

Abstract
Lobar cerebral microbleeds are most often sporadic and associated with Alzheimer's disease. The aim of our study was to identify the underlying genetic defect in a family with cognitive complaints and multiple lobar microbleeds and a positive family history for early onset Alzheimer's disease. We performed exome sequencing followed by Sanger sequencing for validation purposes on genomic DNA of three siblings with cognitive complaints, reduced amyloid‐beta‐42 in CSF and multiple cerebral lobar microbleeds. We checked for the occurrence of the variant in a cohort of 363 patients with early onset dementia and/or microbleeds. A novel frameshift variant (c.236_237delAC) generating a premature stop codon in the CCM2 gene shared by all three siblings was identified. Pathogenicity of the variant was supported by the presence of cerebral cavernous malformations in two of the siblings and by the absence of the variant exome variant databases. Two siblings were homozygous for APOE‐ϵ4; one heterozygous. The cognitive complaints, reduced amyloid‐beta‐42 in CSF and microbleeds suggest preclinical Alzheimer's disease, but the stability of the cognitive complaints does not. We hypothesize that the phenotype in this family may be due to a combination of the CCM2 variant and the APOE status. © 2016 The Authors. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics Published by Wiley Periodicals, Inc.
Keywords: cavernoma, cerebral cavernous malformations, cognitive impairment, familial clustering, genetics
INTRODUCTION
Cerebral microbleeds (CMBs), small round hypointense lesions on hemosiderin sensitive MR sequences, are common in Alzheimer's disease patients but also occur in the general population [Cordonnier and van der Flier, 2011; Shams et al., 2015]. Based on clinical and epidemiological studies, CMBs with a lobar location presumably represent cerebral amyloid angiopathy (CAA), while CMBs with a deep location may represent hypertensive vasculopathy [Cordonnier et al., 2006; Poels et al., 2010; Shams et al., 2015]. CAA is most often sporadic, with age being the most important risk factor [Biffi and Greenberg, 2011]. Specific (founder) mutations in the amyloid‐precursor protein (APP) gene, the cystatin 3 (CST3) gene, and the intergral membrane protein 2B (ITM2B) gene are associated with hereditary CAA [Ghiso et al., 1986; Vidal et al., 1999, 2000]. Mutations in presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes are associated with familial early onset Alzheimer's disease with CAA [Nochlin et al., 1998; Dermaut et al., 2001; Sanchez‐Valle et al., 2007]. The APOE ϵ4 allele is the strongest known genetic risk factor for Alzheimer's disease [van der Flier et al., 2006; Kanekiyo et al., 2014], but is also associated with the incidence and severity of CAA [Esiri et al., 2015] and with the prevalence of CMBs in Alzheimer's disease patients [Benedictus et al., 2013; Yates et al., 2014].
We present a family with CMBs and cognitive complaints with no known genetic predisposition for CAA or Alzheimer's disease, in whom we performed whole exome sequencing in three affected siblings. We detected a deletion leading to a frameshift in the CCM2 gene, a gene associated with familial cerebral cavernous malformations.
MATERIALS AND METHODS
Clinical Ascertainment
We selected a family of which two family members were known at our clinic, the Alzheimer center of the VU University Medical Center, because of cognitive complaints and microbleeds and an autosomal dominant family history for Alzheimer's disease.
All patients visiting the Alzheimer center are offered an extensive standardized dementia assessment including medical history, informant‐based history, a physical examination, routine blood (including glucose) and cerebrospinal fluid (CSF) laboratory tests, neuropsychological testing, electroencephalogram (EEG), and magnetic resonance imaging (MRI) of the brain including susceptibility‐weighted T2* images [van der Flier et al., 2014]. The clinical diagnosis Alzheimer's disease is made by consensus in a multidisciplinary team based on the NINCDS‐ADRDA criteria for Alzheimer's disease [McKhann et al., 1984], and a clinical diagnosis mild cognitive impairment (MCI) based on the Petersen criteria [Petersen, 2004]. Patients are labeled as having subjective complaints when reporting cognitive complaints while cognitive and laboratory investigations are normal and criteria for MCI, dementia or any other neurological or psychiatric disorder associated with cognitive complaints are not met. All patients who give consent for research are included in the Amsterdam Dementia Cohort [van der Flier et al., 2014]. A subset of this cohort, consisting of 363 patients with early onset AD and/or multiple microbleeds, was selected for whole exome sequencing for other research purposes.
