage lesions in the knee joint. Biomarkers 21:146–151. [DOI] [PubMed] [Google Scholar]
- 36. Wei T, Kulkarni NH, Zeng QQ, et al. 2010. Analysis of early changes in the articular cartilage transcriptisome in the rat meniscal tear model of osteoarthritis: pathway comparisons with the rat anterior cruciate transection model and with human osteoarthritic cartilage. Osteoarthritis Cartilage 18:992–1000. [DOI] [PubMed] [Google Scholar]
- 37. Echtermeyer F, Bertrand J, Dreier R, et al. 2009. Syndecan‐4 regulates ADAMTS‐5 activation and cartilage breakdown in osteoarthritis. Nat Med 15:1072–1076. [DOI] [PubMed] [Google Scholar]
- 38. Seemayer CA, Kuchen S, Kuenzler P, et al. 2003. Cartilage destruction mediated by synovial fibroblasts does not depend on proliferation in rheumatoid arthritis. Am J Pathol 162:1549–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murayama MA, Kakuta S, Maruhashi T, et al. 2014. CTRP3 plays an important role in the development of collagen‐induced arthritis in mice. Biochem Biophys Res Commun 443:42–48. [DOI] [PubMed] [Google Scholar]
- 40. Snelling S, Rout R, Davidson R, et al. 2014. A gene expression study of normal and damaged cartilage in anteromedial gonarthrosis, a phenotype of osteoarthritis. Osteoarthritis Cartilage 22:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bushby KM, Collins J, Hicks D. 2014. Collagen type VI myopathies. Adv Exp Med Biol 802:185–199. [DOI] [PubMed] [Google Scholar]
- 42. Watanabe T, Sato T, Amano T, et al. 2008. Dnm3os, a non‐coding RNA, is required for normal growth and skeletal development in mice. Dev Dyn 237:3738–3748. [DOI] [PubMed] [Google Scholar]
- 43. Steck E, Boeuf S, Gabler J, et al. 2012. Regulation of H19 and its encoded microRNA‐675 in osteoarthritis and under anabolic and catabolic in vitro conditions. J Mol Med (Berl) 90:1185–1195. [DOI] [PubMed] [Google Scholar]
- 44. Goncalves CM, Castro MA, Henriques T, et al. 2009. Molecular cloning and analysis of SSc5D, a new member of the scavenger receptor cysteine‐rich superfamily. Mol Immunol 46:2585–2596. [DOI] [PubMed] [Google Scholar]
- 45. Yoshida H, Nagaoka A, Kusaka‐Kikushima A, et al. 2013. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc Natl Acad Sci USA 110:5612–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Balakrishnan L, Bhattacharjee M, Ahmad S, et al. 2014. Differential proteomic analysis of synovial fluid from rheumatoid arthritis and osteoarthritis patients. Clin Proteomics 11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang X, Qiu P, Chen B, et al. 2015. KIAA1199 as a potential diagnostic biomarker of rheumatoid arthritis related to angiogenesis. Arthritis Res Ther 17:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sohaskey ML, Jiang Y, Zhao JJ, et al. 2010. Osteopotentia regulates osteoblast maturation, bone formation, and skeletal integrity in mice. J Cell Biol 189:511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baird AE, Carter SD, Innes JF, et al. 2014. Genome‐wide association study identifies genomic regions of association for cruciate ligament rupture in newfoundland dogs. Anim Genet 45:542–549. [DOI] [PubMed] [Google Scholar]
- 50. Zhang H, Chen X, Sairam MR. 2012. Novel genes of visceral adiposity: identification of mouse and human mesenteric estrogen‐dependent adipose (MEDA)‐4 gene and its adipogenic function. Endocrinology 153:2665–2676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information.
Supporting Information.