

Abstract
Evolocumab binds PCSK9, increasing low‐density lipoprotein cholesterol (LDL‐C) receptors and lowering LDL‐C. Target‐mediated evolocumab elimination is attributable to PCSK9 binding. As circulating PCSK9 and LDL‐C levels are primarily regulated by the liver, we compared evolocumab pharmacokinetics, pharmacodynamics, and safety in individuals with and without hepatic impairment. An open‐label, parallel‐group study evaluated the pharmacokinetics of evolocumab in hepatic‐impaired (Child‐Pugh Class A or B) or healthy adults. Participants were classified as having no, mild, or moderate hepatic impairment (n = 8/group) and received a single 140‐mg evolocumab dose. Assessments of unbound evolocumab and PCSK9 were made predose and postdose. Adverse events were monitored throughout the study. No significant association was observed between baseline PCSK9 and increasing level of hepatic impairment. No difference in extent and time course of PCSK9 or LDL‐C reduction was observed despite an apparent decrease in mean unbound evolocumab exposure with increasing hepatic impairment (Jonckheere‐Terpstra trend test; maximum serum concentration P = .18; area under the curve P = .09). Maximum reductions were observed in moderately impaired subjects vs healthy individuals: mean maximum serum concentration –34%; mean area under the concentration‐time curve (AUC) –47%. On average, unbound PCSK9 serum concentrations fell by >80% at 4 hours after a single evolocumab dose. Mean (95% confidence interval) maximum LDL‐C reductions in the healthy, mild, and moderate groups were –57% (–64% to –48%), –70% (–75% to –63%), and –53% (–61% to –43%), respectively. No safety risks were identified. These results support evolocumab use without dose adjustment in patients with active liver disease and mild or moderate hepatic impairment.
Keywords: evolocumab, AMG 145, hepatic impairment, PCSK9, low‐density lipoprotein, hypercholesterolemia, human monoclonal antibody, pharmacokinetics, pharmacodynamics
Little information is available on the effect of liver impairment on therapeutic monoclonal antibody (mAb) disposition. Current US Food and Drug Administration recommendations focus on small‐molecule drugs, whose elimination is dependent on hepatic metabolism, and offer no guidance for therapeutic proteins.1 Nonetheless, population pharmacokinetic analyses have been conducted for a small number of mAbs used in the oncology setting to treat metastatic liver disease and/or cancer associated with hepatitis. The limited available data suggest that hepatic impairment does not affect the pharmacokinetics of mAbs and are consistent with experimental evidence that multiple tissues other than the liver are also instrumental in eliminating antibodies.2, 3
Evolocumab (AMG 145), a 154‐kDa human mAb with high binding affinity for proprotein convertase subtilisin kexin type 9 (PCSK9), is approved for the treatment of hypercholesterolemia. PCSK9 is primarily synthesized in the liver and secreted into the blood. It acts as a major regulator of circulating low‐density lipoprotein cholesterol (LDL‐C) by binding to hepatic cell surface LDL‐C receptors, directing LDL‐C receptors for degradation, and thereby reducing the clearance of LDL‐C particles.4 Through its PCSK9‐lowering effect, administration of evolocumab increases the density of LDL‐C receptors and significantly reduces LDL‐C levels in patients with hypercholesterolemia.4, 5 In several clinical trials up to 52 weeks in duration, the safety and tolerability profile of evolocumab was similar to that of placebo.6, 7, 8, 9, 10
Like many therapeutic mAbs, evolocumab exhibits target‐mediated elimination via specific binding and complex formation with its target ligand (PCSK9),11, 12 in addition to the usual antibody clearance processes for endogenous immunoglobulin G (IgG) in the reticuloendothelial system.13, 14 Little is known, however, about the effect of hepatic impairment on PCSK9 production or elimination, which might influence the elimination of evolocumab. Therefore, it is unknown whether evolocumab pharmacokinetics, or its effect on lipoproteins, would be affected by hepatic impairment. Furthermore, because statins are contraindicated in patients with active liver disease, it is important to identify alternative therapies for patients with hypercholesterolemia and liver impairment.
This study was conducted to examine the pharmacokinetics of evolocumab, its effect on PCSK9 and LDL‐C levels, and its safety after a single 140‐mg subcutaneous (SC) dose to healthy volunteers or to individuals with mild or moderate hepatic impairment, as defined by Child‐Pugh score classification A or B.
Methods
Study Design
The protocol and study procedures were approved by the institutional review board at the study center. All participants provided written informed consent before study procedures were performed.
This was an open‐label, parallel‐group study evaluating the pharmacokinetics of evolocumab in hepatic‐impaired or healthy volunteers. Participants were assigned to 1 of 3 groups (n = 8 per group), depending on their degree of hepatic impairment (none, mild, or moderate), and each received a single 140‐mg SC dose (1 mL) of evolocumab. The approved dose regimens of evolocumab are 140 mg every 2 weeks (Q2W) and 420 mg monthly. The single dose of 140 mg was selected for evaluation in this study in order to better characterize any effect of hepatic impairment on both the linear and nonlinear (target‐mediated) portions of elimination as the 420‐mg dose is primarily in the linear dose range. Eligible participants were men and women aged 18 to 55 years who were otherwise healthy or had Child‐Pugh Class A or B hepatic impairment. Participants were required to have a calculated LDL‐C value of 70 to 190 mg/dL and a body mass index of 18 to 35 kg/m2 at the time of screening. No other lipid‐lowering treatment during the study was permitted.
On the morning of day 1, after an overnight fast of ≥10 hours, participants received a single 140‐mg SC dose (1 mL) of evolocumab via a prefilled autoinjector/pen. After completion of all study procedures on day 1, participants were discharged, returning at specified timepoints until the end of the study (8 weeks) for collection of blood samples for pharmacokinetic and pharmacodynamic assessments and completion of safety assessments (including blood samples for antidrug antibody analysis).
Investigational Product
Evolocumab was provided as a sterile, preservative‐free solution in a single‐use, disposable, hand‐held mechanical (spring‐based) prefilled autoinjector/pen that delivered a fixed SC dose of 140 mg.
Pharmacokinetic and Pharmacodynamic Assessments
Assessments of unbound evolocumab and unbound PCSK9 were made at predose and 4 hours postdose on day 1 and then on days 2, 3, 4, 6, 8, 11, 15, 22, 29, 43, 50, and 57.
Unbound evolocumab was measured in serum using a validated enzyme‐linked immunosorbent assay (ELISA). The pharmacokinetics ELISA was designed to measure unbound evolocumab in test samples by using highly specific anti‐idiotype antibodies for capture and detection of evolocumab. By virtue of binding to the antigen‐combining site of evolocumab, the anti‐idiotype antibodies bind to evolocumab that is not bound to PCSK9. The validated assay procedure requires accuracy (percentage difference from nominal concentration) and precision (percentage coefficient of variation) of ±15% and ≤15%, respectively, for the standard, and ±20% and ≤15%, respectively, for the quality control (QC) samples. The assay limits of quantification ranged from 0.8 μg/mL to 10 μg/mL, the accuracy (percentage bias) in QC samples ranged from –3% to –1%, and the precision (coefficient of variation) was 5% to 6%.
Unbound PCSK9 was measured in serum using a qualified ELISA method.15 Standard and QC samples were prepared in 100% fetal bovine serum because of the high and variable levels of endogenous PCSK9 in human serum. The QC samples in each run were 45, 140, and 500 ng/mL PCSK9 in fetal bovine serum. The validated assay procedure requires accuracy (percentage difference from nominal concentration) and precision (percentage coefficient of variation) of ±15% and ≤20%, respectively, for the standard and ±25% and ≤20%, respectively, for the QC samples. The assay lower and upper limits of quantification were 15 ng/mL and 1200 ng/mL, respectively, with accuracy in QC samples ranging from 2% to 3% and precision ranging from 6% to 9%.
A complete fasting lipid profile—total cholesterol, triglycerides, high‐density lipoprotein cholesterol, and ultracentrifuged (UC) LDL‐C—was measured in serum according to the pharmacokinetic collection schedule above with the exception of the 4‐hour postdose timepoint. Serum IgG levels and reported serum chemistry values were also analyzed.
Statistical Analysis
Noncompartmental pharmacokinetic analysis methods using Phoenix® WinNonlin® software (Certara, Princeton, New Jersey) were used to estimate the unbound evolocumab pharmacokinetic parameters: maximum unbound serum concentration of evolocumab (Cmax), time to Cmax (tmax), area under the concentration‐time curve from time 0 to the time of the last quantifiable concentration (AUClast), and AUC from time 0 to infinity (AUC∞). Serum concentrations below the lower limit of quantification were set to 0 for the estimation of pharmacokinetic parameters for each participant. AUC∞ was determined in the majority of subjects with the mean extrapolated AUC <20%.
To estimate differences in pharmacokinetic parameters (Cmax, AUClast, and AUC∞) with respect to level of hepatic impairment, an analysis of variance was performed on the log‐transformed pharmacokinetic parameters. Geometric mean ratios and their associated 90% confidence intervals (CIs) between subjects with impaired hepatic function (mild or moderate) and subjects with normal hepatic function were estimated and multiplied by 100 to present as a percentage of the healthy group. To test the association between increasing level of hepatic impairment (ordered: healthy, mild, and moderate impairment) and Cmax, AUClast, or AUC∞, the nonparametric Jonckheere‐Terpstra trend test was used.
Pharmacodynamic LDL‐C and PCSK9 data over time were analyzed using a repeated‐measures mixed‐effect model with level of hepatic impairment, day, and level of hepatic impairment‐by‐day interaction as independent variables, log‐transformed baseline value as covariate, and subject as random effect. An analysis of covariance model was performed on the area under the LDL‐C effect‐time curve (AUEC1‐57) for LDL, with level of hepatic impairment and log baseline LDL‐C as covariate. All LDL‐C, AUEC1‐57 for LDL, and PCSK9 data were log‐transformed before analysis. Trends between baseline LDL‐C or baseline PCSK9 and increasing level of hepatic impairment (ordered: healthy, mild, and moderate impairment) were evaluated with the nonparametric Jonckheere‐Terpstra trend test. A significance level of .05 was considered significant for all analyses.
All statistical analyses were carried out using SAS version 9.2 software (SAS Institute Inc, Cary, North Carolina).
Safety Analysis
The Medical Dictionary for Regulatory Activities version 16.0 was used to code all adverse events to a system organ class and a preferred term. The incidence of adverse events was summarized for all treatment‐emergent, serious treatment‐emergent, treatment‐related, and serious treatment‐related events and those leading to study withdrawal. The severity of each adverse event was graded using Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Anti‐evolocumab antibody formation was assessed just before evolocumab treatment, on day 29 and at the end of the study.
Results
Participants
In total, 24 participants were enrolled, including 8 each in the Child‐Pugh A and B categories and 8 healthy volunteers. All 24 participants received a single dose of evolocumab and completed the study.
Demographic and baseline characteristics of study participants are shown in Table 1. Most participants were men (83%) and white (75%). The mean (standard deviation) age and body mass index of participants at baseline were similar among groups. Mean baseline concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were lowest in the healthy group and higher in the mild and moderate hepatic impairment groups. Mean baseline concentrations of albumin were highest in the healthy group and lowest in the moderate hepatic impairment group. Mean baseline serum IgG values increased with increasing severity of liver impairment. Median Child‐Pugh scores for participants with mild and moderate impairment were at the lower end of their respective defined Child‐Pugh ranges. Overall, 13 of 16 participants (81%) had a history of viral hepatitis as the etiology of their liver impairment. Mean baseline serum PCSK9 values did not appear to be altered in the presence of mild or moderate hepatic impairment, with no significant association observed with increasing severity of liver impairment (P = .43 for the Jonckheere‐Terpstra trend test). Mean baseline serum LDL‐C decreased with increasing severity of liver impairment (P = .12 for the Jonckheere‐Terpstra trend test).
Table 1.
Participant Demographics and Baseline Characteristics
| Healthy Volunteers (n = 8) | Mild Hepatic Impairment (Child‐Pugh Class A) (n = 8) | Moderate Hepatic Impairment (Child‐Pugh Class B) (n = 8) | |
|---|---|---|---|
| Sex, n (%) | |||
| Female | 1 (12.5) | 2 (25.0) | 1 (12.5) |
| Male | 7 (87.5) | 6 (75.0) | 7 (87.5) |
| Race, n (%) | |||
| Black, African‐American | 3 (37.5) | 2 (25.0) | 1 (12.5) |
| White | 5 (62.5) | 6 (75.0) | 7 (87.5) |
| Age (years) | |||
| Mean (SD) | 45.9 (5.1) | 50.5 (4.4) | 49.8 (4.9) |
| Range | 39‐52 | 42‐55 | 42‐55 |
| BMI (kg/m2) | |||
| Mean (SD) | 28.4 (3.8) | 26.7 (3.1) | 28.7 (3.9) |
| Serum albumin (g/dL) | |||
| Mean (SD) | 4.2 (0.4) | 3.9 (0.3) | 3.5 (0.5) |
| Liver function | |||
| Mean (SD) AST (U/L) | 20.1 (4.8) | 48.8 (31.4) | 77.3 (45.1) |
| Mean (SD) ALT (U/L) | 21.9 (10.8) | 67.1 (45.5) | 64.0 (45.8) |
| Mean (SD) AP (U/L) | 60.6 (13.3) | 59.4 (8.1) | 113.0 (52.3) |
| Child‐Pugh score, median (range) | |||
| Median (range) | NA | 5 (5‐6) | 7 (7‐9) |
| History of viral hepatitis, n (%) | |||
| Yes | 0 | 8 (100) | 5 (62.5) |
| No | 8 (100) | 0 | 3 (37.5) |
| Serum IgG (mg/dL) | |||
| Mean (SD) | 1208 (322) | 1348 (460) | 1747 (474) |
| Unbound PCSK9 (ng/mL) | |||
| Mean (SD) | 322 (45) | 390 (73) | 343 (111) |
| Ultracentrifugation LDL‐C (mg/dL) | |||
| Mean (SD) | 121 (32) | 112 (31) | 96 (38) |
ALT, alanine aminotransferase; AP, alkaline phosphatase; AST, aspartate aminotransferase; BMI, body mass index; IgG, immunoglobulin G; LDL‐C, low‐density lipoprotein cholesterol; n, number of participants with the given characteristic; NA, not applicable; PCSK9, proprotein convertase subtilisin kexin type 9; SD, standard deviation.
Serum Unbound Evolocumab Pharmacokinetics
Unbound evolocumab serum concentrations as a function of time are shown in Figure 1, and derived unbound evolocumab pharmacokinetic parameters are presented by treatment group in Figure 2.
Figure 1.
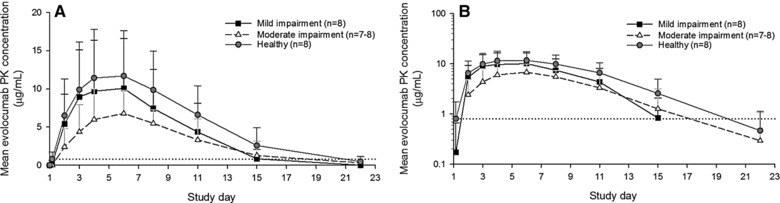
Mean (standard deviation) serum unbound evolocumab concentration‐time profiles from healthy individuals and those with hepatic impairment after a single 140‐mg subcutaneous evolocumab dose, shown as linear‐linear (A) and as log‐linear (B) plots. The horizontal dashed line represents the detection limit for unbound evolocumab (0.8 μg/mL). PK, pharmacokinetic.
Figure 2.
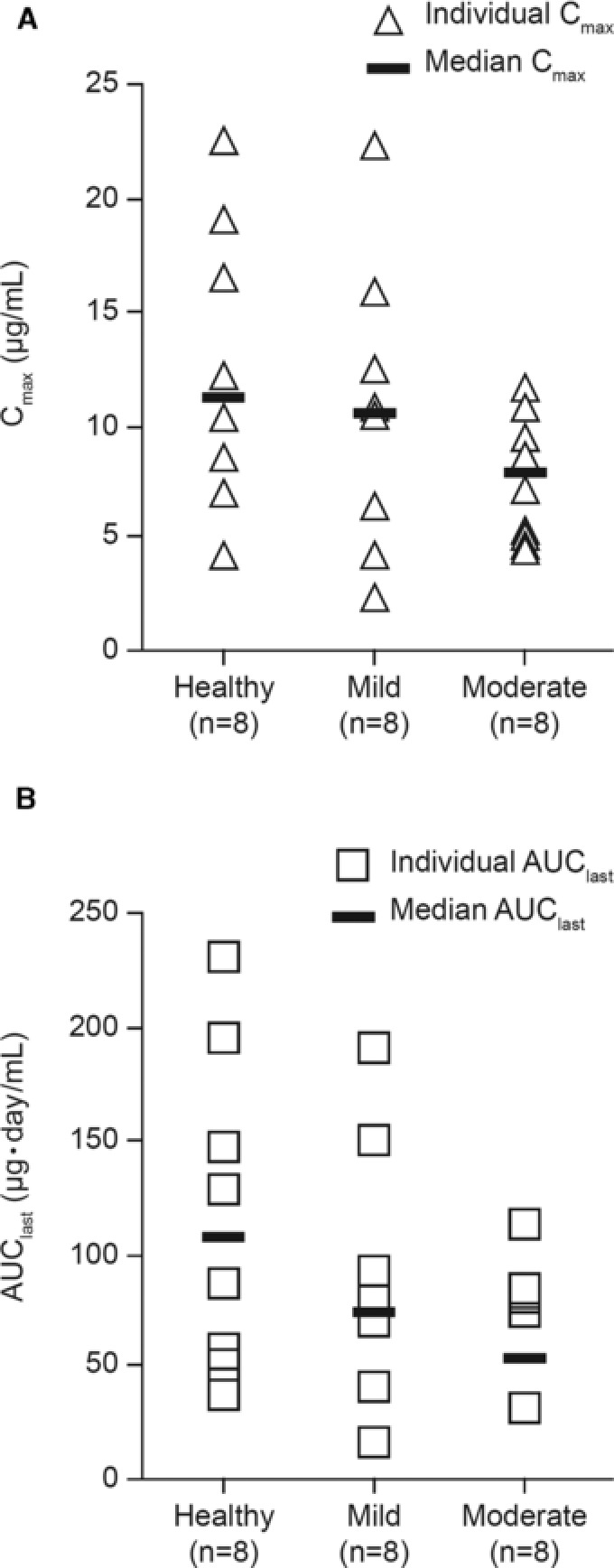
Scatter plots of individual and median values for (A) maximum unbound serum evolocumab concentration (Cmax) and (B) area under the concentration‐time curve from time 0 to the time of the last quantifiable concentration (AUClast).