All participants of this study had at least two MRI's performed on a 3.0 T GE scanner (type HDXT) with SWI sequence. Analysis of small vessel disease was performed according to STRIVE [Wardlaw et al., 2013]. The three participating subjects of the described family gave written informed consent for genetic research specifically prior to inclusion. The research protocol was approved by the ethical review board of our hospital.
Genetic Analysis
DNA of the described participants and of the 363 selected patients of the Amsterdam dementia cohort was derived from peripheral blood. Exomes were captured by the Nimblegen human exome v3 capture kit, and were sequenced with 2 × 100 paired‐end sequencing on the Illumina HiSeq 2000 platform, according to the manufacturer's protocol. Reads were mapped to the human reference genome sequence (UCSC hg19) using the Burrows‐Wheeler Alignment Tool (http://bio-bwa.sourceforge.net) [Li and Durbin, 2009]. Duplicate read removal, local sequence realignment, and base quality recalibration were performed by Picard (http://picard.sourceforge.net) and Genome analysis Tool Kit (GATK, (https://www.broadinstitute.org/gatk/) [McKenna et al., 2010]. Variants were called using the GATK HaplotypeCaller, and filtered using the variant filtration tool. For each variant, we set the filter to PASS if the variant complied with (i) GATK quality score ≥50; (ii) quality over depth ≥1.5; (iii) Strand bias ≤60; (iv) total read depth ≥5.0. Variants were annotated and analyzed with Cartagenia (http://www.cartagenia.com/) filter tree specifically designed to detect variants causative for a trait with an autosomal dominant inheritance pattern. In the described family, variants were selected if (i) absent in the following databases: dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP, build 138), the 1.000 genome project (www.1000genomes.org) or the National Heart Lung Blood Institute Exome Variant Server (EVS) (https://evs.gs.washington.edu/EVS); (ii) prevalent ≤5% in the 363 patients of the Amsterdam Dementia Cohort [van der Flier et al., 2014]; (iii) heterozygote in all three affected individuals; (iv) potential causative based on the possible effects of the variants on the expression or function of the protein and in a morbid OMIM gene.
To find out whether a predisposition for CCMs is common in patients with presumed microbleeds, we searched the exomes of 363 patients of the Amsterdam Dementia Cohort [van der Flier et al., 2014], including 68 patients with microbleeds, for occurrence of exonic and splice site variants in the genes KRIT1, CCM2, and PDCD10. Nucleotides were numbered according to Genbank accession number NM_004912.3 (KRIT1), NM_001029835.2 (CCM2), and NM_007217.3 (PDCD10) with A of initiator ATG numbered as +1. Variants in the CCM genes were analyzed by using the Combined Annotation Dependent Deletion (CADD) scoring tool (http://cadd.gs.washington.edu/) v1.2.We used Sanger sequencing to confirm the novel variant we found with exome sequencing. We submitted the variant in the CCM2 gene to the Leiden Open Variant Database (http://ccm2.lovd.nl).
APOE genotyping was performed by Sanger sequencing of codons 112 and 158 of the APOE gene. For this, a 428 bp fragment was generated from genomic DNA by PCR, checked for size (Fast DNA analysis with QIAxcel), and sequenced (BigDye Terminator v3.1 Cycle Sequencing kit followed by ABI 3130XL Genetic Analyzer).
RESULTS
Characteristics of the Participants
The pedigree is shown in Figure 1. Individuals III‐1, III‐2, and III‐3 participated in our study. They reported early‐onset dementia in one parent (II‐6) and one grandparent (I‐1). Three siblings of the affected parent had symptoms of dementia before the age of 65 years (II‐1, II‐2, and II‐3), another relative (II‐4) died of a stroke of unknown aetiology. Of these reported affected relatives, neither detailed medical information nor DNA was available.
Figure 1.
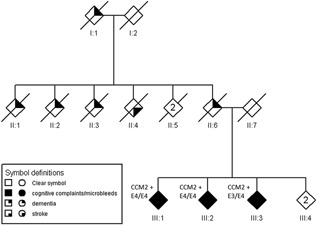
Pedigree of the described family. The sex of each individual is masked and the pedigree scrambled to protect the privacy of the participants. The diamonds indicate individuals with cognitive complaints and microbleeds (filled symbols), dementia (filled upper right quadrant), and stroke (filled lower right quadrant). CCM+ indicates that the subject is positive for the c.236_237delAC variant in the CCM2 gene. E3 indicates an APOE ϵ3 allele, E4 an APOE ϵ4 allele. “n” in a diamond indicates more than one individual. Consequently, the ID under the symbols with an “n” refers to multiple individuals as a group.
Case III‐1
This patient visited our center at the age of 58 years with memory complaints for over 5 years. The MMSE score was 27 out of 30. Physical examination, routine blood tests, neuropsychological testing, and EEG were all normal. Cerebral MRI showed nine lobar CMBs in the absence of cortical atrophy (global cortical atrophy [GCA] score 0) and hippocampal atrophy (medial temporal lobe atrophy [MTA] score 0) (Fig. 2A), a few punctiform white matter lesions (Fazekas 1) and a large T2*‐hypointensity in the pons consistent with a macroscopic hemorrhage. CSF analysis showed decreased amyloid‐beta‐42 and increased tau levels (Aβ: 529 ng/L, reference > 550 ng/L; t‐tau: 673 ng/L, reference ≤ 375 ng/L; ptau‐181: 86 ng/L, reference ≤ 52 ng/L). The patient was homozygous for the APOE ϵ4 allele. Diagnostic DNA‐analysis revealed no mutations in the genes APP (Sanger sequencing and copy number variant analysis), PSEN1, PSEN2, and MAPT.
Figure 2.

Cerebral MR imaging. A: Microbleed (arrow) in individual III‐1 visible on T2‐weighted image. B: Left cerebellar cavernous malformation (arrow) in individual III‐2 on T2‐weighted image. C: Left frontal cavernous malformation (arrow) in individual III‐3 with characteristic central high signal on T2 weighted image and hypointense outer rim.
Since the clinical and neuropsychological exam were within the normal range, the cognitive complaints were labeled as subjective cognitive decline. CMBs were interpreted as suggestive of underlying CAA and the CSF biomarkers indicative of preclinical AD.
During the following 7 years, the patient reported only mild progression of her cognitive dysfunction, and neuropsychological testing remained normal. Repeat MR imaging showed a few new CMBs but still no signs suggestive of neurodegeneration such as cortical atrophy.
Case III‐2
This patient visited our center at the age of 54 years with complaints of long lasting mild memory loss. Physical examination and routine blood tests showed no relevant abnormalities. The patient scored 29 out of 30 on the MMSE, and neuropsychological testing revealed only mild language disturbances. T2*‐weighted MR imaging revealed three lobar CMBs, two larger hematomas (cerebellar and parietal) with a hyperintense center on FLAIR and T1 consistent with a cavernous malformation (Fig. 2B), and a few punctiform vascular white matter lesions (Fazekas 1); cerebral cortical or hippocampal atrophy was absent (GCA 0, MTA 0). The EEG was normal. CSF analysis showed decreased amyloid‐beta‐42 but normal t‐tau and borderline p‐tau levels (Aβ: 418 ng/L, t‐tau: 343 ng/L, ptau‐181: 56 ng/L). The patient was homozygous for the APOE ϵ4 allele. No mutations were identified in the genes PSEN1, PSEN2, and MAPT. The cognitive complaints were labelled as subjective cognitive decline. The CMBs were interpreted as suggestive for underlying CAA.
During the following 7 years, the patient reported a slight worsening of cognitive complaints, which could not be confirmed by neuropsychological testing (criteria for MCI not fulfilled). Repeat MRI showed mild increase of white matter hyperintensities and a few new lobar CMBs.
Case III‐3
The youngest sibling presented with progressive memory loss and executive dysfunction at another hospital at the age of 51 years and was diagnosed with Alzheimer's disease. Detailed information on the test results was not available. No mutations, duplications, or deletions were found in the genes APP and PSEN1.
The patient visited our clinic for a second opinion 4 years later, as symptoms had not worsened. At this time, MMSE was 25/30, and neuropsychological assessment revealed executive dysfunction and mild impairment of memory and naming. MRI showed more than 20 supra‐ and infratentorial lobar CMBs, a few punctiform vascular white matter lesions (Fazekas 1), and a cavernous malformation in the left frontal lobe (Fig. 2C). There was no cortical atrophy (GCA 0) or relevant hippocampal atrophy (MTA 0 on the left and grade 1 on the right). The patient had an APOE ϵ3/ϵ4 genotype. A lumbar puncture was refused by the participant.
The patient did not fulfill the criteria for Alzheimer's disease and was diagnosed with MCI. The CMBs were interpreted as probably due to CAA. Two‐year follow up showed no further clinical deterioration.
Genetic Findings
With exome sequencing, we identified a heterozygous two‐base pair deletion in exon 3, c.236_237delAC, in the CCM2 gene in all three siblings. This deletion creates a frameshift starting at codon Tyr79 resulting in a premature stop codon. Based on the location of the new stop, the transcript is likely to be targeted by nonsense‐mediated mRNA decay, resulting in haploinsufficiency. The variant has not been reported before in literature or in the Dutch genetic biobank GoNL (http://www.nlgenome.nl/) and was not found in 363 patients with early onset Alzheimer's disease and/or multiple microbleeds. Based on the predicted effect of variants and known function of the associated genes, we found no other variants of interest in the family.
To investigate whether mutations in the CCM2‐gene or related genes are common in patients with CMBs, we analyzed the cohort of 363 patients with early onset Alzheimers disease and/or microbleeds for rare variants in the genes KRIT1, CCM2, and PDCD10. We detected two missense variants of unknown significance in the KRIT1 gene with a minor allele frequency of less than 0.5%: One protein modifying variant, c.1882A > C, p.Asn628His was found in an Alzheimer's disease patient without vascular abnormalities on brain imaging. This variant has a CADD score of 23.1 and is found in the Dutch genomic biobank GoNL with an allele frequency of 0.1%. A synonymous variant, c.1752C > T, p.Ile584 = with a CADD score of 17.5 was found in an Alzheimer's disease patient with moderately severe vascular white matter lesions but no CMBs or cavernous malformations. The GoNL biobank reports an allele frequency of close to 0.5% of this variant.
DISCUSSION
We describe a novel variant in the CCM2 gene identified by whole exome sequencing in a family with non‐progressive cognitive symptoms in three siblings at relatively young age, with CCMs in two and multiple lobar CMBs in all three on cerebral MRI. In addition, decreased amyloid beta‐42 levels in CSF were found in both tested individuals. Mutations in the genes APP, PSEN1, and PSEN2 were absent, and at least one APOE ϵ4 allele was present in all three.
The novel frameshift variant generates a premature stop codon in the CCM2 gene, probably resulting in haploinsufficiency by nonsense mediated mRNA decay. Loss of function mutations in the CCM2 gene and in the genes KRIT1 and PDCD10 are associated with familial cerebral cavernous malformations (FCCM) [Laberge‐le et al., 1999; Sahoo et al., 1999; Liquori et al., 2003; Bergametti et al., 2005]. Cerebral cavernous malformations (CCMs) are enlarged, thin walled capillaries in the brain and spinal cord without fibrous support tissue. In familial cases, a combination of a germ line mutation (first hit) and a somatic mutation (second hit) in one of the FCCM genes are associated with CCMs [Akers et al., 2009; Pagenstecher et al., 2009]. The KRIT1‐CCM2‐PDCD10‐complex is considered to interact with the PI3 K/Akt signaling pathway associated with metabolism, growth, proliferation, survival, transcription, and protein synthesis mechanisms [Kar et al., 2015]. Most symptomatic FCCM patients present between the age of 10 and 40 years with seizures, focal neurologic deficits, non‐specific headaches, or acute cerebral hemorrhage [Denier et al., 2006]. Up to 50% of the patients with FCCM remain asymptomatic, although most asymptomatic mutation carriers do have at least one CCM on MRI [Denier et al., 2006].
The presence of CCMs in two of the three siblings (individual III‐1 and III‐2) supports the diagnosis of FCCM in this family. The absence of a CCM in the third individual does not contradict this diagnosis, since FCCM is known to have an incomplete penetrance. However, decreased CSF amyloid beta‐42 has not been reported in FCCM and cognitive complaints and CMBs are not common symptoms of this disease [Denier et al., 2006].
The (subjective) cognitive decline may reflect a concomitant preclinical Alzheimer disease. The abnormal amyloid beta levels in CSF and the presence of APOE ϵ4 in homozygous or heterozygous state in all three affected siblings would support this. However, the stable character of the cognitive complaints over 7 years does not. Poor cognitive function has also been associated with APOE ϵ4 status in the absence of Alzheimer's disease [Small et al., 2004] as well as the presence of lobar CMBs [Poels et al., 2012; Hilal et al., 2014].
Reduced amyloid beta‐42 levels in CSF are strongly correlated with Alzheimer's disease, but have been associated with the APOE ϵ4 genotype regardless of the presence of Alzheimer's disease [Liu et al., 2015], and with several other brain diseases, such as CADASIL, neuroinflammation, and Creutzfeldt‐Jakob Disease [Otto et al., 2000; Formichi et al., 2008; Krut et al., 2013]. No studies have been published on CSF profiles in FCCM patients [Morrison and Akers, 2003].
An intriguing question is whether the occurrence of CMBs in this family is due to FCCM, or whether it should be attributed to a co‐existent disease such as preclinical Alzheimer's disease or isolated CAA. CMBs as such have not been reported in (F)CCM [Morrison and Akers, 2003]. The mean prevalence of CBMs in women aged <70 years is about 5% in the general population [Cordonnier et al., 2007; Sveinbjornsdottir et al., 2008], therefore, normal ageing does not seem a plausible cause in these siblings. Based on the CSF findings, also isolated CAA is unlikely. The aspects and location of the bleeds do not fit with other causes of small bleeds such as vasculitis, hypertensive encephalopathy, or coagulopathy.
Another possibility, however, is that the CMBs are in fact small CCMs. While larger CCMs typically show signs of stagnant blood in the sinusoidal lumen, extravasated blood at varying stages of degradation and a characteristic hemosiderin rim on MRI [Al‐Shahi et al., 2008], the small Zambinski's classification type 4 CCM lesions [Zabramski et al., 1994] are more difficult to distinguish from CMBs. The presence of variants in the FCCM genes in other patients with presumed CMBs would support this hypothesis. We did not find any other variants predicted to result in a loss of function in a cohort of patients with multiple CBMs, however, the number of tested individuals was small.
It is interesting to hypothesize whether ApoE and CCM2 interact. No common pathway has been described. However, APOE ϵ4 has been reported to increase the susceptibility to blood‐brain‐barrier injury [Bell et al., 2012] and, therefore theoretically, this genotype may result in an increased bleeding risk of CCMs.
Taken together, the non‐progressive cognitive complaints, the lobar hypointense lesions on cerebral MRI, and the reduced amyloid beta‐42 levels in CSF may be due to the combination of the CCM2 variant and the APOE ϵ4 genotype, although an early stage of Alzheimer's disease cannot be ruled out. Unfortunately, no other relatives were available for segregation analysis. Further studies on CSF profiles in FCCM patients and mutations in the FCCM genes in patients with multiple CBMs could give more insight into the pathogenic mechanism in this family.
DISCLOSURES
Research of the VUmc Alzheimer center is part of the neurodegeneration research program of the Neuroscience Campus Amsterdam. The VUmc Alzheimer Center is supported by Alzheimer Nederland and Stichting VUmc fonds.
F. Barkhof has received grant support from the Dutch MS Society (EU FP‐7). He has received consultancy/speaker fees from Bayer‐Schering Pharma, Sanofi‐Aventis, Biogen‐Idec, TEVA, Merck‐Serono, Novartis, Roche, Synthon BV, Jansen Research, Genzyme, Serono Symposia Foundation, and MedScape. F. Barkhof is member of the board from Brain, Eur Radiology, Neuroradiology, Multiple Sclerosis Journal, Radiology, and Neurology.
W. M. van der Flier performs contract research for Boehringer Ingelheim and has been an invited speaker at Boehringer Ingelheim. Research programs of W. M. van der Flier have been funded by ZonMW, NWO, EU‐FP7, Alzheimer Nederland, CardioVascular Onderzoek Nederland, stichting Dioraphte, Gieskes‐Strijbis fonds, Boehringer Ingelheim, Piramal Neuroimaging, Roche BV, Janssen Stellar. All funding is paid to her institution.
P. Scheltens has received grant support (for the institution) from GE Healthcare, Danone Research, Piramal, and MERCK. In the past 2 years, he has received consultancy/speaker fees (paid to the institution) from Lilly, GE Healthcare, Novartis, Forum, Sanofi, Nutricia, Probiodrug, and EIP Pharma.
The other authors report no disclosures.
ACKNOWLEDGMENTS
The authors thank the participating patients.
Cohn‐Hokke PE, Holstege H, Weiss MM, van der Flier WM, Barkhof F, Sistermans EA, Pijnenburg YAL, van Swieten JC, Meijers‐Heijboer H, Scheltens P. 2016. A Novel CCM2 Variant in a Family With Non‐Progressive Cognitive Complaints and Cerebral Microbleeds. Am J Med Genet Part B 174B: 220–226.
Conflicts of interest: The authors declare no conflicts of interest.
REFERENCES
- Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. 2009. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two‐hit mechanism of CCM pathogenesis. Hum Mol Genet 18:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Shahi SR, Berg MJ, Morrison L, Awad IA. 2008. Hemorrhage from cavernous malformations of the brain: Definition and reporting standards. Angioma Alliance Scientific Advisory Board. Stroke 39:3222–3230. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. 2012. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedictus MR, Goos JD, Binnewijzend MA, Muller M, Barkhof F, Scheltens P, Prins ND, van der Flier WM. 2013. Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer's disease. Neurobiol Aging 34:2488–2494. [DOI] [PubMed] [Google Scholar]
- Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier‐Lasserve E. 2005. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Greenberg SM. 2011. Cerebral amyloid angiopathy: A systematic review. J Clin Neurol 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordonnier C, Al‐Shahi SR, Wardlaw J. 2007. Spontaneous brain microbleeds: Systematic review, subgroup analyses and standards for study design and reporting. Brain 130:1988–2003. [DOI] [PubMed] [Google Scholar]
- Cordonnier C, van der Flier WM. 2011. Brain microbleeds and Alzheimer's disease: Innocent observation or key player? Brain 134:335–344. [DOI] [PubMed] [Google Scholar]
- Cordonnier C, van der Flier WM, Sluimer JD, Leys D, Barkhof F, Scheltens P. 2006. Prevalence and severity of microbleeds in a memory clinic setting. Neurology 66:1356–1360. [DOI] [PubMed] [Google Scholar]
- Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, Maciazek J, Vicaut E, Brunereau L, Tournier‐Lasserve E. 2006. Genotype‐phenotype correlations in cerebral cavernous malformations patients. Ann Neurol 60:550–556. [DOI] [PubMed] [Google Scholar]
- Dermaut B, Kumar‐Singh S, de Jonghe C, Cruts M, Lofgren A, Lubke U, Cras P, Dom R, de Deyn PP, Martin JJ, Van Broeckhoven C. 2001. Cerebral amyloid angiopathy is a pathogenic lesion in Alzheimer's disease due to a novel presenilin 1 mutation. Brain 124:2383–2392. [DOI] [PubMed] [Google Scholar]
- Esiri M, Chance S, Joachim C, Warden D, Smallwood A, Sloan C, Christie S, Wilcock G, Smith AD. 2015. Cerebral amyloid angiopathy, subcortical white matter disease and dementia: Literature review and study in OPTIMA. Brain Pathol 25:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formichi P, Parnetti L, Radi E, Cevenini G, Dotti MT, Federico A. 2008. CSF levels of beta‐amyloid 1‐42, tau and phosphorylated tau protein in CADASIL. Eur J Neurol 15:1252–1255. [DOI] [PubMed] [Google Scholar]
- Ghiso J, Jensson O, