

Abstract
Unspecific peroxygenases (UPO, EC 1.11.2.1) secreted by fungi open an efficient way to selectively oxyfunctionalize diverse organic substrates, including less‐activated hydrocarbons, by transferring peroxide‐borne oxygen. We investigated a cell‐free approach to incorporate epoxy and hydroxyl functionalities directly into the bulky molecule testosterone by a novel unspecific peroxygenase (UPO) that is produced by the ascomycetous fungus Chaetomium globosum in a complex medium rich in carbon and nitrogen. Purification by fast protein liquid chromatography revealed two enzyme fractions with the same molecular mass (36 kDa) and with specific activity of 4.4 to 12 U mg−1. Although the well‐known UPOs of Agrocybe aegerita (AaeUPO) and Marasmius rotula (MroUPO) failed to convert testosterone in a comparative study, the UPO of C. globosum (CglUPO) accepted testosterone as substrate and converted it with total turnover number (TTN) of up to 7000 into two oxygenated products: the 4,5‐epoxide of testosterone in β‐configuration and 16α‐hydroxytestosterone. The reaction performed on a 100 mg scale resulted in the formation of about 90 % of the epoxide and 10 % of the hydroxylation product, both of which could be isolated with purities above 96 %. Thus, CglUPO is a promising biocatalyst for the oxyfunctionalization of bulky steroids and it will be a useful tool for the synthesis of pharmaceutically relevant steroidal molecules.
Keywords: epoxidation, hydroxylation, oxyfunctionalization, peroxidase, steroid
Introduction
Steroids, ubiquitous in living systems, constitute an important group of terpenoid lipids with four cycloalkane rings arranged in a specific gonane structure. They are found in eukaryotic membranes and have diverse functions as signal molecules (hormones, pheromones). Steroid hormones, for example, are known to control various aspects of cell proliferation, tissue differentiation and signal transduction pathways by binding to specific receptors.1 Therefore, steroids play an important role as pharmaceuticals in, for example, endocrinology, oncology, rheumatology, and gynecology. They represent the second largest category of marketed medical products after antibiotics.2 The biological activity of steroids depends on their structure, the oxidation state of the ring system, and the type, number, regio‐, and stereo‐position of functional groups attached to the gonane nucleus. Even minor chemical changes can substantially influence the physiological activity of steroids. Hydroxylation is one of the most important reactions for modulating steroid function.3, 4 Hydroxylated steroids often have increased biological activity compared to their less‐polar, non‐hydroxylated analogues.1 Chemical synthesis of hydroxylated variants of steroids is challenging as it requires complicated multistep synthetic pathways and has low overall yield and high cost.3 To the best of our knowledge, there is no report on large‐scale preparation of hydroxylated steroids by chemical synthesis.
Over the last years, biotechnological research in pharmacy has focused on the structural modification of bioactive steroids by using various microorganisms and their monooxygenases as transformation agents (whole‐cell biotransformations).5, 6 Members of the cytochrome P450 monooxygenase superfamily (P450s), which catalyzes the transfer of oxygen from O2 to a variety of organic compounds, are responsible for selective steroid oxyfunctionalizations.7 In this context, enzymatic steroid hydroxylation catalyzed by engineered P450 mutants (e.g., of BM3) has gained attention for the preparation of diverse and unique hydroxylated steroids with pharmacological activity.8, ‐9 However, the use of isolated P450 for larger‐scale application is hampered by low activity, catalytic efficiency, and stability outside cells, the requirement for NAD(P)H (as electron donating co‐substrate), and the necessity for electron‐transferring partners.10
For more than ten years, unspecific peroxygenases (UPOs, EC 1.11.2.1) have gained attention in the field of oxyfunctionalization chemistry. They constitute a distinct (super)family of fungal heme‐thiolate proteins that catalyze efficient and selective oxygen‐transfer from peroxides to diverse organic substrates including non‐activated compounds.11 UPOs are relatively stable and versatile biocatalysts, and combine the catalytic cycle of heme peroxidases with the “peroxide shunt” of P450s.12, 13 The first UPO was discovered in the wood‐dwelling agaric fungus Agrocybe aegerita (AaeUPO); two similar enzymes were described in the mushrooms Coprinellus radians (CraUPO) and Marasmius rotula (MroUPO).14, 15, 16 The reactions catalyzed by these UPOs comprise aromatic and aliphatic hydroxylations (including subsequent bond cleavages resulting in N‐ and O‐dealkylations), epoxidation, N‐oxygenation, sulfoxidation, dechlorination, and halide oxidation.11 Frequently, the product patterns of UPOs resemble those of P450s, including various human liver enzymes. UPOs have successfully been used as oxygenation tools for the synthesis of human drug metabolites.17, 18, 19 The first detailed studies on steroid hydroxylation by several UPOs revealed preferred accessibility of the hydrophobic C17 alkyl side chain to the substrate channel; the entrance of whole rings was found to be energetically penalized, in particular when the C3 position was already oxyfunctionalized.17, 20, ‐21 Consequently, substrates, such as testosterone, that lack an alkyl side chain or bearing oxidized C3 were not converted.20
Over 2500 putative UPO sequences from all larger taxonomic groups of fungi can be found in genetic databases; this has encouraged the prospective extension of the UPO toolbox to new synthetic applications including the oxyfunctionalization of bulky steroids.11, 12, 22 A blastp search using known UPO references (e.g. from Agrocybe aegerita B9W4V6) to the recently published genome of the ubiquitous ascomycete fungus Chaetomium globosum (order Sordariales) revealed four sequences encoding putative peroxygenases.23 C. globosum is a cellulolytic fungus that colonizes a variety of materials in terrestrial and marine environments.24
In this study, we investigated the major UPO of C. globosum (CglUPO, XM_001219539.1). We describe its secretion during fungal cultivation and purification, and its molecular and catalytic characterization. Furthermore, oxyfunctionalization of testosterone by CglUPO resulted in the selective epoxidation of the A ring of the gonane nucleus as well as in the hydroxylation of the D ring.