Reductions in the geometric mean ratios of Cmax, AUClast, and AUC∞ were seen in hepatic‐impaired subjects compared with healthy individuals. Maximum observed reductions were 34% for Cmax, 47% for AUClast, and 43% for AUC∞ in subjects with moderate hepatic impairment compared with healthy individuals (Jonckheere‐Terpstra trend test P = .18, P = .09, and P = .06, respectively) (Table 2).
Table 2.
Statistical Analysis of Unbound Evolocumab Pharmacokinetic Parameters in Healthy Individuals and Those With Hepatic Impairment After a Single 140‐mg Subcutaneous Evolocumab Dose
| Healthy Volunteers (n = 8) | Mild Hepatic Impairment (Child‐Pugh Class A) (n = 8) | Moderate Hepatic Impairment (Child‐Pugh Class B) (n = 8) | P‐Valuea | |
|---|---|---|---|---|
| AUClast (μg · day/mL) | ||||
| Least‐squares geometric mean | 96.8 | 58.8 | 51.5 | 0.09 |
| Ratio (impaired to healthy, %) | – | 60.8 | 53.2 | |
| 90% CI of ratio (impaired to healthy, %) | – | (32.1, 115.3) | (28.1, 100.9) | |
| AUC∞ (μg · day/mL) | ||||
| Least‐squares geometric mean | 107.0 | 84.8b | 60.6 | 0.06 |
| Ratio (impaired to healthy, %) | – | 79.3 | 56.6 | |
| 90% CI of ratio (impaired to healthy, %) | – | (45.8, 137.3) | (33.3, 96.2) | |
| Cmax (μg/mL) | ||||
| Least‐squares geometric mean | 11.0 | 8.6 | 7.3 | 0.18 |
| Ratio (impaired to healthy, %) | – | 78.5 | 66.0 | |
| 90% CI of ratio (impaired to healthy, %) | – | (47.8, 129.1) | (40.1, 108.4) | |
| tmax (days) | ||||
| Median | 5.0 | 5.0 | 4.5 | |
| Range | 3.0‐7.0 | 1.0‐5.0 | 3.0‐10 | |
AUC∞, area under the concentration‐time curve from time 0 to infinity; AUClast, area under the drug concentration‐time curve from time zero to time of last quantifiable concentration; CI, confidence interval; Cmax, maximum observed drug concentration; tmax, time to reach Cmax.
Jonckheere‐Terpstra test.
AUC∞ was not estimated in 1 subject, as the rate constant, λz, associated with the terminal elimination phase could not be estimated due to limited quantifiable data.
There was considerable overlap in the individual Cmax and AUClast values between the participants in the healthy group and those in the groups with hepatic impairment. Less variability was seen in values for Cmax and AUClast in participants with moderate hepatic impairment compared with those with less or no hepatic impairment (Figure 2). There was no relationship between AUClast and individual Child‐Pugh components (ascites, albumin, or total bilirubin) or with hepatitis C status, triglycerides, or AST (data not shown). The median time to maximum concentration (tmax) did not appear to be influenced by the degree of hepatic impairment, with values of 5.0 days, 5.0 days, and 4.5 days in healthy individuals, mild hepatic impairment, and moderate hepatic impairment, respectively.
Unbound Serum PCSK9 and UC LDL‐C Pharmacodynamics
The effect of a 140‐mg SC evolocumab dose on unbound serum PCSK9 and LDL‐C determined by UC are shown in Figures 3 and 4, respectively.
Figure 3.
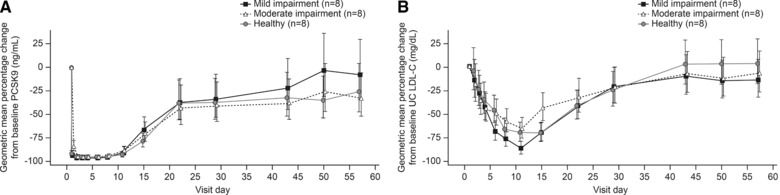
Geometric mean percentage (±95% confidence intervals) changes from baseline in (A) proprotein convertase subtilisin kexin type 9 (PCSK9) and (B) ultracentrifugation (UC) low‐density lipoprotein cholesterol (LDL‐C) by degree of hepatic impairment.
Figure 4.
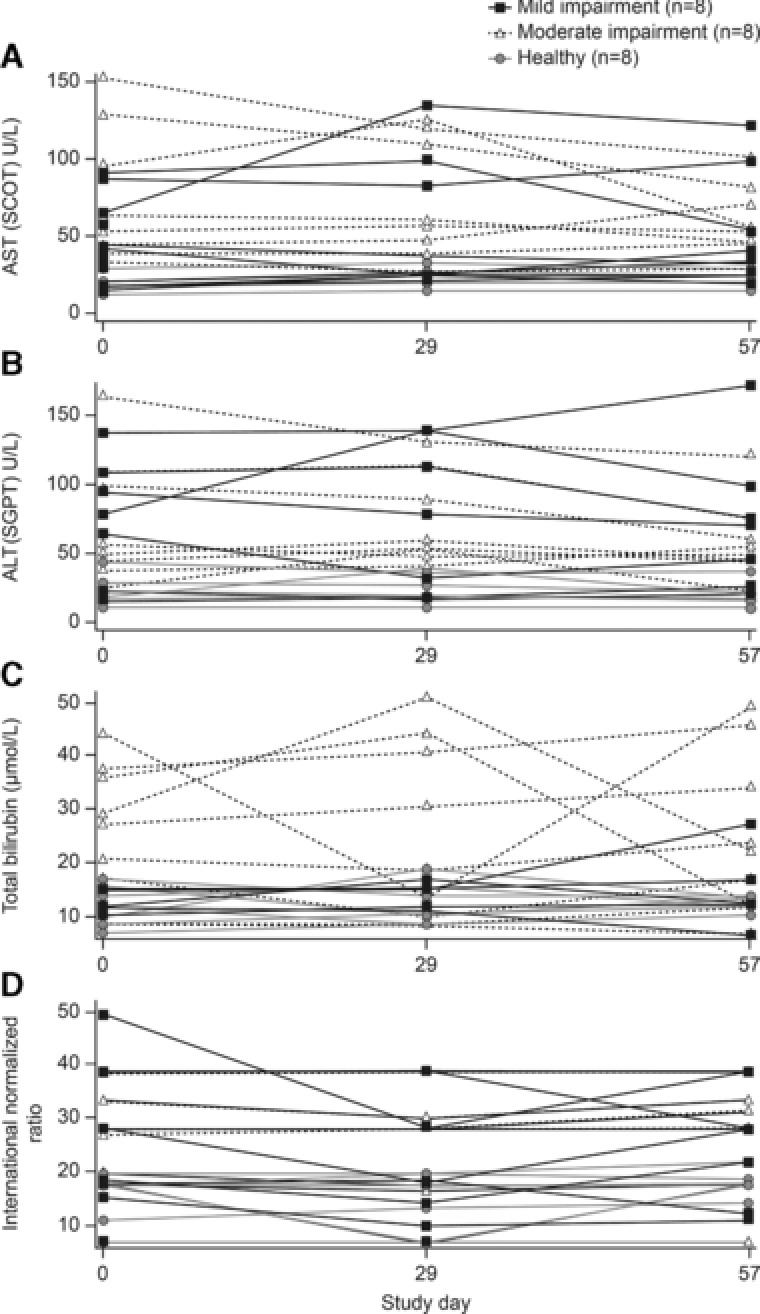
Plots of individual values for (a) aspartate aminotransferase (AST), (b) alanine aminotransferase (ALT), (c) total bilirubin, and (d) international normalized ratio at baseline and on days 29 and 57.
On average, unbound PCSK9 serum concentrations fell by >80% at 4 hours after a single SC dose of evolocumab, reaching a nadir of ≥95% at 24 hours postdose. Suppression of unbound PCSK9 was sustained for 10 days before a gradual return toward baseline levels. The extent and time course of PCSK9 suppression in individuals with hepatic impairment were similar to those seen in healthy volunteers.
LDL‐C levels were lowered after a single 140‐mg SC dose of evolocumab in all 3 groups. The extent and time course of LDL‐C lowering were generally similar between healthy individuals and those with hepatic impairment. The mean LDL‐C nadir occurred between study days 11 and 15 in all 3 groups, followed by a return toward baseline levels. The geometric mean (95% CI) maximum LDL‐C reduction in healthy individuals was –57% (–64%, –48%). The 95% CI of the geometric mean for maximum LDL‐C percentage changes from baseline in patients with mild and moderate hepatic impairment overlapped with those of the healthy individuals (mild, –70% [–75%, –63%]; moderate, –53% [–61%, –43%]), respectively. A measure of the total LDL‐C reduction from study day 1 to day 57 was also estimated by AUEC1‐57. Mean AUEC1‐57 for LDL‐C in patients with mild and moderate hepatic impairment was 4320 day·mg/mL and 4687 day·mg/dL, respectively, and was similar to that in healthy individuals (4684 day·mg/dL). No significant association was observed in AUEC1‐57 for LDL with increasing severity of impairment (Jonckheere‐Terpstra trend test P = .56).
Safety of Evolocumab in Mild and Moderate Hepatic Impairment
Seven participants experienced treatment‐emergent adverse events (healthy volunteers, n = 1 [13%]; mild hepatic impairment, n = 4 [50%]; moderate hepatic impairment, n = 2 [25%]). The most frequent treatment‐emergent adverse event was diarrhea, which occurred in 2 subjects (8.3% overall), 1 each in the mild and moderate hepatic impairment groups. All treatment‐emergent adverse events were mild (CTCAE grade 1) or moderate (CTCAE grade 2) in severity. One serious treatment‐emergent adverse event (depression, requiring voluntary hospital admission) was reported in 1 participant (4%) in the mild hepatic impairment group who had a >40‐year history of depression. This and the other treatment‐emergent adverse events were determined by the investigator to be unrelated to evolocumab treatment, and no participant withdrew from the study as the result of an adverse event.
No trends indicative of clinically important treatment‐related laboratory abnormalities, including serum albumin and international normalized ratio as a measure of hepatic synthetic function, were observed in this study, and no anti‐evolocumab antibodies were detected. No subject met Hy's Law criteria16 for potential hepatotoxicity or drug‐induced liver injury during the study. Six subjects had significant elevations in 1 postbaseline liver function test (Figure 4). No participant had an ALT postbaseline assessment that was >3 × the upper limit of normal (ULN). Three individuals with moderate hepatic impairment had AST >3 × the ULN at a single postbaseline assessment, and 1 individual with mild hepatic impairment had AST levels >3 × the ULN at both postbaseline assessments (Table 3). No consistent pattern of AST elevation was observed in these 4 individuals. In 2 participants, the AST value was >3 × ULN at baseline and declined progressively over the course of the study, falling below 3 × ULN by day 57. In 1 participant, the AST value at baseline was slightly below 3 × ULN, transiently rose slightly above 3 × ULN on day 29, and then declined again to below 3 × ULN by day 57. The remaining participant's AST level was below 3 × ULN at baseline and was then elevated above 3 × ULN on day 29. This individual's AST level remained above 3 × ULN on day 57 but had declined from the day 29 level.
Table 3.
Summary of Participants With Postbaseline AST >3 × Upper Limit of Normal Elevations
| Hepatic | ALT (U/mL) | AST (U/mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| Impairment Group | Viral Hepatitis History? | Concomitant Medications During the Study | Baseline | Day 29 | Day 57 | Baseline | Day 29 | Day 57 |
| Moderate | No | Lactulose 60 mL/day | 45 | 60 | 45 | 98 | 126 | 56 |
| Spironolactone 25 mg/day | ||||||||
| Sertraline 100 mg/day | ||||||||
| Moderate | Yes | Furosemide 40 mg bid | 97 | 88 | 60 | 154 | 121 | 102 |
| Lactulose 10 g/day | ||||||||
| Pantoprazole 60 mg/day | ||||||||
| Phytonadione 5 mg bid | ||||||||
| Spironolactone 100 mg bid | ||||||||
| Rifaximin 550 mg tid | ||||||||
| Thiamine hydrochloride 100 mg/day | ||||||||
| Vitamin B6 100 mg/day | ||||||||
| Tramadol 100 mg prn | ||||||||
| Moderate | Yes | Metformin 500 mg bid | 164 | 130 | 121 | 130 | 110 | 82 |
| Mild | Yes | Hydrochlorothiazide 25 mg/day | 78 | 139 | 98 | 67 | 135 | 123 |
| Loratadine 10 mg/day | ||||||||
| Losartan potassium 50 mg/day | ||||||||
| Human insulin 70 U bid | ||||||||
| Clarithromycin 500 mg bid | ||||||||
ALT, alanine transaminase; AST, aspartate aminotransferase; bid, twice daily; prn, as required; tid, 3 times daily.
Three individuals with moderate hepatic impairment had total bilirubin levels >2 × ULN at a single postbaseline assessment (Table 4).
Table 4.
Summary of Participants With Postbaseline Bilirubin >2 × Upper Limit of Normal Elevations
| Hepatic | ALT (U/mL) | Total Bilirubin (μmol/L) | ||||||
|---|---|---|---|---|---|---|---|---|
| Impairment Group | Viral Hepatitis History? | Concomitant Medications During the Study | Baseline | Day 29 | Day 57 | Baseline | Day 29 | Day 57 |
| Moderate | Yes | Human insulin 70‐76 U bid | 24 | 53 | 23 | 38 | 41 | 46 |
| Human insulin 8 U prn | ||||||||
| Furosemide 60 mg bid | ||||||||
| Omeprazole 20 mg/day | ||||||||
| Propranolol 20 mg tid | ||||||||
| Spironolactone 100 mg bid | ||||||||
| Diphenhydramine 25 mg prn | ||||||||
| Trazodone 50 mg prn | ||||||||
| Cephalexin 500 mg qid | ||||||||
| Moderate | No | Amiloride 5 mg (1 dose) | 56 | 48 | 48 | 29 | 51 | 22 |
| Furosemide 20 mg (1 dose) | ||||||||
| Human insulin 12 U bid | ||||||||
| Pregabalin 75 mg (1 dose) | ||||||||
| Moderate | No | None | 38 | 35 | 50 | 45 | 14 | 50 |
ALT, alanine transaminase; bid, twice daily; tid, 3 times daily; prn, as required; qid, 4 times daily.
Discussion
Although hepatic impairment studies are generally deemed unnecessary for mAbs, the liver is the primary organ regulating circulating levels of both endogenous PCSK9 and LDL‐C. Therefore, a dedicated hepatic impairment study was conducted to examine the pharmacokinetics and pharmacodynamics of a single 140‐mg SC evolocumab dose.
Mean unbound exposures and the observed relatively large variability in the healthy participants from this study were consistent with results from previously conducted single‐dose studies in healthy volunteers.17 Considerable overlap in the Cmax and AUC values was observed between healthy subjects and those with hepatic impairment (Figure 2). Given the variability and the typical sample size of hepatic impairment studies, it is difficult to ascertain the reason for the observed reduction in mean pharmacokinetics associated with hepatic impairment. In addition, no significant trends in either AUClast or Cmax with advancing degree of liver impairment were noted. This is in contrast to typical oral small‐molecule drugs that are often metabolized and/or excreted by the liver, and for which liver impairment may increase drug exposure, often necessitating dose reduction. It is generally accepted, however, that general IgG mAb clearance is mediated by the same processes that recycle and remove endogenous IgG antibodies and, therefore, is not likely to be affected by functional hepatic impairment.18 Estimates from physiologically based pharmacokinetic models suggest that hepatic contribution to mAb elimination ranges from 16% to 40%.3
There is a paucity of pharmacokinetic data for therapeutic mAbs in hepatic impairment. The only other study that we are aware of was recently published and characterized the impact of hepatic impairment on the antibody‐drug conjugate (ADC), brentuximab vedotin in 7 patients.19 In addition, there are population pharmacokinetic modeling analyses from 3 other therapeutic mAbs (cetuximab, panitumumab, and ipilimumab). Based on these analyses, dose adjustments were recommended only for the ADC because of the drug component. However, no dose change was recommended based on antibody pharmacokinetics or in any of the other mAbs in their respective package labeling, consistent with our current findings.2
Evolocumab is eliminated by binding to its target (PCSK9) and by endogenous IgG catabolic routes. Mean baseline serum PCSK9 levels, as well as the extent and time course of unbound PCSK9 lowering by evolocumab, were similar between healthy individuals and those with mild or moderate hepatic impairment. This suggests that the net production and degradation of PCSK9 are not altered in these patients and, therefore, that no changes are expected in the target‐mediated clearance of evolocumab. Evolocumab is also thought to undergo general processes of IgG clearance, which are balanced between endosomal salvage, mediated by a saturable neonatal Fc receptor (FcRN), and lysosomal degradation.13, 20 FcRN can effectively recycle proteins such as IgG and albumin, thereby increasing their residence time in the circulation.21, 22 In the present study, mean baseline serum IgG levels increased with increasing severity of liver impairment, consistent with previously reported studies.23, 24 This represents a potential mechanism by which therapeutic mAb clearance might be increased because of competition for the FcRN salvage pathway, as is hypothesized to occur in patients with myeloma or during the therapeutic use of high‐dose intravenous immunoglobulin (IVIG) therapy in patients with autoimmune disorders.25, 26 The mean IgG levels in the present study (1208‐1747 mg/dL) were considerably lower than those seen in patients with myeloma and gammopathies, in whom levels can exceed 7000 mg/dL, or levels seen during IVIG use, which can result in mean peak IgG levels of 2500 mg/dL.27 In addition, there was no correlation between baseline serum IgG level and unbound evolocumab AUClast (r2 = 0.002) or evolocumab half‐life (r2 = 0.0378). Further evidence for a lack of general FcRN impairment in patients with hepatic impairment comes from a study of IgG and albumin turnover in patients with chronic active liver disease. In these patients the fractional clearance rates of both IgG and albumin were not significantly different from those of healthy individuals, and changes in serum levels were attributed to changes in production rates.28 Collectively, these data are consistent with the lack of effect on unbound evolocumab clearance in patients with hepatic impairment and with the pharmacokinetic findings in this study. Finally, no subject developed anti‐evolocumab antibodies as a potential mechanism for increasing evolocumab clearance. Overall, the small number of participants and, as noted above, the known large variability in evolocumab pharmacokinetics are likely explanations for the mean pharmacokinetic observations. There were no findings to explain the reduction in evolocumab pharmacokinetics with increasing degree of liver impairment. However, similar antibody (ADC) pharmacokinetic findings occurred with brentuximab vedotin, where 6 lymphoma patients with moderate hepatic impairment had a 35% mean reduction in the AUC.19 No mechanism for this change was identified, although the authors hypothesized that lower albumin might be somehow associated with the pharmacokinetic changes. We saw no such relationship between serum albumin concentrations and AUC.
In addition to clearance, a modest change in the extent of absorption (absolute bioavailability) after SC injection may be a potential cause for a reduced exposure. The understanding of presystemic processes that might affect absorption are poorly understood. Low colloid osmotic pressure in the circulation of these patients is one possible identified factor that might affect absorption of a mAb from an SC site.29 However, the lower AUC observed with brentuximab vedotin after intravenous administration19 suggests that there is a change in systemic clearance of mAbs in hepatic disease, although its cause remains obscure.30
The timing and magnitude of LDL‐C lowering after a dose of evolocumab were consistent with those seen in other studies in patients and healthy volunteers.5, 6, 8, 17, 31 For example, the randomized, double‐blind, placebo‐controlled, phase 2 MENDEL study showed that monotherapy with evolocumab 140 mg Q2W in 45 patients with hypercholesterolemia (baseline LDL‐C 3.6 mmol/L) reduced LDL‐C from baseline by 51% (95% CI –56% to –46%) after 12 weeks of treatment.31 Similarly, the phase 3 placebo‐controlled trial MENDEL‐2 showed that evolocumab 140 mg Q2W used as monotherapy in 153 patients with primary hypercholesterolemia reduced LDL‐C from baseline by 57% (95% CI –59.5% to –54.6%) after 12 weeks.8 In the present study evolocumab was able to lower LDL‐C effectively in patients with mild or moderate hepatic impairment, and removal of LDL‐C through this mechanism was functional in these individuals. Mean baseline LDL‐C levels decreased with increasing liver impairment, consistent with reports from patients with chronic liver disease and individuals with hepatitis C.30, 32 These data suggest that evolocumab may be effective in treating individuals with primary hyperlipidemia or mixed dyslipidemia who have active liver disease, a population in whom statin use is contraindicated.
Safety and tolerability profiles in the present study were consistent with previous studies of evolocumab6, 7, 8, 9, 10 and did not identify any risks. No anti‐evolocumab antibodies were detected in any participant. Importantly, there was no evidence of any adverse effect on hepatic safety in individuals with pre‐existing hepatic impairment.
Conclusions
The unbound evolocumab pharmacokinetic data from the present study demonstrated a mean decrease in exposure among subjects with mild or moderate hepatic impairment. The magnitude of mean lower exposure with the expected robust LDL‐C and PCSK9 reductions observed in this study indicates that any real pharmacokinetic changes were of little clinical significance. This observation is consistent with the dose‐response relationship observed in the phase 2 MENDEL and LAPLACE studies, where the 140‐mg Q2W dose of evolocumab approached the flat upper part of the dose‐response curve.5, 31 For example, in the LAPLACE study, a 30% increase in evolocumab dose from 105 mg SC Q2W to 140 mg SC Q2W resulted in an additional ∼5% reduction in LDL‐C at week 12.5 As a consequence, changes to the LDL‐C‐lowering effect are relatively insensitive to changes in pharmacokinetics. Baseline PCSK9 and its suppression via a single 140‐mg SC evolocumab dose in individuals with hepatic impairment were similar to those seen in healthy individuals, suggesting that PCSK9 turnover rates are not different. The observed LDL‐C lowering in these individuals was consistent with other experience in healthy volunteers and in patients with primary hyperlipidemia or mixed dyslipidemia. No safety risks were identified in this population. Collectively, no dose adjustment is warranted in patients with mild or moderate hepatic impairment as indicated by the results of this study. These data suggest that evolocumab may be effective in treating primary hyperlipidemia or mixed dyslipidemia in patients who have active liver disease, in whom statins are contraindicated. Individuals with severe hepatic impairment were not included, and so the results from this study cannot be extrapolated to that population.
Author Contributions
M.G.E., J.G.S., S.M.W., L.H., J.P.G., M.G., R.X., and C.S.D. wrote and edited the manuscript. M.G.E., J.G.S., J.P.G., L.H., O.E., and J.J. performed data analysis. C.S.D., S.M.W., O.E., M.G., J.J., R.X., and M.G.E. designed the research.
Declarations of Conflicting Interests
All authors were employees and stockholders of Amgen Inc. at the time this work was conducted. Dan Booth of Bioscript Medical Ltd provided medical writing assistance.
Acknowledgments
The authors thank Thomas Marbury for his contribution to this study. Dan Booth of Bioscript Medical Ltd provided medical writing assistance. Funding for this medical writing support was provided by Amgen (Europe) GmbH. Editorial support was provided by Carine Thual and Lucy Hyatt of Amgen (Europe) GmbH and Shauna Hutton of Amgen Inc.
References
- 1. US Department of Health and Human Services , Food and Drug Administration , Center for Drug Evaluation and Research (CDER) , Center for Biologics Evaluation and Research (CBER). Guidance for industry. Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. 2003. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072123.pdf. Accessed September 2014.
- 2. Yang J, Shord S, Zhao H, Men Y, Rahman A. Are hepatic impairment studies necessary for therapeutic proteins? Clin Ther. 2013;35(9):1444–1451. [DOI] [PubMed] [Google Scholar]
- 3. Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically‐based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet. 2013;52(2):83–124. [DOI] [PubMed] [Google Scholar]
- 4. Davidson MH. Emerging low‐density lipoprotein therapies: targeting PCSK9 for low‐density lipoprotein reduction. J Clin Lipidol. 2013;7(suppl 3):S11–S15. [DOI] [PubMed] [Google Scholar]
- 5. Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL‐C Assessment with Proprotein Convertase Subtilisin Kexin Type 9 Monoclonal Antibody Inhibition Combined with Statin Therapy (LAPLACE)‐Thrombolysis in Myocardial Infarction (TIMI) 57 trial. Circulation. 2013;128(9):962–969. [DOI] [PubMed] [Google Scholar]
- 6. Blom DJ, Hala T, Bolognese M, et al. A 52‐week placebo‐controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370(19):1809–1819. [DOI] [PubMed] [Google Scholar]
- 7. Hirayama A, Honarpour N, Yoshida M, et al. Effects of evolocumab (AMG 145), a monoclonal antibody to PCSK9, in hypercholesterolemic, statin‐treated Japanese patients at high cardiovascular risk—primary results from the phase 2 YUKAWA study. Circ J. 2014;78(5):1073–1082. [DOI] [PubMed] [Google Scholar]
- 8. Koren MJ, Lundqvist P, Bolognese M, et al. Anti‐PCSK9 monotherapy for hypercholesterolemia: the MENDEL‐2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2531–2540. [DOI] [PubMed] [Google Scholar]
- 9. Robinson JG, Nedergaard BS, Rogers WJ, et al. Effect of evolocumab or ezetimibe added to moderate‐ or high‐intensity statin therapy on LDL‐C lowering in patients with hypercholesterolemia: the LAPLACE‐2 randomized clinical trial. JAMA. 2014;311(18):1870–1882. [DOI] [PubMed] [Google Scholar]
- 10. Stroes E, Colquhoun D, Sullivan D, et al. Anti‐PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS‐2 randomized, placebo‐controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2541–2548. [DOI] [PubMed] [Google Scholar]
- 11. Stein EA, Wasserman SM, Dias C, Scott R, Raal F. AMG‐145. Drugs Future. 2013;38(7):451–459. [Google Scholar]
- 12. Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006;11(1‐2):81–88. [DOI] [PubMed] [Google Scholar]
- 13. Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1–110. [DOI] [PubMed] [Google Scholar]
- 14. Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2‐microglobulin‐containing neonatal intestinal transport receptor. Proc Natl Acad Sci USA. 1996;93(11):5512–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Colbert A, Umble‐Romero A, Prokop S, Xu R, Gibbs J, Pederson S. Characterization of a quantitative method to measure free proprotein convertase subtilisin/kexin type 9 in human serum. MAbs. 2014;6(4):1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reuben A. Hy's law. Hepatology. 2004;39(2):574–578. [DOI] [PubMed] [Google Scholar]
- 17. Dias CS, Shaywitz AJ, Wasserman SM, et al. Effects of AMG 145 on low‐density lipoprotein cholesterol levels: results from 2 randomized, double‐blind, placebo‐controlled, ascending‐dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60(19):1888‐1898. [DOI] [PubMed] [Google Scholar]
- 18. Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–659. [DOI] [PubMed] [Google Scholar]
- 19. Zhao B, Chen R, O'Connor OA, et al. Brentuximab vedotin, an antibody‐drug conjugate, in patents with CD30‐positive haematologic malignancies and hepatic or renal impairment. Br J Clin Pharmacol. 2016;82(3):696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuo TT, Aveson VG. Neonatal Fc receptor and IgG‐based therapeutics. MAbs. 2011;3(5):422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7(9):715–725. [DOI] [PubMed] [Google Scholar]
- 22. Anderson CL, Chaudhury C, Kim J, Bronson CL, Wani MA, Mohanty S. Perspective—FcRn transports albumin: relevance to immunology and medicine. Trends Immunol. 2006;27(7):343‐348. [DOI] [PubMed] [Google Scholar]
- 23. Sherlock S. Immunological changes in liver disease. Proc R Soc Med. 1977;70(12):851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka S, Okamoto Y, Yamazaki M, Mitani N, Nakqjima Y, Fukui H. Significance of hyperglobulinemia in severe chronic liver diseases—with special reference to the correlation between serum globulin/IgG level and ICG clearance. Hepatogastroenterology. 2007;54(80):2301–2305. [PubMed] [Google Scholar]
- 25. Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man. J Clin Invest. 1970;49(4):673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reilly MP, McKenzie SE. Mechanisms of action of IVIg: physiology of Fc receptors. Vox Sang. 2002;83(suppl 1):57–63. [DOI] [PubMed] [Google Scholar]
- 27. Bonagura VR. Using intravenous immunoglobulin (IVIG) to treat patients with primary immune deficiency disease. J Clin Immunol. 2013;33(Suppl 2):S90–S94. [DOI] [PubMed] [Google Scholar]
- 28. Westergaard H, Jarnum S, Ramsoe K, Ranek L. Albumin and immunoglobulin turnover in patients with chronic active liver disease treated with prednisone. Scand J Gastroenterol. 1972;7(7):623–630. [DOI] [PubMed] [Google Scholar]
- 29. Richter WF, Jacobsen B. Subcutaneous absorption of biotherapeutics: knowns and unknowns. Drug Metab Dispos. 2014;42(11):1881–1889. [DOI] [PubMed] [Google Scholar]
- 30. Cicognani C, Malavolti M, Morselli‐Labate AM, Zamboni L, Sama C, Barbara L. Serum lipid and lipoprotein patterns in patients with liver cirrhosis and chronic active hepatitis. Arch Intern Med. 1997;157(7):792–796. [PubMed] [Google Scholar]
- 31. Koren MJ, Scott R, Kim JB, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double‐blind, placebo‐controlled, phase 2 study. Lancet. 2012;380(9858):1995–2006. [DOI] [PubMed] [Google Scholar]
- 32. Bassendine MF, Sheridan DA, Bridge SH, Felmlee DJ, Neely RD. Lipids and HCV. Semin Immunopathol. 2013;35(1):87‐100. [DOI] [PubMed] [Google Scholar]