

Abstract
Bio‐orthogonal labelling schemes based on inverse‐electron‐demand Diels–Alder (IEDDA) cycloaddition have attracted much attention in chemical biology recently. The appealing features of this reaction, such as the fast reaction kinetics, fully bio‐orthogonal nature and high selectivity, have helped chemical biologists gain deeper understanding of biochemical processes at the molecular level. Listing the components and discussing the possibilities and limitations of these reagents, we provide a recent snapshot of the field of IEDDA‐based biomolecular manipulation with special focus on fluorescent modulation approaches through the use of bio‐orthogonalized building blocks. At the end, we discuss challenges that need to be addressed for further developments in order to overcome recent limitations and to enable researchers to answer biomolecular questions in more detail.
Keywords: bio-orthogonal building blocks, cycloaddition, dienophiles, fluorescence, inverse-electron-demand Diels–Alder reaction, tetrazines
1. Introduction
Small‐molecule‐based manipulation of biomatter by means of bio‐orthogonal transformations has brought remarkable advances in the exploration of biological processes at the molecular level. Generally, the goals of these biocompatible and chemoselective modulation schemes are to install various kinds of labels onto some biomolecule of interest or to furnish these biologically relevant species with artificial functionalities. The concepts of bio‐orthogonal chemistry were delineated by Bertozzi and Saxon in the early 2000s.1 Since then, several chemical reactions between unnatural functions fulfilling the criteria of bio‐orthogonality have been discovered or, most of the time, rediscovered.2 This was also the case with tetrazines and strained ring systems. Boger and Sauer described the chemical nature of tetrazines in the 1980s; however, it took about 30 years until Fox and Weissleder almost simultaneously discovered the great potential that this chemistry offers from the synthetic biological point of view.3, 4, 5 Amongst bio‐orthogonal reactions, [4+2] cycloaddition between 1,2,4,5‐tetrazines (s‐tetrazines, Tz) and various dienophiles (inverse‐electron‐demand Diels–Alder (IEDDA) cycloaddition, also often termed the Carboni–Lindsey reaction) deserves a special credit (Scheme 1).6
Scheme 1.
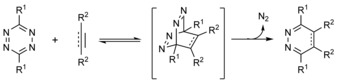
General scheme of IEDDA reactions.
Although the synthesis of the reagents is often laborious, the excellent reaction rates, biocompatibilities and selectivity make up for the pain and provide a catalyst‐free chemistry that really meets the demands of chemical‐biology‐driven exploration of molecular biology processes. There are several publications that provide guides to different segments of the world of tetrazine‐dienophile‐based bioconjugation techniques.7 Here we place special emphasis on tetrazine‐based schemes for labelling of biopolymers with manipulated building blocks and do not go into detailed explanations of, for example, reaction kinetics or synthetic advancements in reagents because these are summarized well in excellent recent reviews.7
1.1. Tetrazines
Over the years numerous tetrazines have been developed. The driving force behind these developments was to find the fine line between reactivity and physiological stability. Detailed theoretical and synthetic studies investigated the reactivities of tetrazines. Although the pioneering works always discussed these reactions strictly within the context of IEDDA reactions, recent theoretical studies questioned the validity of this view by pointing out that reactions between tetrazines and suitable dienophiles can also proceed in a normal electron‐demand manner.8 However, in line with the frontier molecular orbital (FMO) theory of IEDDA reactions, the reactivity of tetrazines is increased when they possess lowered LUMO energies as a result of the introduction of electron‐withdrawing substituents at their 3‐ or 6‐positions, or both.7 Despite the large number of tetrazines developed, only a few biologically stable, routinely accessible tetrazine frames are used. Scheme 2 summarizes a set of bio‐orthogonally relevant tetrazine derivatives (1–6) that are routinely applied in labelling schemes. Besides electronic features, steric effects should also be taken into account, as recently pointed out by others and by ourselves.9 There are cases in which steric demand of the substituents overrules the predicted order of reactivities.10 It should also be noted that despite the fact that IEDDA reactions are only slightly affected by the polarity or ionic strength of the solvent, the reaction is greatly accelerated in aqueous media, due to hydrophobic effects.
Scheme 2.
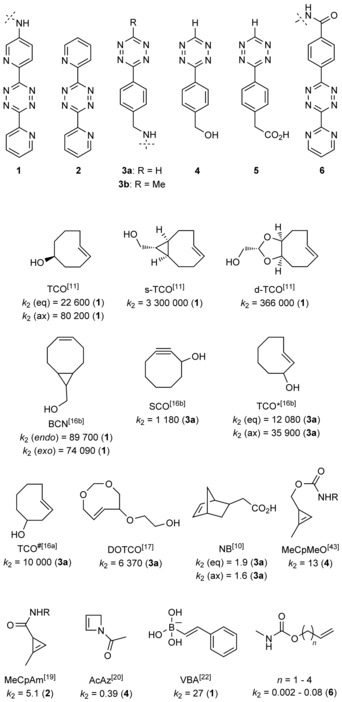
Dienophiles, with second‐order rate constants (k 2 [m −1 s−1] in water) measured with tetrazines indicated in parentheses.
1.2. Dienophiles
According to the FMO theory of IEDDA reactions, use of dienophiles with raised HOMO energies accelerates reaction rates.7 This is generally achieved by placing electron‐donating substituents on the dienophile or by introducing strain. It can be said that fine tuning reactivity and stability in IEDDA schemes in combination with the few generally applied tetrazines is easier from the dienophile side. In general, strained alkenes or alkynes are used (e.g., norbornenes, cyclooctynes, trans‐cyclooctenes (TCOs) and cyclopropenes); however, vinylboronic acids have recently also been reported to be suitable moieties for dienophiles. Just like tetrazines, these dienophiles should meet the criteria for stability and reactivity under physiological conditions. Detailed kinetic investigation by Sauer in the late 1990s indicated that the fastest IEDDA reactions are offered by dienophiles with strained eight‐membered ring systems.3a
As a consequence, the largest group of dienophiles is represented by cyclooctynes and TCOs. Besides participating in IEDDA reactions, these strained ring systems are also prone to react with nucleophiles, especially with free thiols such as GSH, not to mention that, for instance, TCOs can isomerize to their corresponding, much less reactive cis forms either spontaneously or by a thiol‐mediated route. This sometimes suggests the use of less reactive dienophiles in combination with more reactive tetrazines or vice versa.
Nevertheless, the physiological stability of dienophiles should always be investigated individually prior to use for a given purpose. The very first bio‐orthogonal examples were presented almost simultaneously by the Weissleder and the Fox groups. The former group applied a commercially accessible norbornene scaffold in IEDDA‐based labelling schemes with reaction rates at around k 2=1.9 and 1.6 m −1 s−1 for the two isomers of norbornene.5 Fox and co‐workers, on the other hand, used functionalized dipyridyl‐tetrazine 1 (Scheme 2) and a diastereomeric mixture of 5‐hydroxy‐TCO (TCO), which showed impressive second‐order rate constants [k 2=(22 600±40) and (80 200±200) m −1 s−1 for the two diastereomers].4, 11 They also developed a continuous‐flow photochemical apparatus for the efficient conversion of cis‐cyclooctenes into trans‐cyclooctenes.12 Later, the same authors discovered that forcing the crown/“half‐chair” conformational equilibrium of the TCO ring towards the more reactive “half‐chair” conformation through cis‐ring fusion (to give s‐TCO, Scheme 2, a bicyclo[6.1.0]non‐4‐ene) greatly enhanced reaction speed [k 2=(3 300 000±40 000) m −1 s−1].11, 13 A more water‐soluble bicyclic TCO in which the TCO ring was fused to a dioxolane unit (d‐TCO) was also introduced by the Fox group.11 Very importantly, the two accessible diastereomers of d‐TCO did not show such large differences in reaction rates towards the tetrazine as diastereomers of the original TCO (Scheme 2).
Although the use of these TCOs was demonstrated on chemically modified isolated proteins, the need for TCO‐bearing noncanonical amino acids (ncAAs) initiated the development of new TCO derivatives. At this point cyclooctyne (e.g., SCO, Scheme 2) and bicyclo[6.1.0]non‐4‐yne (BCN) frames, formerly developed for strain‐promoted azide–alkyne click reactions (SPAAC) also started to play a role in IEDDA schemes.14, 15, 16 To increase stability and applicability in biological assays, Lemke and Schultz developed new variants of the original TCO frame. These derivatives carried the hydroxy substitution closer to the double bond (TCO* and TCO#) than in TCO.16 Stability studies indicated that TCO* is much more stable than TCO#. Kinetic studies showed that TCO* reacts with a sterically less hindered tetrazine (H‐Tz, 3 a) with second‐order rate constants of around 35 900 and 12 080 m −1 s−1 for the axial and equatorial atropisomers, respectively.16b With a sterically more hindered tetrazine (Me‐Tz, 3 b) this was found to be two orders of magnitude slower for the more reactive (yet more stable) isomer, which could be accessed selectively.16b
Although not closely related to the topic of this review, it is worth mentioning that TCO* has also been used as a decaging scaffold in the context of IEDDA‐based release of a chemotherapeutic agent or in the site‐specific unmasking of the ϵ‐amino group of Lys residues in proteins.16c,16d
Hydrophilicity of TCOs is a quite an issue, especially in live‐cell labelling experiments because removal of unincorporated excess hydrophobic TCO‐bearing tags from the cytosol requires intensive and numerous washing cycles. This is in particular a problem when high concentrations of TCOs are used: that is, during metabolic or genetic‐code‐expansion‐based incorporation of modified building blocks. Labelling of the remaining TCOs bound non‐specifically to hydrophobic surfaces leads to background signal, thus lowering signal‐to‐noise ratios. In a joint work with the Lemke group we have demonstrated that use of the hydrophilic TCO DOTCO (Scheme 2) can eliminate this problem.17 It is worth mentioning that DOTCO showed only moderate reactivity towards the reactive 3 a [k 2=(6370±80) m −1 s−1], which could be partly due to reduced hydrophobic effects.
The requirement for lower perturbation of biological structures raised the need for the development of smaller dienophiles. This led to the synthesis of cyclopropenes for use as dienophiles in IEDDA schemes. Devaraj and, later, Prescher investigated the structure–reactivity relationships of differently functionalized cyclopropene scaffolds.18, 19, 43 Cyclopropenes tend to polymerize and also to be prone to attack by nucleophiles. Both groups concluded that 1‐methyl substitution of the cyclopropene framework overcomes this problem efficiently, providing suitable platforms without compromising reactivity, although the IEDDA reactivity of cyclopropenes towards tetrazines is orders of magnitudes lower than that of eight‐membered ring systems. A further dienophile moiety is represented by acylazetines, in which the strain is less dominant, but the enamine moiety facilitates reaction with tetrazines to reach reaction rates similar to those achieved with cyclopropenes.20
In some instances, simple vinyl‐derived building blocks have also been found to be suitable for biomolecular tagging schemes with tetrazines.21
A very recent work by Bonger et al. demonstrated that vinylboronic acids are versatile scaffolds in IEDDA chemistry.22 The reactivity of the vinyl group is boosted by the weakly electron‐donating boronic acid moiety, and this is even more profound in aqueous media, in which the boron hybridizes to a negatively charged, strongly electron‐donating group. Detailed mechanistic and kinetic studies revealed a vinylboronic acid derivative that showed a second‐order rate constant k 2=(27±2) m −1 s−1 in combination with a derivative of 6. Moreover, the boronic acid moiety renders this dienophile stable, hydrophilic and nontoxic. It should be noted, however, that vinylboronic acids have not yet been applied in live‐cell labelling experiments, due to their possible side reactions with vicinal diols (e.g., sugars). Scheme 2 summarizes the structures of these dienophiles together with second‐order rate constants of IEDDA reactions with the indicated tetrazines.