Results
UPO production by C. globosum
C. globosum produced an unspecific peroxygenase (CglUPO) in carbon‐ and nitrogen‐rich liquid medium. Changes in medium composition led to considerable changes in productivity and enzyme yield (data not shown). The maximum activity (530 U L−1, based on a veratryl alcohol assay) was achieved after 21 days of cultivation in 42 g L−1 glucose, 18 g L−1 peptone, and 4.5 g L−1 yeast extract (secretion ∼40 mg L−1 UPO protein). Afterwards, UPO activity rapidly decreased within a few days (Figure 1). During the cultivation process, C. globosum progressively alkalified its medium: from pH 6.0 to pH 7.1 (day 11) and up to pH 8.9 on the last day of cultivation (day 27). Fungal biomass consisted of pellets (3–5 mm diameter) with a dry mass of 7.2 g L−1 around day 20. In contrast to known UPO‐producing strains, an accompanied laccase activity was not detected under these culture conditions.14, 15, 16 Laccase was measured because it can hamper UPO purification.
Figure 1.
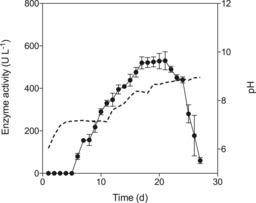
Time‐course of UPO production (circles) by C. globosum in 4.2 % glucose, 1.8 % soybean peptone, and 0.45 % (w/v) yeast extract. CglUPO activity was measured with a veratryl alcohol assay at pH 7.14 Data are mean±SD from three culture flask assays. The dotted line shows pH.
Purification and characterization of CglUPO
Two fractions (I and II) obtained after elution of the concentrated culture liquid of 21‐day‐old cultures from a Mono Q column (Figure S1 A in the Supporting Information) showed H2O2‐dependent activity towards veratryl alcohol at pH 7. The final total activities were 191 and 292 U, respectively; SDS‐PAGE revealed a molecular mass of 36 kDa for both fractions (Figure S1 B). As the specific activity of fraction II (12 U mg−1) was three times higher than that of fraction I, it was used for physical and catalytic characterization. The UV/Vis spectrum of the enzyme's resting state demonstrated a typical UPO maximum at 422 nm (Soret band) with α‐ and β‐bands at 570 and 540 nm (Figure S1 C).
The published C. globosum genome sequence contains four UPO genes.23 We identified CglUPO as CHGG_00 319 (XM_001219539.1) as the purified enzyme by using a proteomic approach (peptide mapping). The CglUPO gene contains two introns (the predicted gene model includes a falsely annotated intron, which excluded resulted in a coding sequence of 780 bp) and encodes a protein of 259 amino acids with a calculated molecular mass of 29.2 kDa and a theoretical pI of 5.59. Nine different peptide fragments of CglUPO obtained by peptide mapping matched perfectly with the deduced amino acid sequence (45.95 % sequence coverage). Several conserved amino acids (e.g., the proximal heme binding region (PCP motif) and the distal binding site for a magnesium ion (EHD motif)) were found in the translated protein sequence (Figure 2).
Figure 2.

Amino acid sequence of the corrected CglUPO protein (XM_001219539.1). Predicted signal peptide is in italics; red letters represent peptides identified by peptide mapping; blue letters show putative N‐glycosylation sites; proximal heme‐binding region (PCP motif) and distal binding‐site for a magnesium ion (EHD motif) are underlined.
CglUPO oxidized typical peroxidase substrates like ABTS (2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulfonic acid)) and DMP (2,6‐dimethoxyphenol) to characteristic radicals and coupling products, and aryl alcohols such veratryl alcohol and benzyl alcohol into the corresponding aldehydes (Figure S2). The enzyme also catalyzed the H2O2‐dependent conversion of naphthalene to 1‐naphthol (via naphthalene oxide), and of 5‐nitro‐1,3‐benzodioxole (NBD) to 4‐nitrocatechol. This unambiguously demonstrates its true peroxygenase nature. In general, CglUPO was active over pH 4.5 to 9 (substantial activity loss outside this pH range; Figures S2–S4). CglUPO was not capable of oxidizing bromide or chloride, as no bromo‐ or chlorophenols were detectable by HPLC after exposure of phenol to UPO in the presence of the respective halides.
Enzymatic conversion of testosterone
In contrast to the UPOs from A. aegerita (AaeUPO), M. rotula (MroUPO), Coprinopsis cinerea (rCciUPO), and Humicola insolens (rHinUPO), which did not convert testosterone, CglUPO accepted testosterone as a substrate (total turnover number (TTN) 7000 under the conditions used). Because of the moderate turnover number (k cat=0.26 s−1), the catalytic efficiency (k cat/K m=1.60×103 s−1 m −1) for testosterone was about two orders of magnitude lower than for the other peroxidase and peroxygenase substrates tested (Table S2).
Testosterone (1) was oxidized into two products, 1 a and 1 b, in the ratio 9:1 (according to LC‐MS). Both compounds exhibited a mass shift of m/z+16, thus indicating incorporation of oxygen (Figure S6). Product 1 a lost the UV‐absorption characteristics of testosterone and was more hydrophobic (Figure 3 A), whereas 1 b retained the UV characteristics and displayed increased hydrophilicity. In order to determine the exact structure of the testosterone metabolites, larger amounts of 1 a and 1 b were enzymatically synthesized starting from 100 mg of 1. The time course of the reaction (Figure 3 B) shows that the products formed concomitantly and accumulated in the reaction mixture. After isolation and purification, 65 mg of 1 a and 7 mg of 1 b were obtained (purities >96 %). 13C‐ and 1H‐NMR studies revealed formation of the 4,5‐epoxide of testosterone in β‐configuration (17β‐hydroxy‐4,5‐epoxy‐5β‐androstan‐3‐one, 1 a) and 16α‐hydroxytestosterone (16α,17β‐dihydroxyandrost‐4‐en‐3‐one, 1 b; Scheme 1). Both oxygenations occurred with high selectivity (diastereomeric ratios >98 %).
Figure 3.
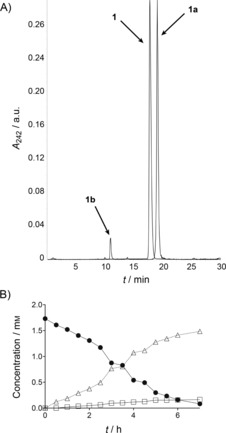
A) HPLC chromatogram and B) time‐course of CglUPO‐catalyzed conversion of testosterone (1). Overlaid LC‐MS elution profiles were recorded at 247 nm for •: testosterone; single ion t [M+H]+ 305 for the products (▵: 1 a, □: 1 b). B displays the reaction progress of the 100 mg approach.
Scheme 1.
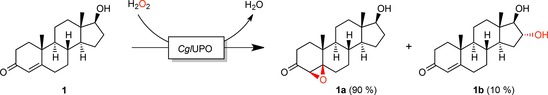
Conversion of testosterone (1) by CglUPO to testosterone 4,5β‐epoxide (1 a) and 16α‐hydroxytestosterone (1 b).
Discussion
The ascomycetous fungus C. globosum (order Sordariales) is a strong cellulolytic mold and is well known for its ability to produce various secretory enzymes and secondary metabolites with a wide range of biological activity.24 Production of an unspecific peroxygenase (CglUPO) by this fungus was achieved in a complex medium rich in carbon and nitrogen. The corresponding UPO sequence was assigned in the genome of the fungus (GenBank XM_001219539.1) and verified by secretomic analysis. According to these data, it belongs to the “short” type UPOs of group I; these lack an internal disulfide bridge.12 The purified enzyme catalyzed the oxidation of classic peroxidase substrates such as ABTS and DMP, it cleaved NBD (demethylenation, a special case of O‐dealkylation), and it selectively incorporated peroxide‐borne oxygen into naphthalene and the bulky gonane structure of testosterone.
CglUPO is the fourth wild‐type UPO to be described, and the first to be isolated from an ascomycetous fungus. The spectral data of the resting state resemble those of basidiomycetous UPOs (AaeUPO, CraUPO, MroUPO), with the Soret band at around 420 nm (typical for heme‐thiolate proteins including numerous P450s).36 The α and β maxima at 570 and 540 nm are almost identical to the respective bands of reported UPOs.12 The molecular mass of CglUPO, 36 kDa, is between those of AaeUPO (43–46 kDa) and MroUPO (32 kDa). The difference to the molecular mass calculated on the basis of the CglUPO gene (29.2 kDa) can be explained by glycosylation (19 %), as has been demonstrated for all other characterized UPOs (14–44 %).12 Four potential N‐glycosylation sites (Asn residues) were identified in the amino acid sequence of CglUPO, thus strongly supporting this assumption (Figure 2). The final purification step in an anion exchanger (MonoQ column) resulted in two fractions with the same molecular mass, thus indicating the presence of two isoforms. Isoforms with almost identical molecular masses were reported for other UPOs (e.g., UPOs of A. aegerita and C. radians) and fungal heme peroxidases (e.g., manganese peroxidases of Ceriporiopsis subvermispora).14, 15, 16, 37
A distinctive feature of CglUPO is its relatively poor stability at acidic pH. AaeUPO and MroUPO are active over a remarkably broad pH range (pH 2–9), whereas CglUPO showed substantial activity loss below pH 4.5 (∼50 % after 4 h). Thus, the enzyme is poorly suited to an acidic reaction environment, and, additionally, its purification by ion exchange chromatography (e.g., on MonoQ columns) is hampered because of the use of acidic eluents. Another feature of CglUPO is the lack of halogenating activity. In contrast to AaeUPO and CraUPO, which efficiently oxidize bromide, CglUPO did not catalyze halide oxidation; in this context it resembles MroUPO.14, 15, 16 Interestingly, CglUPO and MroUPO belong to group I UPOs (“short” peroxygenases); however, the strongly halogenating chloroperoxidase (CPO) is also in the same subfamily.12
Without doubt, the oxidation of testosterone is the most remarkable catalytic feature of CglUPO, by the transfer of peroxide‐borne oxygen to the gonane ring system. Neither MroUPO, nor AaeUPO, nor recombinant UPOs (rCciUPO, rHinUPO) were able to convert testosterone substantially. Recently, this inability was proposed to be related to the absence of an alkyl side chain at C17 (D ring), which is the preferred UPO oxidation site of steroids, as the bulky cholesteryl caprylate was the only steroid (besides testosterone) that was not attacked (three UPOs and 15 steroidal substrates).20 Although AaeUPO, MroUPO, and rCciUPO preferentially oxidize hydrophobic molecules, including relatively bulky substrates,17, 19, 20, 38 size and geometry of the gonane core seemingly impedes diffusion of complete steroid molecules through the heme‐access channel to the active site. In contrast, the substrate channel of CglUPO must be broader or more flexible, so sterically demanding compounds such as testosterone can enter the active site in a way that allows correct positioning and subsequent oxygenation.
The conversion of testosterone (1) by CglUPO resulted in the formation of two oxygenated products: an epoxide (1 a) and 16α‐hydroxytestosterone (1 b). The higher hydrophobicity of 1 a (compared to 1) can be explained by the epoxidation of 1 in the 4,5‐position, thus altering the spatial structure and rigidity of the steroidal A ring, and in turn the polarity of 1. Epoxidation of the A ring also explains the loss of UV absorption by 1 a; this is not typical for oxyfunctionalizations, and led to the breakup of the chromophoric enone structure of 1. However, with its additional secondary alcohol functionality, 1 b is more hydrophilic, and thus has the physicochemical properties of a typical oxyfunctionalization product. The product ratio (1 a/1 b, 9:1) might reflect the probability of ring A or ring D of testosterone entering the heme channel first. A plausible explanation for why CglUPO prefers the oxidation of ring A over ring D is not currently clear, particularly as other UPOs are not able to oxidize enone structures.26, 31 Computational docking studies or co‐crystallization experiments revealing the exact position of testosterone in the active site of CglUPO could help to solve this problem. With regard to a shift of the ratio in favor of D ring hydroxylation, the affinity of the A ring to the enzyme (and hence the formation of 1 a) could be reduced by introducing appropriate protective groups (e.g., acetals, oximes) into the enone structure. Another approach to alter the product ratio might be protein engineering, such as rational design (which would require comprehensive structure–function data, including crystal structures) or directed evolution. For example, a peroxygenase from A. aegerita was recently engineered for increased selectivity and TTN in the conversion of naphthalene into 1‐naphthol, as well as in a diminished peroxidative activity.39
Compound 1 a, a steroid containing a 4,5‐epoxy‐3‐oxo moiety, is of special relevance for synthetic applications and biological activities, as epoxy steroids are involved in the regulation of cell proliferation and cholesterol homeostasis.40 The synthetic versatility of epoxides has established them as useful precursors to obtain a variety of polyfunctional derivatives or rearranged compounds.41, 42, 43, 44, 45, 46, 47 Cyclic α‐epoxide enones are found in a number of natural products, but asymmetric epoxidation of their precursors is notoriously difficult.48 The synthesis of β‐epoxy steroids has been achieved by using different chemical oxidants, such as chromyl diacetate, potassium permanganate salts, chiral ketones combined with oxone, and transition metal complexes in the presence of molecular oxygen.49 Selective biocatalytic oxygen incorporation by UPOs to form β‐epoxides under mild conditions could supplement these existing chemical methods.
The second product of CglUPO‐catalyzed testosterone conversion, 1 b (16α‐hydroxytestosterone), is a metabolite of oxidative steroid metabolism in liver cells. Its formation was shown to be catalyzed by membrane‐bound cytochrome P450s in human and rat liver microsomes;50 beyond that, the hydroxylation of testosterone at the 16α‐position is an important step in the formation of estriol in late pregnancy.51, 52 Soluble P450s of Gram‐positive bacteria, such as CYP154C3 from Streptomyces griseus, P450 BM3 (CYP102A1, variant M01 A82W S72I) from Bacillus megaterium, and CYP145C5 from Nocardia farcinica, were reported to selectively hydroxylate testosterone and related steroids at the 16α‐position.8, 53, 54 Crude preparations of the latter enzyme were particularly efficient (as an isolated P450) and achieved TTNs of 500 to 2300 for this reaction.54 We achieved a TTN above 7000 for testosterone oxidation, yet, in contrast to P450s, CglUPO requires neither reduced nicotinamide cofactors nor any regenerating system—hydrogen peroxide alone was sufficient for activation and function.
If the challenge of heterologous expression of CglUPO in a suitable host (e.g., in Aspergillus oryzae, Saccharomyces cerevisiae, or Pichia pastoris) can be overcome, a powerful synthetic tool would be available for the oxyfunctionalization of steroidal structures, and this could be subjected to protein engineering. Such an approach was recently described for an A. aegerita UPO that was expressed in S. cerevisiae and mutated in nine positions, and then optimized for recombinant protein production and secretion in P. pastoris.25, 55, 56 Moreover, modern protein engineering techniques might help to design tailor‐made UPOs for specific steroid hydroxylations and to overcome catalytic bottlenecks such as solvent and peroxide instability. Concerning reaction design, UPO immobilization (for example, by encapsulation) and gentle co‐substrate generation will contribute to improved CglUPO performance.11, 57 These and other approaches are currently under investigation.
Experimental Section
Material and reagents: Testosterone (17β‐hydroxy‐4‐androsten‐3‐one), yeast extract, ABTS and naphthalene were purchased from AppliChem (Arheilgen, Germany); H2O2 (30 % w/v), soybean peptone, agar, α‐d‐glucose, benzyl alcohol, phenol, and sodium azide were from Carl Roth; malt extract, 2‐chlorophenol, and 4‐chlorophenol were obtained from Merck; glucose oxidase from Aspergillus niger was purchased from Sigma–Aldrich (specific activity 215 U mg−1). All other chemicals were purchased from Sigma–Aldrich at the highest purity available.
Peroxygenases of A. aegerita (AaeUPO) and M. rotula (MroUPO) were produced and purified as described previously;14, 16 recombinant UPOs from C. cinerea (rCciUPO) and H. insolens (rHinUPO=rnovo) were gifts from Novozymes A/S (Copenhagen, Denmark).25, 26, 27, 28
The specific activities of AaeUPO and MroUPO were 63.5 U mg−1 and 48.1 U mg−1, respectively (1 U is the oxidation of 1 μmol veratryl alcohol to veratraldehyde per 1 min at 23 °C).14
Cultivation of C. globosum: C. globosum (strain DSM 62110) was purchased from the German Collection of Microorganisms and Cell cultures (Braunschweig, Germany) and was routinely grown on malt extract agar medium (malt extract (20 g L−1) and agar (15 g L−1)) at 24 °C. For enzyme production, the fungus was cultured in 500 mL Erlenmeyer flasks containing carbon‐ and nitrogen‐rich basic liquid medium (200 mL; glucose (42 g L−1), peptone (18 g L−1), yeast extract (4.5 g L−1) in deionized water) on a rotary shaker (120 rpm) at 24 °C for four weeks. Liquid cultures were inoculated with a mycelial suspension (5 % v/v) obtained by homogenization of the content of two agar plates fully covered with fungal mycelium in sterile sodium chloride (100 mL, 0.9 % w/v).
Enzyme assays: UPO activities were measured photometrically by monitoring the oxidation of veratryl alcohol (5 mm) into veratraldehyde at 310 nm (ϵ 310=9300 m −1 cm−1) in McIlvaine buffer at pH 7.14 Reaction was started by the addition of hydrogen peroxide (2 mm). Laccase activity during cultivation was determined by the oxidation of ABTS to the corresponding ABTS cation radical at 420 nm (ϵ 420=36 000 m −1 cm−1) in McIlvaine buffer at pH 4.5 in the absence of H2O2.29 The specific ring‐hydroxylating activity of CglUPO was monitored by the oxygenation of naphthalene (1 mm) to naphthalene oxide and 1‐naphthol at 303 nm (ϵ 303=2030 m −1 cm−1) in McIlvaine buffer at pH 6.0; the reaction was started by adding hydrogen peroxide (2 mm).30
Purification and characterization of Cgl UPO: All purification steps were carried out at room temperature. Enzyme fractions were assayed for UPO activity, and protein content was determined with a Pierce BCA protein assay kit (Thermo Fisher) with bovine serum albumin as standard. Protein purification was carried out by using ammonium sulfate precipitation and fast protein liquid chromatography (FPLC) on Q‐Sepharose FF (IEC), Superdex75 (SEC), and Mono Q columns (IEC), successively. All chromatographic steps were accomplished with an ÄKTA purifier FPLC system (GE Healthcare).
The molecular mass of purified CglUPO was analyzed by SDS‐PAGE by using a 10 % Bolt Bis‐Tris Gel (Thermo Fisher Scientific). The separated protein bands were visualized with a Colloidal Blue Staining Kit (Generon Ltd, Berkshire, UK, order code GEN‐QC‐Stain‐1L); a protein marker (#26616, Thermo Fisher Scientific) was used as standard.
Proteomic enzyme identification was performed at the Helmholtz‐Centre for Environmental Research—UFZ, Department of Molecular Systems Biology (Leipzig, Germany). For detailed information (peptide mapping), see the Supporting Information.
Kinetic constants (K m, k cat) of CglUPO and pH optima were determined for veratryl alcohol, benzyl alcohol, DMP, ABTS, NBD (pH 7),31 and naphthalene (pH 6; Supporting Information). Halogenating activity was tested by incubating CglUPO (0.2 U mL−1, 0.46 μm) in potassium phosphate buffer (100 mm, pH 3 and pH 7) in the presence of phenol (0.1 mm), potassium bromide or chloride (10 mm) and H2O2 (2 mm).32 After 10 min, the reaction mixture was analyzed by HPLC for the formation of bromo‐ and chlorophenols against authentic standards.
Enzymatic conversion of testosterone: The reaction mixture (total volume 0.5 mL) contained purified AaeUPO (2 U mL−1, 0.7 μm), MroUPO (2 U mL−1, 1.3 μm) or CglUPO (0.2 U mL−1, 0.46 μm) in potassium phosphate buffer (20 mm, pH 7) with testosterone (5 mm), α‐d‐glucose (2 %, w/v), and acetone (5 %, v/v). Reactions were started by addition of glucose oxidase (GOx, 0.02 U mL−1) and stirred at room temperature for 24 h (after this time, no residual activity of CglUPO was detectable). Kinetic data were determination for CglUPO (2 U mL−1, 4.8 μm) with testosterone (5 mm) in potassium phosphate buffer (20 mm, pH 7). Reactions were initiated by the addition of hydrogen peroxide (2 mm) and stopped after 2 min by adding sodium azide (1 mm). Higher concentrations of hydrogen peroxide were not applied, in order to prevent enzyme inactivation from heme bleaching and the disproportionate increase in the UPO intrinsic catalase activity (both have been reported for other UPOs and heme peroxidases).33, 34, 35 Products were recovered with reversed‐phase SPE cartridges (Strata‐X 33u, Phenomenex), with elution in methanol, and analyzed by HPLC.
At preparative scale, a 500 mL flask was filled with testosterone (100 mg, 0.35 mmol), acetone (10 mL), water (140 mL), potassium phosphate buffer (40 mL 0.1 m, pH 7), and CglUPO stock solution (20 mL 400 U in potassium phosphate buffer (0.1 m, pH 7)). The reaction mixture was stirred at room temperature while hydrogen peroxide (100 mm, 4 mL h−1) was continuously supplied by a syringe pump. Hydrogen peroxide was used instead of glucose/GOx, in order to ensure constant peroxide dosage and to avoid impurities in the reaction mixture (glucose and gluconolactone); the syringe pump system was as effective as the GOx‐based H2O2 generation system. Samples (50 μL) were taken from the reaction mixture every 30 min, and the reaction (in the samples) was stopped by adding acetonitrile (50 μL) and sodium azide (10 μL, 10 mm). The samples were centrifuged, and the supernatants were analyzed by HPLC (below). After 7 h, thin layer chromatography (in ethyl acetate/n‐hexane, 9:1) indicated complete conversion of testosterone. The reaction mixture was extracted three times with ethyl acetate (50 mL), then the combined organic fractions were dried with Na2SO4 and evaporated to dryness to give 91 mg of crude products 1 a (R f 0.67) and 1 b (R f 0.11). The compounds were purified by chromatography on silica gel with ethyl acetate/n‐hexane (9:1) as the eluent to obtain 65 mg (61.1 %) of 1 a (96.3 % purity) and 7 mg (6.6 %) of 1 b (98.7 % purity).
Analytical methods: The HPLC‐MS system (Waters) comprised a 2690 separation module, a 2996 photo diode array detector, and a Micromass ZMD 2000 single quadrupole mass spectrometer. Separation was on a LiChrospher C18 column (125×4 mm, 5 μm, Phenomenex) with mobile phases A (formic acid (0.1 %)) and B (acetonitrile) and at stepwise gradient (20 % B (3 min), increase to 55 % B (20 min), increase to 90 % B (3 min)). The final level was maintained until all analytes had been eluted from the column (flow‐rate 1 mL min−1, column temperature 30 °C). Reaction products were identified by comparison to authentic standards based on retention time, UV absorption spectrum and mass spectra [M+H]+ or [M−H]− ions, and quantified by total peak area by using response factors of the same or similar compounds. Data from replicates were averaged. Standard deviations were below 5 % of the mean in all cases.
1H (400 MHz) and 13C (100 MHz) NMR spectra of testosterone and its enzymatic conversion products were obtained on Bruker spectrometer (Bruker Avance II 400 MHz) in the solvent indicated.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the European Union, projects INDOX (KBBE‐2013‐7‐613549) and EnzOx2 (H2020‐BBI‐PPP‐2015‐2‐1–720297). We would also like to thank Tino Koncz and Josephine Lippert for their assistance as well as Julia Althammer (JenaBios GmbH) for NMR support.
J. Kiebist, K.-U. Schmidtke, J. Zimmermann, H. Kellner, N. Jehmlich, R. Ullrich, D. Zänder, M. Hofrichter, K. Scheibner, ChemBioChem 2017, 18, 563.