From these examples it can be concluded that the vast variety of possible tetrazine/dienophile combinations allows researchers to choose the ones that best suit the scope of the given investigation.
1.3. Small‐molecule probes in IEDDA labelling schemes
The beneficial features of reactions between tetrazines and, mainly, strained alkenes/alkynes have borne fruit in various biomolecular labelling schemes. The fast and efficient reactions enable the use of labels with short half‐lives and even the tagging of species with short circulation times or rapid turnovers. Different non‐invasive imaging methods have used Diels–Alder‐based bio‐orthogonal labelling schemes in cells or in live animals to identify or localize biomolecules, to visualize proteins at super‐resolution level, to detect miRNA, to image tumours or to track systemic metabolism processes.
Bio‐orthogonal labelling methods follow a two‐step, sequentially driven scheme. Firstly, a bifunctional unnatural handle, called a chemical reporter, is installed onto the biomolecule of interest. The chemical reporter that carries a bio‐orthogonal function (i.e., a dienophile or a tetrazine in the context of this review) can be incorporated into the biomolecule of interest either synthetically (e.g., in the case of pretargeting antibodies) or by means of enzymatic tagging, genetic code expansion or the metabolic machinery of the system investigated. After attachment of the bio‐orthogonal function, the biomolecule modified with the chemical reporter tag can be selectively targeted with small‐molecule probes incorporating the complementary bio‐orthogonal function.
Radiolabels and fluorescent labels are generally used in IEDDA‐based bio‐orthogonal labelling schemes; however, some examples also use this chemistry to introduce magnetic resonance (MR) contrast agents. In the case of radiolabelled markers, tetrazine chemistry has been used by several groups to install 18F‐ or 11C‐labelled PET tracers onto bioactive moieties or drugs.23 The fast reaction speeds have allowed straightforward introduction of the radiolabels in low dosages. Other radiolabels such as metallated probes (e.g., 64Cu, 111In, 177Lu, 68Ga) have also been applied. In these cases, together with that of Gd‐based MR contrast agents, chelators (e.g., DOTA) are conjugated to cyclooctenes or tetrazines.24
Amongst imaging modes, however, fluorescent labelling by means of bio‐orthogonal installation of small‐molecule fluorophores is by far the dominant method of choice. The popularity of fluorescence‐based detection could be due to its relatively easy and cheap nature or to its excellent sensitivity, together with its good spatial and temporal resolution. The poorer contrast of fluorescence images can be improved either by combination with, for example, PET imaging or by means of super‐resolution fluorescence technology. In the context of fluorescence imaging techniques, the sensitivity and resolution of imaging are most commonly limited by autofluorescence of naturally occurring fluorophores and nonspecific background fluorescence of unreacted probes.25 More and more attempts to overcome these problems have appeared lately, as well as attempts to design fluorescent labels that are suitable for super‐resolution technologies. Fluorescent probes possessing large Stokes shifts or those that can be excited in the red/near‐infrared regime can diminish autofluorescence, whereas so‐called fluorogenic dyes efficiently reduce nonspecific background signal. Besides simple probes in which a tetrazine unit is linked flexibly to a fluorescent label, the tetrazine moiety is also suitable for the construction of fluorogenic dyes because it is capable, in a suitably designed setup, of efficiently quenching fluorescence.16, 26 Upon reaction with their target dienophile the quenching effect is abolished and the fluorescence is reinstated. Although the exact quenching mechanisms in these examples were only assumed in the works by the authors, and the exact means of quenching are still debated, two mechanisms are proposed in general. Tetrazines are able to diminish the fluorescence of fluorescent cores either by FRET or by a through‐bond energy transfer (TBET) process (Scheme 3).27, 28
Scheme 3.
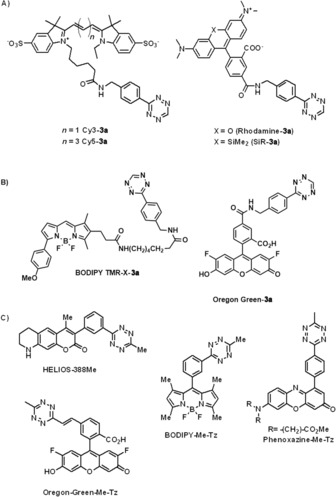
Some representative examples of synthetic small‐molecule fluorescent tetrazine probes. A) No spectral constraints between the fluorophore and the tetrazine linked together with a flexible linker. B) Fluorogenic FRET probes in which the spectrally matching fluorophore is linked to the tetrazine through a flexible linker. C) Fluorogenic TBET probes in which the fluorophore is linked to the tetrazine through a rigid linker.
In FRET‐based quenched systems the fluorescent core is linked to the tetrazine through a flexible linker. Besides other criteria (e.g., distance, alignment of transition moments), FRET systems require considerable overlap between the emission spectrum of the fluorophore and the absorption band of the tetrazine (typically falls between 520–540 nm). In TBET‐based fluorogenic probes the fluorophore and the tetrazine are attached together through a rigid, conjugated, but otherwise electronically decoupled (twisted) linker. Most importantly, quenching through a TBET mechanism does not require any spectral match between the fluorophore and the quencher, so virtually any kind of fluorophore can be used. Although the exact mechanism of TBET is still disputed and predictions are limited, a TBET mechanism, with optimal design, is able to reduce fluorescence more efficiently than a FRET one, and the increase in fluorescence signal upon conjugation can reach as much as three orders of magnitude (10–50‐fold vs. 100–10 000‐fold enhancements of fluorescence through FRET and TBET, respectively). Finally, it should be noted that in TBET‐based systems a parallel FRET effect is also existent, though this is a much slower process.
In the subsequent sections we show recent advancements in the use of tetrazine‐ or dienophile‐modified biopolymeric building blocks as chemical reporters, together with examples of complementarily functionalized labels, with emphasis on fluorescent labels.
2. Nucleotide Labelling
Nucleic acids (DNA and RNA) have essential roles in biological systems; they carry the genetic material of each cell of a living organism, so their study, detection and modification is a major goal of chemical biology. Nucleic acid modifications by means of solid‐phase oligonucleotide synthesis from phosphoramidite building blocks, of polymerase‐assisted biochemical pathways or of post‐synthetic methodologies are versatile tools in exploring, elucidating and imaging DNA/RNA functions.
The field of IEDDA reactions with bio‐orthogonalized nucleic acids was pioneered by the Jäschke group, who described the very first examples of DNA modification through IEDDA reactions based on norbornene‐modified deoxyuridine dienophiles (e.g., building block 7, Scheme 4) that were effectively incorporated into oligonucleotides during solid‐phase synthesis.
Scheme 4.
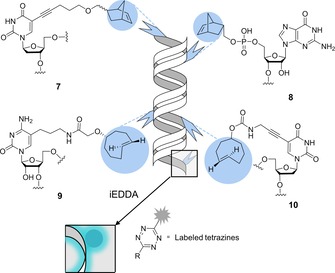
Bio‐orthogonalized building blocks for chemical or enzymatic incorporation of strained dienophiles. Post‐synthetic modification of DNA and RNA by IEDDA cycloaddition with various tetrazines bearing fluorophores, biotin or boronic acid groups.
These manipulated oligonucleotides were used as primers to amplify a double‐stranded 109‐mer DNA through a polymerase chain reaction (PCR) approach. The workflow yielded products modified with one, two or three norbornene‐based dienophiles that served as handles for biotinylation with a biotinylated conjugate of tetrazine 6. This approach was further expanded to label RNA, in an approach based on norbornene‐guanosine building blocks (e.g., building block 8 in Scheme 4) that were incorporated either chemically by solid‐phase synthesis into a 19‐mer RNA or enzymatically by a 5′‐initiation protocol with T7 RNA polymerase.29 The same group also reported the first combination of the fully orthogonal CuAAC and IEDDA for simultaneous site‐specific double‐labelling of DNA oligonucleotides, in a work that emphasized the superior reactivity of TCO‐based dienophiles.30
Kath‐Schorr et al. demonstrated that IEDDA could be effectively performed on RNA in mammalian cells. Their work included the incorporation of a norbornene‐guanosine derivative (building block 8, Scheme 4) as a phosphoramidite at specific positions in RNA during solid‐phase synthesis. To validate the concept, cells were transfected with a manipulated small interfering RNA (siRNA; the sense strand bearing the norbornene moiety, the antisense strand containing an ATTO647 fluorophore) and subsequently incubated with a fluorescent conjugate of tetrazine 3. Showing that the reactivity depends not only on the tetrazine derivatives but also on steric effects, diffusion and cell permeability, the labelling of the fixed cells proved to be successful because the product of the click reaction resulted in strong turn‐on fluorescence that colocalized with the ATTO647‐labelled siRNA. They concluded that the methodology might be a useful tool for detection of RNA functions in cells.31
Royzen et al. described another general approach to fluorescent labelling of RNA strands using a cytidine analogue derivatized with the TCO group (building block 9, Scheme 4). The TCO moiety was attached to position 5 of the nucleobase to preserve enzymatic recognition, and the resulting derivative was incorporated into the model strand by use of T7 RNA polymerase. Fluorescein‐modified tetrazine 3 b served as tag to label the synthetic RNA strand.32
Polymerase‐mediated incorporation of TCO‐modified (at position 5′) thymidine triphosphate (building block 10, Scheme 4) into DNA was reported by Wang et al. Through IEDDA reactions with suitably derivatized tetrazines, incorporation of a boronic acid group or a fluorophore was also demonstrated. Notably, boronic‐acid‐labelled DNA can be a new candidate for aptamer selection against carbohydrates and glycoproteins.33 With a similar synthetic approach, Brown et al. incorporated two TCO‐modified deoxyuridine moieties with different linkers between the TCO and uracil base along with a 5‐azidomethyl‐2′‐deoxyuridine unit into DNA. On comparison, the 5‐azidomethyl derivative proved to be a better substrate for polymerases than the two TCO‐bases; however, the efficiency of the IEDDA reactions between the TCOs and Cy3‐3 dye was found to be much more satisfactory than that of the strain‐promoted azide–alkyne click reaction between the azide and a BCN‐Cy3 fluorophore.34
The most compelling argument for the addition of TCO or norbornene motives into DNA/RNA is the remarkable reaction kinetics these dienophiles can undoubtedly offer. Still, incorporation of these rather bulky substituents into nucleosides might inhibit their natural cellular metabolism and thus cause DNA damage. Luedtke et al. addressed this challenge by identifying 5‐vinyl‐2′‐deoxyuridine (VdU, building block 12, Scheme 5) as the smallest possible moiety that could transform nucleosides into efficient dienophiles and thus be a potential metabolic label for DNA without perturbing any original function or causing cell cycle arrest. Although the rate of the reaction between the vinyl group and the tetrazine was found to be slower than those reported for strained olefins, it was quite comparable with those of SPAAC reactions. When added to the medium, the probe was selectively incorporated by endogenous enzymes into the genomes of replicating cells, where it was detected by alkene–tetrazine ligation with a TAMRA‐1 conjugate, resulting in rapid intranuclear staining. It was also established that VdU is incorporated selectively into cellular DNA and not RNA, because there was no evidence of detectable VdU labelling in the presence of the DNA synthesis inhibitor aphidicolin. The results underlined that strained cycloalkenes are not always prerequisites for effective intracellular labelling.35 Marx et al. reported a similar methodology, by synthesising deoxyuridine (dvinU) and deoxydeazaadenosine (dvinA) derivatives each bearing a vinyl group that points towards the major groove of the DNA duplex to be accessible by labelling reagents. DvinA (building block 11, Scheme 5) was found to be more reactive than dvinU, having a second‐order rate constant of k 2=0.010 m −1 s−1, thus implying that IEDDA reaction of vinyl groups are comparable with SPAAC or Bertozzi–Staudinger ligations in terms of kinetics. DvinA was demonstrated to substitute natural dATP in PCR completely, and the DNA product was effectively tagged with a biotin‐6 conjugate.36
Scheme 5.
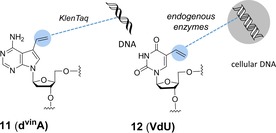
Vinyl‐type building blocks. VdU (12) was demonstrated to be an efficient metabolic probe for cellular DNA imaging. DNA modified with DvinA (11) was tagged with biotin‐tetrazine conjugate.
Like vinyl derivatives, cyclopropenes are also small moieties; however, they possess better reactivity, which makes them potent candidates for DNA/RNA building blocks.
Kath‐Schorr and Eggert described the first cyclopropene‐modified ribonucleotide, which was introduced into RNA at predefined sequence positions. The fully replicable and transcribable unnatural TPT3‐dNAM base pair system was modified with a methylcyclopropenyl system on the ribose TPT3 unit (TPT3CP, building block 13, Scheme 6).37 Enzymatic in vitro transcription led to successful site‐specific incorporation of the cyclopropene moiety, followed by labelling with a fluorophore–3 a conjugate. In vitro transcription and post‐transcriptional labelling of a tRNA in the anticodon loop was also demonstrated.38
Scheme 6.
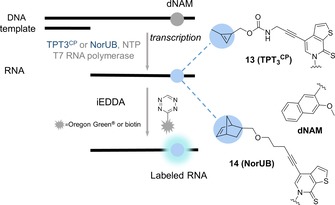
Building blocks from the toolbox of unnatural base pair genetic code expansion.
A similar, T7‐assisted in vitro transcription based on the same unnatural hydrophobic base pairing system was used to modify RNA oligonucleotides with norbornene moieties (NorUB, building block 14, Scheme 6). Both of these examples could offer a facile tool for studying RNA constructs with complex folding pathways, such as ribozymes and aptamers.39
As illustrated by the above examples, nucleic acid labelling through IEDDA cycloaddition mostly proceeds through incorporation of the dienophile into the DNA or RNA, followed by post‐synthetic attachment of a tetrazine probe. The reason behind this is the general instability of tetrazines under the conditions of chemical DNA synthesis. Still, the incorporation of tetrazines into DNA is an open possibility, as demonstrated by Wagenknecht and Kele in a study that revealed that steric demand of substituents plays a prominent role in the Diels–Alder reactivity of electron‐deficient tetrazines. The tetrazine‐modified building block 15 (Scheme 7) was incorporated into a DNA sequence, followed by on‐bead labelling with cyclooctyne‐modified fluorescein.10 The same authors also reported that tetrazine building blocks 16 and 17 can be incorporated as 2′‐deoxyuridine derivatives into oligonucleotides by using standard DNA polymerases (Hemo KlenTaq, KOD XL, Vent, Deep Vent). The resulting full‐length primer extension products were labelled with cyclooctyne‐modified fluorophores.40
Scheme 7.
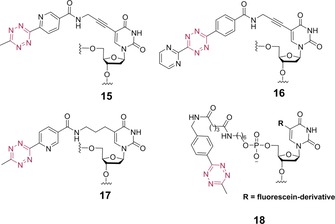
Tetrazine‐based nucleic acid building blocks.
The area of oligonucleotide‐templated ligation reactions has been extended to IEDDA applications by the Devaraj group. They developed DNA/RNA template‐dependent fluorogenic ligation between oligonucleotides capped at the 5′‐end with a quenched fluorophore‐tetrazine unit (tetrazine building block 18, Scheme 7) and oligonucleotides bearing a cyclopropene unit (building block 19, Scheme 8) at the 3′‐end. The close proximity of the reactive counterparts and the presence of an antisense oligonucleotide template resulted in an increase in the effective molarity (50–120 mm), a condition that allowed reactions to proceed rapidly only in the presence of sequence‐specific targets, thus resulting in large increases in fluorescence.41
Scheme 8.
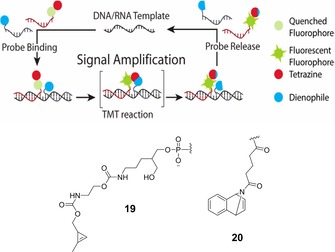
Schematic and building blocks for template‐driven oligonucleotide labelling through modified DNA ligation, catalysed by hybridization against a template strand. Reprinted from ref. 42, with permission. Copyright: 2014, American Chemical Society.
An interesting aspect of this template‐driven IEDDA application is that the product of cycloaddition has higher affinity for the oligo template than the corresponding precursors, thus impeding further signal amplification or reaction turnover. Regarding this disadvantageous aspect as requiring further improvement, Devaraj et al. used a 7‐azabenzonorbornadiene derivative (building block 20, Scheme 8) with an alkenyl‐tetrazine fluorogenic probe; this demonstrated better performance, enabling imaging of microRNA in living human cancer cell lines (Scheme 8).42
3. Fluorescent Labelling of Lipids
Lipids (waxes, sterols, vitamins, glycerides and phospholipids, among others) are important mainly in energy storage and in signalling pathways and in acting as structural elements of cell membranes. Although several examples have described the use of bio‐orthogonal chemistry in various lipid imaging schemes, the number of those involving IEDDA reactions is surprisingly low.
Devaraj et al. studied the suitability of stabilized, yet reactive cyclopropene tags (building block 21, Scheme 9) for live‐cell imaging, by labelling modified phospholipids with fluorogenic dyes. The cyclopropene handles elicited quick fluorescent responses from the quenched tetrazine dye BODIPY–4 a, making it possible to visualize efficiently the distribution of the tagged phospholipids in SKBR3 cells.43
Scheme 9.
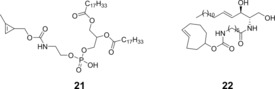
Lipid building blocks for tagging phospholipids (left) or visualising the Golgi apparatus (right).
A remarkable lipid‐based approach to visualizing the structure and dynamics of the Golgi apparatus also relied on the IEDDA reaction. This strategy used the TCO‐modified ceramide lipid Cer‐TCO (building block 22, Scheme 9) and fluorophore‐tetrazine conjugate SiR–3 a together with their click product, the extremely photostable “vital dye” Cer‐SiR. The study concluded that Cer‐TCO accumulated in the Golgi without disturbing cell morphology and then reacted with SiR‐3 a, thus enabling selective visualization of the Golgi apparatus in HeLa cells co‐expressing GFP‐modified fusion protein GaLNAcT2 as Golgi marker for colocalization.
Monitoring of trafficking both through and within the Golgi by means of a ratiometric inside/out assay verified that the organelle remained functional after IEDDA because the addition of Cer‐TCO and SiR‐3 a had no effect on the mobility of proteins inside the Golgi or on cargo traffic from the ER to the plasma through the apparatus. Cer‐SiR also proved to be extremely photostable and thus an ideal candidate for stimulated emission depletion (STED) microscopy (Figure 1).26b
Figure 1.
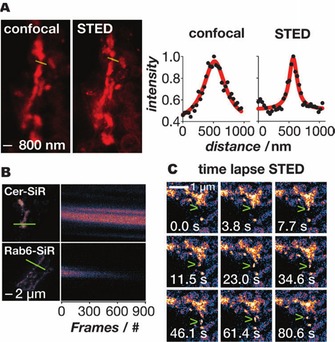
Super‐resolution imaging of the Golgi in live cells with the aid of Cer‐Sir. A) Confocal and STED images of the Golgi in live cells treated with Cer‐TCO and SiR‐3 a. Line traces through the Golgi (yellow) show the greatly improved resolving power of STED (right‐hand panel). B) Kymo‐graphs (line profile vs. time) of fixed cells imaged by STED in which the Golgi were labelled with Cer‐SiR or Rab6‐SNAP‐SiR (Rab6 is a Golgi‐targeted protein); note that the signal decays much more quickly when the protein is labelled with SiR. C) Time‐lapse STED of vesicle budding and trafficking out of the Golgi (green arrowhead). Reproduced from ref. 26b, with permission. Copyright: 2014, John Wiley & Sons, Inc.
4. IEDDA Labelling of Proteins through Genetic Encoding of Noncanonical Amino Acids
Proteins play central roles in the majority of biological processes: they function as receptors and transporters and also serve as enzymes or structural elements. Current methodologies for fluorescent labelling of protein targets rely mostly on fluorescent proteins (GFP, CFP, YFP, mCherry etc.). However, due to their large size (>20 kDa) and only moderate photostability, there is more and more interest in approaches based on small‐molecule fluorescent systems. A convenient method is the use of other fusion proteins such as self‐labelling tags or enzymes (e.g., SNAP‐tag, Halo‐tag, CLIP‐tag, tetracysteine‐tag). They are smaller than GFP analogues and enable the use of small‐molecule fluorescent probes with high specificity. An alternative labelling method makes use of engineered enzymes that recognize peptide sequences fused to the proteins of interest (POIs; e.g., phosphopantheinyl‐transferases, biotin ligase, lipoic acid ligase, sortase, etc.). Although these methods are suitable for site‐specific modification of proteins, introduction of shorter or longer peptide sequences into a POI can disturb its structure and/or function.44
Modification of proteins through incorporation of designer amino acids provides an increasingly useful tool for studying protein structure and function. Incorporation of amino acids modified with bio‐orthogonal functions provides a versatile platform for fluorescent labelling through Diels–Alder cycloaddition. Amino acids bearing unconventional functional groups have been incorporated into proteins by a range of methods, including solid‐phase synthesis, native chemical ligation, and in vitro translation.45 However, these methods are limited only to in vitro applications. In live cells, modified amino acids can be incorporated into proteins in a residue‐ or a site‐specific manner. Numerous applications of copper‐catalysed 1,3‐cycloadditions in residue‐specific unnatural amino acid incorporation to label whole proteomes of cells have been reported. To the best of our knowledge, however, IEDDA reactions have not yet been applied in this context.46 Here we review recent developments in site‐specific IEDDA protein labelling through genetic encoding of ncAAs.
In genetic code expansion technology, amino acids modified with a unnatural—bio‐orthogonal, for example—function are introduced into the growing polypeptide chain during translation in response to an assigned, usually amber, STOP codon. To insert large functional groups appended from the amino acid, engineered aminoacyl tRNA synthetase/tRNA pairs are used. The method and the development of genetic code expansion have been extensively reviewed by others, so we focus only on IEDDA‐related works.44b, 47 Encoding of ncAAs into proteins is achieved with the aid of several engineered tRNA synthetase(RS)/tRNA pairs: the Methanococcus jannaschii tyrosyl‐RS/tRNA pair, the Escherichia coli tyrosyl‐RS/tRNA pair and the E. coli leucyl‐RS/tRNA pair. These pairs each recognize one of the 20 natural amino acids; therefore, their active sites have to be mutated for specificity towards the particular ncAA by directed evolution approaches.
On the other hand, pyrrolysine RS/tRNA (PylRS/tRNAPyl) from the prokaryote Methanosarcina species customarily recognizes pyrrolysine, an amino acid not used by most prokaryotic and eukaryotic organisms, and it is orthogonal in bacteria, yeast, eukaryotic cells and animals. Wild‐type and engineered PylRS/tRNAPyl pairs with expanded binding pockets are used to incorporate numerous bio‐orthogonalized ncAAs into proteins.48
The first example of genetic encoding of a strained‐dienophile‐modified amino acid was reported in 2011 by Plass et al.49 Simple cyclooctynyllysine (simple cyclooctye, SCO) derivatives 23 and 24 (for structures of ncAAs, see Scheme 10) were encoded into GFP modified with an amber STOP codon (GFPY39TAG) by using an engineered Y306A/Y384F mutant PylRSAF/tRNAPyl pair in E. coli. This two‐site mutation was introduced to support recognition of the bulkier side chain on the amino acid.50 The first IEDDA reactions with genetically encoded ncAAs were presented in 2012 by the same group. Cyclooctyne (23), norbornene (25 and 26) and trans‐cyclooctene (27) residues were encoded into GFP by use of the PylRSAF/tRNAPyl pair in E. coli.51 Intracellular fluorescent labelling was performed inside fixed mammalian cells on maltose binding protein (MBP) modified with 27 and fluorescent tetrazine conjugate Cy5‐6. Later, live‐cell labelling of intracellular GFPY39TAG→27 was established as well with fluorogenic siliconrhodamine SiR‐3 b derivative in E. coli.52 The fluorogenicity of SiR probes relies on spirolactone formation of the carboxylate group, which quenches the fluorescence in a polarity‐dependent manner. Not only the above mutant, but also wild‐type PylRS/tRNAPyl could also be used to encode 25.53 Lang et al. described live‐cell membrane labelling of 25‐modified epidermal growth factor receptor (EGFR128TAGGFP) on the surfaces of mammalian HEK293 cells by using FRET‐type fluorogenic TAMRA‐1. Kaya et al. described the use of a triple‐mutant Y384F/Y306G/I405R PylRSFGR/tRNAPyl pair to encode 26 into human polymerase κ (hPolκ).54 In addition to reactions with nitrile imines and tetrazoles, they performed IEDDA labelling schemes on purified 26‐hPolκ.
Scheme 10.
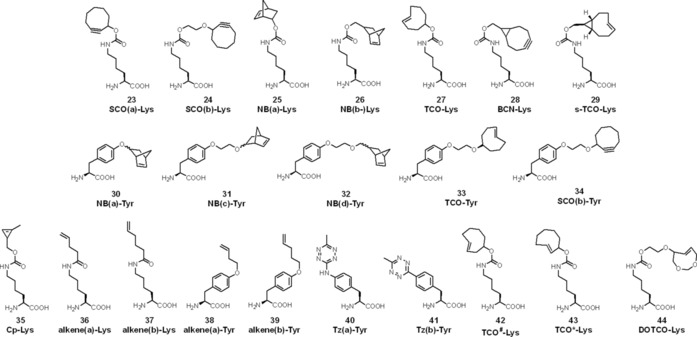
Structures of noncanonical amino acids used as building blocks for fluorescent protein labelling in IEDDA.
The Chin group discovered further engineered PylRS/tRNAPyl pairs capable of encoding TCO‐Lys (27) and BCN‐Lys (28).27b The lysine derivative of the fastest known TCO to date—s‐TCO‐Lys (29)—was also included in the study; however, due to its isomerization to the unreactive cis form in the thiol‐rich cellular environment, it was not possible to apply it in live‐cell labelling. TCO‐Lys (27), however, was successfully applied to label EGFR128TAG with TAMRA‐1, and BCN‐Lys (28)55 was also used in an intracellular labelling scheme of transcription factor protein jun with a cell‐permeable fluorescein–3 probe in HEK293 cell nuclei.
Although lysine is the most commonly used amino acid to which IEDDA tags are conjugated, tyrosine derivatives of norbornene, SCO and TCO are also known (30–34). The Tyr derivatives were genetically encoded into GFP by employing an evolved PylRS/tRNAPyl pair (Y306A, N346A, C348A, Y384F) in E. coli and applied in IEDDA with fluorescein–3 as well as in 1,3‐cycloaddition with hydrazonoyl chloride.56
The smallest strained dienophile system that has been encoded into proteins is cyclopropene, in Cp‐Lys (35).57 Although this system has slower reaction kinetics than cyclooctynes or TCOs, it was found to be applicable in proteomics because it causes minimal perturbation to protein structures and synthesis. Elliott et al. developed SORT‐M (stochastic orthogonal recording of translation with chemoselective modification) by use of tRNAPyls with modified sense‐decoding anticodons to perform proteome‐wide labelling.58 This approach was applied to Drosophila melanogaster to label and to image proteins with Alexa647‐1 in specific tissues at precise developmental stages. It allowed the identification of proteins synthesized in germ cells of the fly ovary without dissection. Genetic encoding of unstrained olefins was reported by Lee et al.59 Fluorescent labelling of terminal alkenyllysine (36 and 37) and ‐tyrosine (38 and 39) derivatives with fluorescein‐3 a probe was performed on E. coli outer membrane protein OmpX, though with sluggish reaction rate constants (≈3×10−2 m −1 s−1). In the array of biological building blocks suitable for IEDDA, dienophile‐appended derivatives are mostly the reaction partners of choice, due to their generally higher stability over tetrazines. Genetic encoding of stable tetrazine‐containing ncAAs, however, was described by the Mehl group. They developed the M. jannaschii tyrosyl tRNA synthetase/tRNACUA pair to incorporate 40 into GFP150TAG in E. coli.60 Kinetic studies showed moderate reaction rates with s‐TCO (880 m −1 s−1 in vitro and 330 m −1 s−1 in vivo) and d‐TCO isomers (95 and 99 m −1 s−1 in vitro), measured by the increment of quenched GFP fluorescence caused by FRET between the GFP and the tetrazine moiety (λ abs=520 nm).11, 60 Conjugate 40–GFP was labelled with diacetyl‐fluorescein‐s‐TCO. Removal of the amine linkage between the phenyl ring and the tetrazine led to the development of 41, with greatly improved stability and reactivity (72 500 m −1 s−1 in vivo with s‐TCO).61 The 41‐GFP conjugate was selectively labelled with TAMRA‐s‐TCO in E. coli cell lysate.
In spite of their high reactivity and stability, there are no reports on live‐cell labelling with tetrazine‐ncAAs. However, 40 was successfully applied in vitro in a site‐specific double‐labelling scheme.62 To encode different amino acids at different sites in a single protein, an orthogonal ribosome (ribo‐Q1) that recognizes quadruplet‐decoding PylRS/tRNA and amber‐decoding MjTyrRS/tRNA was developed.63 This enabled genetic incorporation of 40 and 25 into calmodulin (modified at position 1 with UAG amber and at position 40 with AGTA quadruplet codon) by using evolved MjTyrRS/tRNACUA and PylRS/tRNAUACU, respectively.62 The ncAAs did not cross‐react with each other in the protein and permitted selective double labelling with BODIPY‐TMR‐X‐BCN and BODIPY‐FL‐1 at the tetrazine and norbornene residues, respectively. FRET between the two fluorescent labels in the presence of increasing concentrations of urea allowed probing of calmodulin's structure and dynamics (Figure 2). This example demonstrated the power of using two IEDDA reactions in parallel fashion for site‐specific modification of proteins on purified samples.
Figure 2.
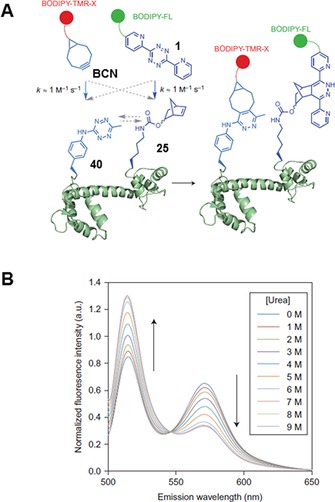
Double labelling of calmodulin. A) Amber suppression is used to encode 40, whereas 25 is incorporated in response to a quadruplet codon. Double labelling is performed with dyes based on BCN and 1, respectively. B) Probing calmodulin dynamics by detecting FRET in the presence of increasing urea concentrations. Reprinted in part and with permission from ref. 62. Copyright: 2014, Nature Publishing Group.
Nikić et al. applied dual‐colour labelling to live cells by taking advantage of different reaction kinetics of two sets of dienophile/tetrazine pairs.64 They synthesized TCO‐ncAAs (TCO*‐Lys, TCO#‐Lys) that showed improved stability and reactivity towards tetrazine probes 3 a and 3 b. Initial attempts combining strain‐promoted 1,3‐dipolar cycloaddition (BCN/azide) and IEDDA (TCO*/tetrazine) reactions for dual labelling led to high background caused by the slow reaction kinetics of azide–alkyne cycloaddition. However, combining TCO*‐Lys and SCO(a)‐Lys encoded in a time‐dependent manner in response to amber stop codons paved the way for dual‐colour IEDDA labelling by using fluorescent tetrazine conjugates. In pulse‐chase labelling experiments, Cy5‐3 b reacts selectively with TCO*‐Lys and shows no substantial reactivity with SCO(a)‐Lys, whereas Atto532‐3 a reacts with SCO(a)‐Lys rapidly.
The reactivity difference between SCO/TCO* and 3 a/3 b is due to the steric hindrance of the methyl group at the tetrazine moiety as well as the different carbamate conjugation positions.8 Furthermore, the two dyes allowed dual‐colour super‐resolution microscopy study of insulin receptors and virus‐like nanoparticles expressed at different times on the surfaces of mammalian HEK293T cells (Figure 3 A).
Figure 3.
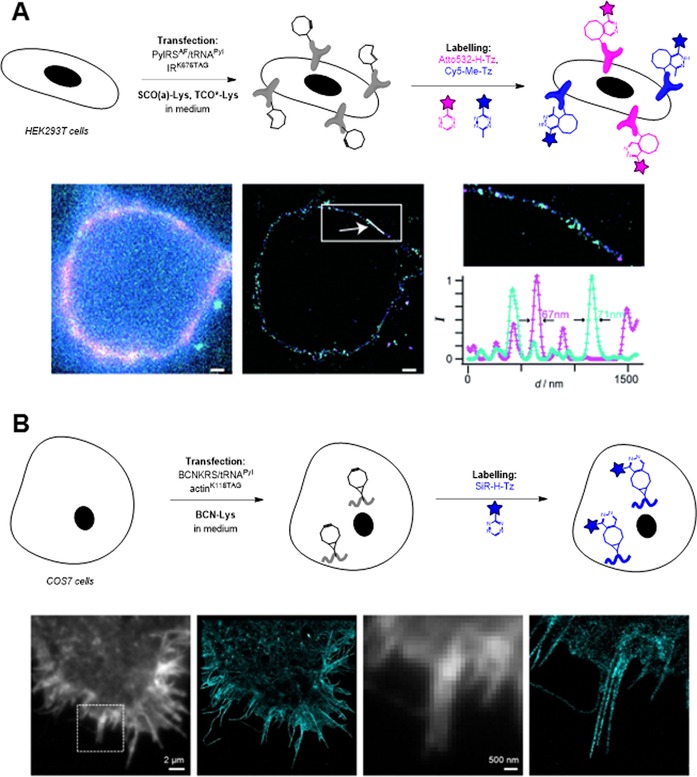
Site‐specific fluorescent labelling of proteins through IEDDA cycloaddition enables super‐resolution microscopy. A) Cell‐membrane dual‐labelling of insulin receptors: schematic two‐step cell labelling protocol and GSDIM images of double‐labelled insulin receptors. B) Intracellular labelling of actin filaments and subsequent STORM imaging. Figure 3 A is reproduced in part and with permission from ref. 64. Copyright: 2014, John Wiley & Sons, Inc. Figure 3 B is reprinted with permission from ref. 65. Copyright: 2015, American Chemical Society.
As mentioned above, IEDDA‐mediated protein labelling has been used to study cell‐surface proteins in several cases. Intracellular applications, however, are limited, mainly due to the hydrophobicity of the dienophile‐ncAAs. Residual ncAAs in the cytoplasm and the nuclei of live cells cause high background in fluorescent tetrazine labelling. To address this problem, Uttamapinant et al. applied BCN‐Lys in intracellular labelling of skeletal proteins actin and vimentin in combination with a fluorogenic SiR‐3 a dye (Figure 3 B).65 BCN‐Lys is slightly more hydrophilic than TCO*‐Lys, and this resulted in better clearance of the non‐incorporated ncAA from the intracellular compartments, but prolonged (>6 h) washes before fluorescent labelling were still required.
To extend the applicability of IEDDA cycloaddition in intracellular labelling, our group developed a set of new hydrophilic TCO and cyclooctyne derivatives, of which dioxo derivative DOTCO‐Lys (44, Scheme 10) showed high incorporation efficiency with PylRSAF/tRNAPyl.17 We performed a washout experiment to detect residual ncAAs in the cytoplasm (Figure 4). Efficient removal of hydrophobic TCO*‐Lys from the cytoplasm was not possible, even after 6 h of washing. Slightly more hydrophilic BCN‐Lys gave almost no background, but only after 6 h of washing. Hydrophilic DOTCO‐Lys, on the other hand, was easily removed from the cytoplasm after 5 min of washing. The greatly improved hydrophilicity of DOTCO‐Lys enabled fluorescent labelling of intracellular lymphocytic protein SLP76‐YFP40TAG with Cy5‐3 after only 15 min washing. Another approach to achieve low background in intracellular labelling involves application of fluorogenic tetrazine probes. We developed six phenoxazine‐tetrazine fluorogenic probes based on TBET‐quenching through phenyl or vinyl linkers (e.g., phenoxazine‐Me‐Tz, Scheme 3). Site‐specifically TCO*‐modified GFP tagging was established with these probes in vivo in mammalian HEK293T cells with very low background signal.28c
Figure 4.
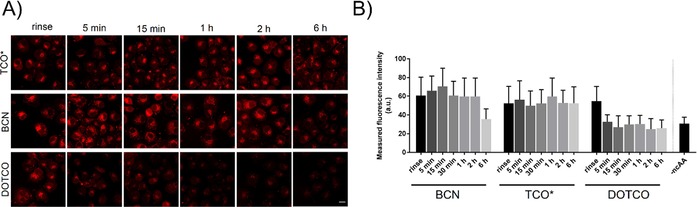
Washout assay of ncAAs: experimental demonstration that nonspecific background in intracellular labelling is directly linked to the hydrophilicity of the ncAA. A) Confocal microscopy images of non‐transfected COS7 cells washed for different times before labelling with TAMRA‐3. B) Quantitative analysis. Reproduced in part and with permission from ref. 17. Copyright: 2016, John Wiley & Sons, Inc.
A highly appealing advantage of using genetic code expansion technology combined with IEDDA cycloaddition is that it paves the way to imaging of proteins, which previously were impossible to visualize. In a very recent paper, Peng and Hang used the axial isomer of TCO*‐Lys to image interferon‐inducible transmembrane protein 3 (IFITM3), a vesicle‐associated membrane protein involved in host restriction of viruses.66 IFITM3 contains only 137 amino acids and so is only around half the size of fluorescent proteins such as GFP. Thus, it is not surprising that it has not been possible to image IFITM3 in live cells with GFP because fusion proteins disrupted IFITM3 cellular localization and antiviral activity. However, genetic encoding of TCO*‐Lys and subsequent labelling with BODIPY‐FL‐3 a led to successful imaging of localization and trafficking of IFITM3 in live cells (Figure 5).
Figure 5.
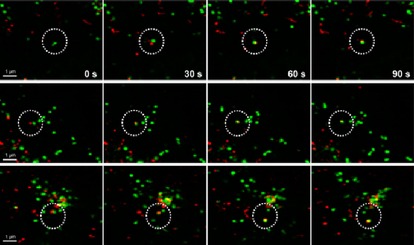
Timelapse imaging of the fusion process of IFITM‐containing vesicles with dextran particles. IFITM3‐TCO*‐Lys was labelled with BODIPY‐FL‐3 a, and dextran particles were labelled with pHrodo Red. Reprinted with permission from ref. 66. Copyright: 2016, American Chemical Society.
5. Fluorescent Glycan Labelling through Metabolic Engineering
Sugar‐modified proteins—that is, glycans—mediate several biological processes in cellular physiology: they play key roles in molecular recognition, protein trafficking, bacterial and viral infection and intercellular communication.67 It is known that the structures of glycans, which decorate all eukaryotic cell surfaces, change with the onset of cancer and inflammation.68 Unlike protein and nucleic acid production, which is genetically encoded, glycosylation is a posttranslational modification. Thus, techniques developed to label nucleic acids and proteins are not directly transferrable to label glycans. Lectins (receptor proteins for glycans) and antibodies can be used to probe glycosylation at a global level. However, they are limited by their low specificity, low affinity and lack of permeability. Besides, they are restricted to in vitro applications. We provide a brief overview of the recent developments in chemical probing of glycans through IEDDA cycloaddition, which have enabled studies in living systems through modification of monosaccharide residues.
Metabolic oligosaccharide engineering (MOE) involves chemically synthesizing a monosaccharide analogue and feeding cells with this unnatural monosaccharide, which is processed similarly to its native counterpart and integrated into cellular glycans. This approach depends on the selection of a reporter group that can readily be installed onto monosaccharide substrates with minimal perturbation to their structures. The reporter is detected in a second step through bio‐orthogonal chemistry. While powerful, this strategy has been limited to only a handful of functional groups. There are numerous examples of azide‐, alkyne‐ and ketone‐modified unnatural sugars and their subsequent bio‐orthogonal labelling with alkynes, azides and soft nucleophiles (e.g., through Bertozzi–Staudinger ligation), respectively.69 Because of their higher reaction rates, IEDDA reactions have found numerous applications in glycan labelling schemes as well. The first IEDDA reaction performed on glycans was employed by Patterson et al.,18 who showed that methylcyclopropene units could be directly conjugated to N‐acetylneuraminic acid (NeuAc) and incorporated into cell‐surface glycans. Jurkat cells were incubated with 9‐Cp‐NeuAc for 24–48 h and subsequently treated with a biotin‐1 conjugate (100 μm for 1 h; for structures of unnatural sugars, see Scheme 11). Subsequent avidin staining allowed examination of glycan labelling by flow cytometry. Although the measured reaction rates were slow—in the 0.05 m −1 s−1 range, for example—they are comparable with those of the SPAAC and Bertozzi–Staudinger ligation reactions.
Scheme 11.
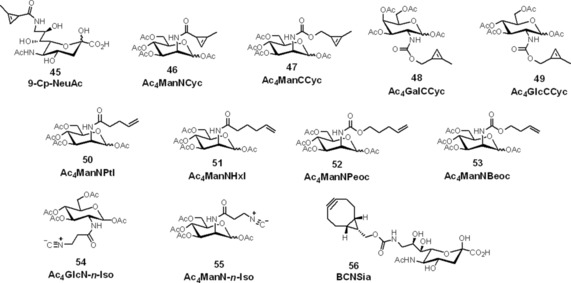
Structures of bio‐orthogonalized monosaccharides for fluorescent glycan labelling through IEDDA cycloaddition enabled by metabolic oligosaccharide engineering.
Shortly after, Cole et al. reported on a cyclopropenylated derivative of mannosamine, a metabolic precursor for sialic acid.70 Peracetylated N‐acyl cyclopropene mannosamine (Ac4ManNCyc, 46, Scheme 11) was metabolized and efficiently incorporated by human cancer cells. Cyclopropene‐tagged cell‐surface glycans could subsequently be labelled with AlexaFluor488‐3 a and visualized by confocal microscopy.
These first examples described IEDDA‐mediated glycan labelling; however, in both reports low incorporation efficiency of cyclopropene‐sugars was a limiting factor. Indeed, the N‐acyl derivatization in 9‐Cp‐NeuAc and Ac4ManNCyc is located at the β‐carbon atom, though it has been reported that β‐N‐acyl substitution is not well tolerated by the sialic acid biosynthetic machinery.71 Moreover, electron‐withdrawing groups (e.g., amides) at C3 reduce reaction rates of cyclopropenes in IEDDA cycloaddition.
To improve cyclopropene glycan tagging rates, Späte et al. and Patterson et al. simultaneously reported cyclopropene‐mannosamine derivative Ac4ManCCyc (47, Scheme 11), with a carbamate linker.72 This slight change in the sugar structure greatly improved the metabolic incorporation efficiency, and the IEDDA reaction rate as well (0.99 m −1 s−1), leading to a 130‐fold improvement in fluorescent labelling efficiency. Furthermore, derivatization through direct conjugation of aminosugars with an activated cyclopropene unit improved synthetic availability. Visualization of Ac4ManCCyc‐modified glycans in mammalian cell surfaces by confocal microscopy was possible after only 5 min staining with 25 μm Cy3‐6. The IEDDA tagging scheme was extended to other monosaccharides. For example, visualization of mucin‐type O‐glycoproteins with Ac4GalCCyc and of O‐GlcNAcylated proteins with Ac4GlcCCyc were also performed; however, efficiency rates lagged behind those of sialic acid labelling.
Recently, Doll et al. used Ac4ManCCyc to study the glycosylation states of specific intracellular proteins [O‐GlcNAc transferase (OGT), transcription factor Foxo1, tumour suppressor p53, serine/threonine kinase Akt1, actin‐binding protein vinculin and calcium/calmodulin‐dependent protein kinase CAMK4] in live cells.73 The target proteins were tagged with GFP and overexpressed in HEK293T cells incubated in Ac4ManCCyccontaining medium. FRET between GFP and TAMRA‐3‐tagged glycan was detected by fluorescence lifetime imaging microscopy (FLIM). This approach revealed nuclear localization and regulation of Akt1 by O‐GlcNAcylation. This was the first demonstration of examination of protein glycosylation states within living cells, stressing the power of bio‐orthogonal chemistry.
A great advantage of cyclopropene‐tetrazine labelling is that it is reported to be orthogonal with azide–alkyne cycloaddition. This was exploited in targeting unique subsets of cellular glycans by dual‐colour labelling. For example, Späte et al. treated HEK293T cells with both Ac4ManCCyc (to target sialated structures) and Ac4GlcNAz (an azido derivative of GlcNAc).74a The cells were treated concurrently with Cy3‐6 and DIBO‐488 for 15 min. Clear membrane staining in both IEDDA and SPAAC channels indicated the orthogonality of the two reactions in glycan labelling (Figure 6). The same authors recently reported a series of norbornene‐tagged mannosamine derivatives suitable for metabolic engineering of glycan structures. They also demonstrated the dual‐colour labelling potential of these derivatives in combination with SPAAC chemistry.74b
Figure 6.
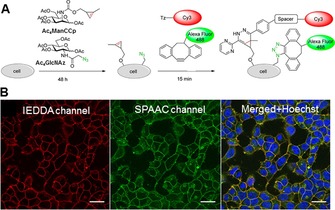
Double labelling of cell‐surface glycans through IEDDA and SPAAC. A) Labelling strategy, based on metabolic incorporation of Ac4ManCCp and Ac4GlcNAz and subsequent labelling with Cy3‐6 and AF488‐DIBO. B) Confocal microscopy images of doubly labelled cell‐surface glycans on HEK293T cells. Reprinted with permission from ref. 74a. Copyright: 2014, American Chemical Society.
To expand the scope of dienophiles, terminal alkenes were employed to visualize cell‐surface glycans by Niederweisser et al.75 Mannosamine derivatives with a pentenoyl (Ac4ManNPtl, Ac4ManNPeoc) or hexenoyl (Ac4ManNHxl) side chain were accepted by sialic acid biosynthetic enzymes and incorporated on the surfaces of HEK293T cells. Live‐cell tagging with biotin‐6 was performed, although the low reaction rates (0.02–0.04 m −1 s−1) required high concentrations (1 mm) of the tagging agent and a long incubation period (i.e., 6 h) before fluorescent labelling with AlexaFluor647‐biotin. The authors also synthesized carbamate‐linked monosaccharides (Ac4ManNPeoc), which were tolerated by the biosynthetic enzymes. In a later report, the same group optimized the alkene side chain of the carbamate‐linked mannosamine derivative to a butenyl moiety (Ac4ManNBeoc).21 Still, reaction rates could be tuned only up to 0.074 m −1 s−1 with tetrazine 6.
Isonitriles were also installed onto monosaccharides for IEDDA labelling applications with tetrazines by the Leeper group.76 Primary isonitrile‐modified glucosamine (Ac4GlcN‐n‐Iso) and mannosamine (Ac4ManN‐n‐Iso) were successfully incorporated into glycans of Lewis lung carcinoma (LL2) cells and labelled sequentially with biotin‐1 and NA647. Interestingly, the reaction could not be extended to tertiary isonitrile‐modified sugars, either due to the instability of this species or because of incompatibility with the metabolic machinery. Although isonitrile‐tetrazine and unstrained alkene‐tetrazine glycan labelling approaches were both orthogonal with azide–alkyne labelling in dual‐colour imaging applications, these approaches did not find wider applications, due to their low reaction rates.
Despite advancements in IEDDA‐based glycan‐labelling, eight‐membered strained alkenes or alkynes were thought to be impossible to utilize as glycobiology building blocks, due, in part, to their large size and incompatibility with many endogenous biosynthetic pathways. In 2015, however, Agarwal et al. exploited sialic acid biosynthetic enzymes' fortuitous tolerance of bulky substituents at the C5 and C9 positions.77 They prepared a BCN‐functionalized sialic acid derivative (BCNSia) and showed by flow cytometry that BCNSia is incorporated into mammalian glycoproteins and can be labelled by tetrazine probes with higher labelling efficiency than SPAAC of azidosugars. This is most probably due to the advantageous features of IEDDA labelling schemes, because the incorporation efficiencies of BCN‐ylated sugars are lower than those of smaller tags (e.g., azide). In zebrafish embryos, strong BCNSia‐dependent labelling was observed on injection of a TBET‐based fluorogenic Oregon green vinyltetrazine probe (Scheme 3, Figure 7). This enabled mapping of the sialyation pattern of zebrafish embryos and hence the identification of several new sialylated structures in developing zebrafish embryos, raising questions about tissue‐specific sialyl‐transferase activity.
Figure 7.
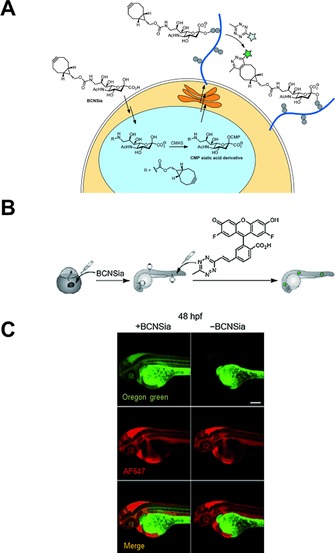
Metabolic incorporation of BCNSia into zebrafish embryos and subsequent labelling with fluorogenic Oregon green tetrazine dye. A) Schematic representation of expected metabolic pathway for the incorporation of BCNSia into cell‐surface glycans and subsequent labelling. CMAS: cytidine monophosphate N‐acetylneuraminic acid. CMP: cytidine monophosphate. B) Schematic representation of labelling protocol. Zebrafish embryos were injected with BCNSia and then with fluorogenic tetrazine in the caudal vein. C) Projection images of 48 hpf (hours post‐fertilisation) embryos treated with BCNSia or vehicle; fluorogenic tetrazine was coinjected with AF647‐NH2 to map the vasculature. Scale bar: 200 μm. Reproduced in part, with permission, from ref. 77. Copyright: 2015, John Wiley & Sons, Inc.
6. Concluding Remarks
Inverse‐electron‐demand Diels–Alder reactions between tetrazines and various dienophiles have been shown to represent a very efficient approach in bio‐orthogonally driven manipulation of biomolecules. Several detailed studies and developments resulted in an array of tetrazine and dienophile scaffolds that can be used for selective and fast labelling of virtually any kind of biomolecule in vivo. This is in contrast to other methods that are limited either to species directly encoded in the genome (e.g., fusion‐protein‐based labelling techniques) or suitable only for in vitro or extracellular labelling studies (e.g., Ig‐based labelling schemes). The different reactivities and stabilities of these reagents can be exploited by using the most suitable tetrazine/dienophile combinations, allowing us to choose those that suit the best for the needs of the particular chemical biology task. Here we have put special emphasis on applications directed towards fluorescent modification of various biopolymers by use of unnatural building blocks. The examples presented here aim to show the great potential that lies behind IEDDA‐based bio‐orthogonal fluorescent labelling schemes. Selective and site‐specific or even multicolour labelling of nucleic acids, proteins, lipids or glycan structures became possible with this rediscovered chemistry.
Although the use of IEDDA labelling schemes can overcome many of the obstacles that might corrupt fluorescent imaging of live cells, for example, by enabling the use of lower probe concentrations or the use of fluorogenic NIR probes, there is still room for more improvements. For specific, site‐selective in vivo tagging schemes, special attention should be devoted to the development of intracellularly applicable, nontoxic, membrane‐permeable, IEDDA‐suitable labels. To provide finer details of investigated biomolecular processes, an increase in the number of probes that enable super‐resolution imaging is also required. Besides being suitable for IEDDA tagging such probes need to be bright, photostable, reversibly switchable or fluorogenic.
Another approach to lowering background signal requires the development of hydrophilic dienophiles with suitable reactivity. This mainly involves the development of hydrophilic TCO derivatives because these offer the highest reaction speeds and are limited mostly by their hydrophobic nature. An appreciably large set of hydrophilic TCO derivatives also makes more likely the development of TCO‐modified noncanonical amino acids that are recognized by orthogonal tRNARS/tRNA pairs or of TCO‐tagged small‐molecule metabolites that are tolerated by the metabolic machinery of the particular system.
As presented in the form of a few examples, combination of mutually orthogonal chemistries—IEDDA with SPAAC or two IEDDA reactions with different reactivities, for example—enables two‐colour labelling schemes. Development of more orthogonal reactions would allow multicolour imaging applications. The large number of dienophiles and their finely tuneable reactivities in combination with tetrazines with different reactivities suggest that this chemistry should be suitable to explore multiply orthogonal transformations.
Although not covered in this review, a promising field in bio‐orthogonal chemistry is the development of nanoparticle systems that allow combined, multimodal imaging (e.g., fluorescence‐PET, fluorescence‐MRI, etc.). Such nanoparticles could also be used as platforms for targeting elements and cargos for drugs simultaneously. Installation of such multiple design elements becomes possible with the use of multiple orthogonal chemistries. Different tetrazine/dienophile pairs can offer such multi‐functionalization in the context of the same chemistry; however, IEDDA in combination with, for example, SPAAC can also be considered.
Briefly, existing tetrazine‐based fluorescent or fluorogenic probes in combination with dienophile‐modified building blocks bring us closer to a more comprehensive understanding of living systems. Seeing the whole picture more clearly permits us to understand the essence of biomolecular processes.
Acknowledgements
This work was supported by the Hungarian Scientific Research Fund (OTKA, NN‐116265) and the “Lendület” Program of the Hungarian Academy of Sciences (LP2013‐55/2013).
E. Kozma, O. Demeter, P. Kele, ChemBioChem 2017, 18, 486.
References
- 1. Saxon E., Bertozzi C. R., Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7108–7133; [Google Scholar]
- 2b. Patterson D. M., Nazarova L. A., Prescher J. A., ACS Chem. Biol. 2014, 9, 592–605; [DOI] [PubMed] [Google Scholar]
- 2c. Ramil C. P., Lin Q., Chem. Commun. 2013, 49, 11007–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Sauer J., Heldmann D. K., Hetzenegger J., Krauthan J., Sichert H., Schuster J., Eur. J. Org. Chem. 1998, 2885–2896; [Google Scholar]
- 3b. Boger D. L., Sakya S. M., J. Org. Chem. 1988, 53, 1415–1423; [Google Scholar]
- 3c. “1, 2,4,5-Tetrazines”, J. Sauer in Comprehensive Heterocyclic Chemistry II, Vol. 6 (Series eds.: A. R. Katritzky, C. W. Rees, E. F. V. Scriven), Pergamon, Oxford, 1996, pp. 901–957; [Google Scholar]
- 3d. Boger D. L., Coleman R. S., Panek J. S., Yohannes D., J. Org. Chem. 1984, 49, 4405–4409. [Google Scholar]
- 4. Blackman M. L., Royzen M., Fox J. M., J. Am. Chem. Soc. 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Devaraj N. K., Weissleder R., Hilderbrand S. A., Bioconjugate Chem. 2008, 19, 2297–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carboni R. A., Lindsey R. V., J. Am. Chem. Soc. 1959, 81, 4342–4346. [Google Scholar]
- 7.
- 7a. Knall A.-C., Slugovc C., Chem. Soc. Rev. 2013, 42, 5131–5142; [DOI] [PubMed] [Google Scholar]
- 7b. Wu H., Devaraj N., Top. Curr. Chem. 2016, 374, 1–22; [DOI] [PubMed] [Google Scholar]
- 7c. Prokhorov A. M., Kozhevnikov D. N., Chem. Heterocyc. Compd. 2012, 48, 1153–1176. [Google Scholar]
- 8. Wagner J. A., Mercadante D., Nikić I., Lemke E. A., Gräter F., Chem. Eur. J. 2015, 21, 12431–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liang Y., Mackey J. L., Lopez S. A., Liu F., Houk K. N., J. Am. Chem. Soc. 2012, 134, 17904–17907. [DOI] [PubMed] [Google Scholar]
- 10. Cserép G. B., Demeter O., Bätzner E., Kállay M., Wagenknecht H.-A., Kele P., Synthesis 2015, 2738–2744. [Google Scholar]
- 11. Darko A., Wallace S., Dmitrenko O., Machovina M. M., Mehl R. A., Chin J. W., Fox J. M., Chem. Sci. 2014, 5, 3770–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Royzen M., Yap G. P. A., Fox J. M., J. Am. Chem. Soc. 2008, 130, 3760–3761. [DOI] [PubMed] [Google Scholar]
- 13. Taylor M. T., Blackman M. L., Dmitrenko O., Fox J. M., J. Am. Chem. Soc. 2011, 133, 9646–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agard N. J., Prescher J. A., Bertozzi C. R., J. Am. Chem. Soc. 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- 15. Dommerholt J., Schmidt S., Temming R., Hendriks L. J., Rutjes F. P., van Hest J. C., Lefeber D. J., Friedl P., van Delft F. L., Angew. Chem. Int. Ed. 2010, 49, 9422–9425; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9612–9615. [Google Scholar]
- 16.
- 16a. Nikić I., Plass T., Schraidt O., Szymański J., Briggs J. A., Schultz C., Lemke E. A., Angew. Chem. Int. Ed. 2014, 53, 2245–2249; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2278–2282; [Google Scholar]
- 16b. Hoffmann J. E., Plass T., Nikić I., Aramburu I. V., Koehler C., Gillandt H., Lemke E. A., Schultz C., Chem. Eur. J. 2015, 21, 12266–12270; [DOI] [PubMed] [Google Scholar]
- 16c. Versteegen R. M., Rossin R., ten Hoeve W., Janssen H. M., Robillard M. S., Angew. Chem. Int. Ed. 2013, 52, 14112–14116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14362–14366; [Google Scholar]
- 16d. Li J., Lia S., Chen P. R., Nat. Chem. Biol. 2014, 10, 1003–1005; [DOI] [PubMed] [Google Scholar]
- 16e. Fan X., Ge Y., Lin F., Yang Y., Zhang G., Ngai W. S. C., Lin Z., Zheng S., Wang J., Zhao J., Li J., Chen P. R., Angew. Chem. Int. Ed. 2016, 55, 14046–14050; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14252–14256. [Google Scholar]
- 17. Kozma E., Nikić I., Varga B. R., Aramburu I. V., Kang J. H., Fackler O. T., Lemke E. A., Kele P., ChemBioChem 2016, 17, 1518–1524. [DOI] [PubMed] [Google Scholar]
- 18. Yang J., Liang Y., Šečkutė J., Houk K. N., Devaraj N. K., Chem. Eur. J. 2014, 20, 3365–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patterson D. M., Nazarova L., Xie B., Kamber D., Prescher J. A., J. Am. Chem. Soc. 2012, 134, 18638–18643. [DOI] [PubMed] [Google Scholar]
- 20. Engelsma S. B., Willems L. I., van Paaschen C. E., van Kasteren S. I., van der Marel G. A., Overkleeft H. S., Filippov D. V., Org. Lett. 2014, 16, 2744–2747. [DOI] [PubMed] [Google Scholar]
- 21. Späte A.-K., Schart V. F., Schöllkopf S., Niederwieser A., Wittmann V., Chem. Eur. J. 2014, 20, 16502–16508. [DOI] [PubMed] [Google Scholar]
- 22. Eising S., Lelivelt F., Bonger K. M., Angew. Chem. Int. Ed. 2016, 55, 12243–12247; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12431–12435. [Google Scholar]
- 23.
- 23a. Denk C., Svatunek D., Filip T., Wanek T., Lumpi D., Fröhlich J., Kuntner C., Mikula H., Angew. Chem. Int. Ed. 2014, 53, 9655–9659; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9810–9814; [Google Scholar]
- 23b. Denk C., Svatunek D., Mairinger S., Stanek J., Filip T., Matscheko D., Kuntner C., Wanek T., Mikula H., Bioconjugate Chem. 2016, 27, 1707–1712; [DOI] [PubMed] [Google Scholar]
- 23c. Devaraj N. K., Thurber G. M., Keliher E. J., Marinelli B., Weissleder R., Proc. Natl. Acad. Sci. USA 2012, 109, 4762–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Zeglis B. M., Sevak K. K., Reiner T., Mohindra P., Carlin S. D., Zanzonico P., Weissleder R., Lewis J. S., J. Nucl. Med. 2013, 54, 1389–1396; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Evans H. L., Carroll L., Aboagye E. O., Spivey A. C., J. Labelled Compd. Radiopharm. 2014, 57, 291–297; [DOI] [PubMed] [Google Scholar]
- 24c. Nichols B., Qin Z., Yang J., Vera D. R., Devaraj N. K., Chem. Commun. 2014, 50, 5215–5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cserép G. B., Herner A., Kele P., Methods Appl. Fluoresc. 2015, 3, 042001. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Liu D. S., Tangpeerachaikul A., Selvaraj R., Taylor M. T., Fox J. M., Ting A. Y., J. Am. Chem. Soc. 2012, 134, 792–795; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Erdmann R. S., Takakura H., Thompson A. D., Rivera-Molina F., Allgeyer E. S., Bewersdorf J., Toomre D. K., Schepartz A., Angew. Chem. Int. Ed. 2014, 53, 10242–10246; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10407–10412. [Google Scholar]
- 27.
- 27a. Devaraj N. K., Hilderbrand S., Upadhyay R., Mazitschek R., Weissleder R., Angew. Chem. Int. Ed. 2010, 49, 2869–2872; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2931–2934; [Google Scholar]
- 27b. Lang K., Davis L., Wallace S., Mahesh M., Cox D. J., Blackman M. L., Fox J. M., Chin J. W., J. Am. Chem. Soc. 2012, 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Carlson J. C., Meimetis L. G., Hilderbrand S. A., Weissleder R., Angew. Chem. Int. Ed. 2013, 52, 6917–6920; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7055–7058; [Google Scholar]
- 28b. Wu H., Yang J., Šečkutė J., Devaraj N. K., Angew. Chem. Int. Ed. 2014, 53, 5805–5809; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5915–5919; [Google Scholar]
- 28c. Meimetis L. G., Carlson J. C., Giedt R. J., Kohler R. H., Weissleder R., Angew. Chem. Int. Ed. 2014, 53, 7531–7534; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7661–7664; [Google Scholar]
- 28d. Knorr G., Kozma E., Herner A., Lemke E. A., Kele P., Chem. Eur. J. 2016, 22, 8972–8979; [DOI] [PubMed] [Google Scholar]
- 28e.A. Wieczorek, P. Werther, J. Euchner, R. Wombacher, Chem. Sci 2017; DOI: 10.1039/C6SC03879D. [DOI] [PMC free article] [PubMed]
- 29.
- 29a. Schoch J., Wiessler M., Jäschke A., J. Am. Chem. Soc. 2010, 132, 8846–8847; [DOI] [PubMed] [Google Scholar]
- 29b. Schoch J., Ameta S., Jäschke A., Chem. Commun. 2011, 47, 12536–12537. [DOI] [PubMed] [Google Scholar]
- 30. Schoch J., Staudt M., Samanta A., Wiessler M., Jäschke A., Bioconjugate Chem. 2012, 23, 1382–1386. [DOI] [PubMed] [Google Scholar]
- 31. Pyka A., Domnick C., Braun F., Kath-Schorr S., Bioconjugate Chem. 2014, 25, 1438–1443. [DOI] [PubMed] [Google Scholar]
- 32. Asare-Okai P. N., Agustin E., Fabris D., Royzen M., Chem. Commun. 2014, 50, 7844–7847. [DOI] [PubMed] [Google Scholar]
- 33. Wang K., Wang D., Ji K., Chen W., Zheng Y., Dai C., Wang B., Org. Biomol. Chem. 2015, 13, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ren X., El-Sagheer A. H., Brown T., Analyst 2015, 140, 2671–2678. [DOI] [PubMed] [Google Scholar]
- 35. Rieder U., Luedtke N. W., Angew. Chem. Int. Ed. 2014, 53, 9168–9172; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9322–9326. [Google Scholar]
- 36. Bußkamp H., Batroff E., Niederwieser A., Abdel-Rahman O. S., Winter R., Wittmann V., Marx A., Chem. Commun. 2014, 50, 10827–10829. [DOI] [PubMed] [Google Scholar]
- 37. Li L., Degardin M., Lavergne T., Malyshev D. A., Dhami K., Ordoukhanian P., Romesberg F. E., J. Am. Chem. Soc. 2014, 136, 826–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eggert F., Kath-Schorr S., Chem. Commun. 2016, 52, 7284–7287. [DOI] [PubMed] [Google Scholar]
- 39. Domnick C., Eggert F., Kath-Schorr S., Chem. Commun. 2015, 51, 8253–8256. [DOI] [PubMed] [Google Scholar]
- 40. Merkel M., Arndt S., Ploschik D., Cserép G. B., Wenge U., Kele P., Wagenknecht H.-A., J. Org. Chem. 2016, 81, 7527–7538. [DOI] [PubMed] [Google Scholar]
- 41. Šečkutė J., Yang J., Devaraj N. K., Nucleic Acids Res. 2013, 41, e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu H., Cisneros B. T., Cole C. M., Devaraj N. K., J. Am. Chem. Soc. 2014, 136, 17942–17945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang J., Šečkutė J., Cole C. M., Devaraj N. K., Angew. Chem. Int. Ed. 2012, 51, 7476–7479; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7594–7597. [Google Scholar]
- 44.
- 44a. Jung D., Min K., Jung J., Jang W., Kwon Y., Mol. BioSyst. Mol. Biosys. 2013, 9, 862–872; [DOI] [PubMed] [Google Scholar]
- 44b. Lang K., Chin J. W., Chem. Rev. 2014, 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- 45.
- 45a. Kimmerlin T., Seebach D., J. Pept. Res. 2008, 65, 229–260; [DOI] [PubMed] [Google Scholar]
- 45b. Dawson P. E., Kent S. B., Annu. Rev. Biochem. 2000, 69, 923–960; [DOI] [PubMed] [Google Scholar]
- 45c. Noren C. J., Anthony-Cahill S. J., Griffith M. C., Schultz P. G., Science 1989, 244, 182–188. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Link A. J., Tirrell D. A., J. Am. Chem. Soc. 2003, 125, 11164–11165; [DOI] [PubMed] [Google Scholar]
- 46b. Dieterich D. C., Lee J. J., Link A. J., Graumann J., Tirrell D. A., Schuman E. M., Nat. Protoc. 2007, 2, 532–540; [DOI] [PubMed] [Google Scholar]
- 46c. Beatty K. E., Xie F., Wang Q., Tirrell D. A., J. Am. Chem. Soc. 2005, 127, 14150–14151. [DOI] [PubMed] [Google Scholar]
- 47.
- 47a. Liu C. C., Schultz P. G., Annu. Rev. Biochem. 2010, 79, 413–444; [DOI] [PubMed] [Google Scholar]
- 47b. Lemke E. A., ChemBioChem 2014, 15, 1691–1694; [DOI] [PubMed] [Google Scholar]
- 47c. Chin J. W., Annu. Rev. Biochem. 2014, 83, 379–408; [DOI] [PubMed] [Google Scholar]
- 47d. Neumann-Staubitz P., Neumann H., Curr. Opin. Struct. Biol. 2016, 38, 119–128. [DOI] [PubMed] [Google Scholar]
- 48. Wan W., Tharp J. M., Liu W. R., Biochim. Biophys. Acta Proteins Proteomics 2014, 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Plass T., Milles S., Koehler C., Schultz C., Lemke E. A., Angew. Chem. Int. Ed. 2011, 50, 3878–3881; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3964–3967. [Google Scholar]
- 50. Yanagisawa T., Ishii R., Fukunaga R., Kobayashi T., Sakamoto K., Yokoyama S., Chem. Biol. 2008, 15, 1187–1197. [DOI] [PubMed] [Google Scholar]
- 51. Plass T., Milles S., Koehler C., Szymanski J., Mueller R., Wiessler M., Schultz C., Lemke E. A., Angew. Chem. Int. Ed. 2012, 51, 4166–4170; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4242–4246. [Google Scholar]
- 52. Lukinavičius G., Umezawa K., Olivier N., Honigmann A., Yang G., Plass T., Mueller V., Reymond L., Corrêa I. R., Luo Z.-G., Schultz C., Lemke E. A., Heppenstall P., Eggeling C., Manley S., Johnsson K., Nat. Chem. 2013, 5, 132–139. [DOI] [PubMed] [Google Scholar]
- 53. Lang K., Davis L., Torres-Kolbus J., Chou C., Deiters A., Chin J. W., Nat. Chem. 2012, 4, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaya E., Vrabel M., Deiml C., Prill S., Fluxa V. S., Carell T., Angew. Chem. Int. Ed. 2012, 51, 4466–4469; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4542–4545. [Google Scholar]
- 55. Borrmann A., Milles S., Plass T., Dommerholt J., Verkade J. M. M., Wiessler M., Schultz C., van Hest J. C., van Delft F. L., Lemke E. A., ChemBioChem 2012, 13, 2094–2099. [DOI] [PubMed] [Google Scholar]
- 56. Kurra Y., Odoi K. A., Lee Y.-J., Yang Y., Lu T., Wheeler S. E., Torres-Kolbus J., Deiters A., Liu W. R., Bioconjugate Chem. 2014, 25, 1730–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu Z., Pan Y., Wang Z., Wang J., Lin Q., Angew. Chem. Int. Ed. 2012, 51, 10600–10604; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10752–10756. [Google Scholar]
- 58. Elliott T. S., Townsley F. M., Bianco A., Ernst R. J., Sachdeva A., Elsässer S. J., Davis L., Lang K., Pisa R., Greiss S., Lilley K. S., Chin J. W., Nat. Biotechnol. 2014, 32, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee Y., Kurra Y., Yang Y., Torres-Kolbus J., Deiters A., Liu W. R., Chem. Commun. 2014, 50, 13085–13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Seitchik J. L., Peeler J. C., Taylor M. T., Blackman M. L., Rhoads T. W., Cooley R. B., Refakis C., Fox J. M., Mehl R. A., J. Am. Chem. Soc. 2012, 134, 2898–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Blizzard R. J., Backus D. R., Brown W., Bazewicz C. G., Li Y., Mehl R. A., J. Am. Chem. Soc. 2015, 137, 10044–10047. [DOI] [PubMed] [Google Scholar]
- 62. Wang K., Sachdeva A., Cox D. J., Wilf N. W., Lang K., Wallace S., Mehl R. A., Chin J. W., Nat. Chem. 2014, 6, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Neumann H., Wang K., Davis L., Garcia-Alai M., Chin J. W., Nature 2010, 464, 441–444. [DOI] [PubMed] [Google Scholar]
- 64. Nikić I., Plass T., Schraidt O., Szymański J., Briggs J. A. G., Schultz C., Lemke E. A., Angew. Chem. Int. Ed. 2014, 53, 2245–2249; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2278–2282. [Google Scholar]
- 65. Uttamapinant C., Howe J. D., Lang K., Beránek V., Davis L., Mahesh M., Barry N. P., Chin J. W., J. Am. Chem. Soc. 2015, 137, 4602–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Peng T., Hang H. C., J. Am. Chem. Soc. 2016, 138, 14423–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.
- 67a. Essentials of Glycobiology, 2nd ed. (Eds.: A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, M. E. Etzler), Cold Spring Harbor Laboratory Press, New York, 2009; [PubMed] [Google Scholar]
- 67b. Prescher J. A., Bertozzi C. R., Cell 2006, 126, 851–854. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a. Mackiewicz A., Mackiewicz K., Glycoconjugate J. 1995, 12, 241–247; [DOI] [PubMed] [Google Scholar]
- 68b. Meezan E., Wu H. C., Black P. H., Robbins P. W., Biochemistry 1969, 8, 2518–2524; [DOI] [PubMed] [Google Scholar]
- 68c. Dube D. H., Bertozzi C. R., Nat. Rev. Drug Discovery 2005, 4, 477–488. [DOI] [PubMed] [Google Scholar]
- 69.
- 69a. Soriano del Amo D., Wang W., Jiang H., Besanceney C., Yan A., Levy M., Liu Y., Marlow F. L., Wu P., J. Am. Chem. Soc. 2010, 132, 16893–16899; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69b. Besanceney-Webler C., Jiang H., Zheng T., Feng L., Soriano del Amo D., Wang W., Klivansky L. M., Marlow F. L., Liu Y., Wu P., Angew. Chem. Int. Ed. 2011, 50, 8051–8056; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8201–8206. [Google Scholar]
- 70. Cole C. M., Yang J., Šečkutė J., Devaraj N. K., ChemBioChem 2013, 14, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bond M. R., Zhang H. C., Kim J., Yu S. H., Yang F., Patrie S. M., Kohler J. J., Bioconjugate Chem. 2011, 22, 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.
- 72a. Späte A.-K., Schart V. F., Häfner J., Niederwieser A., Mayer T. U., Wittmann V., Beilstein J. Org. Chem. 2014, 10, 2235–2242; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72b. Patterson D. M., Jones K. A., Prescher J. A., Mol. BioSyst. 2014, 10, 1693. [DOI] [PubMed] [Google Scholar]
- 73. Doll F., Buntz A., Späte A.-K., Schart V. F., Timper A., Schrimpf W., Hauck C. R., Zumbusch A., Wittmann V., Angew. Chem. Int. Ed. 2016, 55, 2262–2266; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2303–2308. [Google Scholar]
- 74.
- 74a. Späte A.-K., Bußkamp H., Niederwieser A., Schart V. F., Marx A., Wittmann V., Bioconjugate Chem. 2014, 25, 147–154; [DOI] [PubMed] [Google Scholar]
- 74b. Späte A.-K., Dold J. E. G. A., Batroff E., Schart V. F., Wieland D. E., Baudendistel O. R., Wittmann V., ChemBioChem 2016, 17, 1374–1383. [DOI] [PubMed] [Google Scholar]
- 75. Niederwieser A., Späte A.-K., Nguyen L. D., Jüngst C., Reutter W., Wittmann V., Angew. Chem. Int. Ed. 2013, 52, 4265–4268; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4359–4363. [Google Scholar]
- 76.
- 76a. Stairs S., Neves A. A., Stöckmann H., Wainman Y. A., Ireland-Zecchini H., Brindle K. M., Leeper F. J., ChemBioChem 2013, 14, 1063–1067; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76b. Wainman Y. A., Neves A. A., Stairs S., Stöckmann H., Ireland-Zecchini H., Brindle K. M., Leeper F. L., Org. Biomol. Chem. 2013, 11, 7297–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Agarwal P., Beahm B. J., Shieh P., Bertozzi C. R., Angew. Chem. Int. Ed. 2015, 54, 11504–11510; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11666–11672. [Google Scholar]