References
- 1. Donova M. V., Egorova O. V., Appl. Microbiol. Biotechnol. 2012, 94, 1423–1447; [DOI] [PubMed] [Google Scholar]; Rupprecht R., Psychoneuroendocrinology 2003, 28, 139–168. [DOI] [PubMed] [Google Scholar]
- 2. Tong W.-Y., Dong X., Recent Pat. Biotechnol. 2009, 3, 141–153. [DOI] [PubMed] [Google Scholar]
- 3. Fernandes P., Cruz A., Angelova B., Pinheiro H. M., Cabral J. M. S., Enzyme Microb. Technol. 2003, 32, 688–705. [Google Scholar]
- 4. Beneventi E., Ottolina G., Carrea G., Panzeri W., Fronza G., Lau P. C. K., J. Mol. Catal. B 2009, 58, 164–168. [Google Scholar]
- 5. Bhatti H. N., Khera R. A., Steroids 2012, 77, 1267–1290. [DOI] [PubMed] [Google Scholar]
- 6. Kristan K., Rižner T. L., J. Steroid Biochem. 2012, 129, 79–91. [DOI] [PubMed] [Google Scholar]
- 7. Urlacher V. B., Girhard M., Trends Biotechnol. 2012, 30, 26–36. [DOI] [PubMed] [Google Scholar]
- 8. Venkataraman H., de Beer S. B. A., van Bergen L. A. H., van Essen N., Geerke D. P., Vermeulen N. P. E., Commandeur J. N. M., ChemBioChem 2012, 13, 520–523. [DOI] [PubMed] [Google Scholar]
- 9. Rea V., Kolkman A. J., Vottero E., Stronks E. J., Ampt K. A. M., Honing M., Vermeulen N. P. E., Wijmenga S. S., Commandeur J. N. M., Biochemistry 2012, 51, 750–760. [DOI] [PubMed] [Google Scholar]
- 10. Bernhardt R., Urlacher V. B., Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. [DOI] [PubMed] [Google Scholar]
- 11. Hofrichter M., Ullrich R., Curr. Opin. Chem. Biol. 2014, 19, 116–125. [DOI] [PubMed] [Google Scholar]
- 12. Hofrichter M., Kellner H., Pecyna M. J., Ullrich R., Adv. Exp. Med. Biol. 2015, 851, 341–368. [DOI] [PubMed] [Google Scholar]
- 13. Wang X., Peter S., Kinne M., Hofrichter M., Groves J. T., J. Am. Chem. Soc. 2012, 134, 12897–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ullrich R., Nüske J., Scheibner K., Spantzel J., Hofrichter M., Appl. Environ. Microbiol. 2004, 70, 4575–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anh D. H., Ullrich R., Benndorf D., Svatoś A., Muck A., Hofrichter M., Appl. Environ. Microbiol. 2007, 73, 5477–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gröbe G., Ullrich R., Pecyna M. J., Kapturska D., Freidrich S., Hofrichter M., Scheibner K., AMB Express 2011, 1, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poraj-Kobielska M., Kinne M., Ullrich R., Scheibner K., Kayser G., Hammel K. E., Hofrichter M., Biochem. Pharmacol. 2011, 82, 789–796. [DOI] [PubMed] [Google Scholar]
- 18. Poraj-Kobielska M., Atzrodt J., Holla W., Sandvoss M., Gröbe G., Scheibner K., Hofrichter M., J. Labelled Compd. Radiopharm. 2013, 56, 513–519. [DOI] [PubMed] [Google Scholar]
- 19. Kiebist J., Holla W., Heidrich J., Poraj-Kobielska M., Sandvoss M., Simonis R., Gröbe G., Atzrodt J., Hofrichter M., Scheibner K., Bioorg. Med. Chem. 2015, 23, 4324–4332. [DOI] [PubMed] [Google Scholar]
- 20. Babot E. D., del Rio J. C., Cañellas M., Sancho F., Lucas F., Guallar V., Kalum L., Lund H., Gröbe G., Scheibner K., Ullrich R., Hofrichter M., Martinez A. T., Gutiérrez A., Appl. Environ. Microbiol. 2015, 81, 4130–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poraj-Kobielska M., Scheibner K., Gröbe G., Kiebist J., Grün M., Ullrich R., Hofrichter M., 2014, DE 102014005371 A1.
- 22. Hofrichter M., Ullrich R., Pecyna M. J., Liers C., Lundell T., Appl. Microbiol. Biotechnol. 2010, 87, 871–897. [DOI] [PubMed] [Google Scholar]
- 23. Cuomo C. A., Untereiner W. A., Ma L.-J., Grabherr M., Birren B. W., Genome Announc. 2015, 3, e00021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X. W., Lombard L., Groenewald J. Z., Li J., Videira S. I. R., Samson R. A., Liu X. Z., Crous P. W., Persoonia-Mol. Phylogeny Evol. Fungi 2015, 36, 83–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Babot E. D., del Río J. C., Kalum L., Martínez A. T., Gutiérrez A., Biotechnol. Bioeng. 2013, 110, 2323–2332. [DOI] [PubMed] [Google Scholar]
- 26. Babot E. D., del Río J. C., Kalum L., Martínez A. T., Gutiérrez A., ChemCatChem 2015, 7, 283–290. [Google Scholar]
- 27. Peter S., Karich A., Ullrich R., Gröbe G., Scheibner K., Hofrichter M., J. Mol. Catal. B 2014, 103, 47–51. [Google Scholar]
- 28. Lund H., Kalum L., Hofrichter M., Sebastian P., Epoxidation Using Peroxygenase, 2013, WO 2013144105 A1.
- 29. Majcherczyk A., Johannes C., Hüttermann A., Appl. Microbiol. Biotechnol. 1999, 51, 267–276. [DOI] [PubMed] [Google Scholar]
- 30. Kluge M., Ullrich R., Scheibner K., Hofrichter M., Appl. Microbiol. Biotechnol. 2007, 75, 1473–1478. [DOI] [PubMed] [Google Scholar]
- 31. Poraj-Kobielska M., Kinne M., Ullrich R., Scheibner K., Hofrichter M., Anal. Biochem. 2012, 421, 327–329. [DOI] [PubMed] [Google Scholar]
- 32. Ullrich R., Hofrichter M., FEBS Lett. 2005, 579, 6247–6250. [DOI] [PubMed] [Google Scholar]
- 33. Karich A., Scheibner K., Ullrich R., Hofrichter M., J. Mol. Catal. B 2016, 134, 238–246. [Google Scholar]
- 34. Park J.-B., Clark D. S., Biotechnol. Bioeng. 2006, 93, 1190–1195. [DOI] [PubMed] [Google Scholar]
- 35. Hernández-Ruiz J., Arnao M. B., Hiner A. N. P., García-Cánovas F., Acosta M., J. Biochem. 2001, 354, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Omura T., Biochem. Biophys. Res. Commun. 2005, 338, 404–409. [DOI] [PubMed] [Google Scholar]
- 37. Urzúa U., Larrondo L. F., Lobos S., Larraín J., Vicuña R., FEBS Lett. 1995, 371, 132–136. [DOI] [PubMed] [Google Scholar]
- 38. Barková K., Kinne M., Ullrich R., Hennig L., Fuchs A., Hofrichter M., Tetrahedron 2011, 67, 4874–4878. [Google Scholar]
- 39. Molina-Espeja P., Cañellas M., Plou F. J., Hofrichter M., Lucas F., Guallar V., Alcalde M., ChemBioChem 2016, 17, 341–349. [DOI] [PubMed] [Google Scholar]
- 40. Murphy R. C., Johnson K. M., J. Biol. Chem. 2008, 283, 15521–15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Misharin A. Y., Mehtiev A. R., Morozevich G. E., Tkachev Y. V., Timofeev V. P., Bioorg. Med. Chem. 2008, 16, 1460–1473. [DOI] [PubMed] [Google Scholar]
- 42. Uyanik C., Malay A., Ayna A. S., Hanson J. R., Hitchcock P. B., Steroids 2005, 70, 71–75. [DOI] [PubMed] [Google Scholar]
- 43. Jankowska R., Liu H.-J., Mhehe G. L., Chem. Commun. 1999, 1581–1582. [Google Scholar]
- 44. Nowrouzi F., Janetzko J., Batey R. A., Org. Lett. 2010, 12, 5490–5493. [DOI] [PubMed] [Google Scholar]
- 45. Salvador J. A. R., Leitão A. J. L., Sá e Melo M. L., Hanson J. R., Tetrahedron Lett. 2005, 46, 1067–1070. [Google Scholar]
- 46. Jennings B. H., Bengtson J. M., Steroids 1978, 31, 49–68. [DOI] [PubMed] [Google Scholar]
- 47. Michne W. F., Schroeder J. D., Bailey T. R., Neumann H. C., Cooke D., Young D. C., Hughes J. V., Kingsley S. D., Ryan K. A., Putz H. S., Shaw L. J., Dutko F. J., J. Med. Chem. 1995, 38, 3197–3206. [DOI] [PubMed] [Google Scholar]
- 48. Cussó O., Cianfanelli M., Ribas X., Klein Gebbink R. J. M., Costas M., J. Am. Chem. Soc. 2016, 138, 2732–2738. [DOI] [PubMed] [Google Scholar]
- 49. Carvalho J. F. S., Cruz Silva M. M., Sá e Melo M. L., Tetrahedron 2009, 65, 2773–2781. [Google Scholar]
- 50. Agematu H., Matsumoto N., Fujii Y., Kabumoto H., Doi S., Machida K., Ishikawa J., Arisawa A., Biosci. Biotechnol. Biochem. 2006, 70, 307–311. [DOI] [PubMed] [Google Scholar]
- 51. Ryan K. J., J. Biol. Chem. 1959, 234, 2006–2008. [PubMed] [Google Scholar]
- 52. Diczfalusy E., Acta Endocrinol. 1969, 61, 649–664. [DOI] [PubMed] [Google Scholar]
- 53. Makino T., Katsuyama Y., Otomatsu T., Misawa N., Ohnishi Y., Appl. Environ. Microbiol. 2014, 80, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bracco P., Janssen D. B., Schallmey A., Microb. Cell Fact. 2013, 12, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Molina-Espeja P., Ma S., Mate D. M., Ludwig R., Alcalde M., Enzyme Microb. Technol. 2015, 73, 29–33. [DOI] [PubMed] [Google Scholar]
- 56. Molina-Espeja P., Garcia-Ruiz E., Gonzalez-Perez D., Ullrich R., Hofrichter M., Alcalde M., Appl. Environ. Microbiol. 2014, 80, 3496–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Poraj-Kobielska M., Peter S., Leonhardt S., Ullrich R., Scheibner K., Hofrichter M., Biochem. Eng. J. 2015, 98, 144–150. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary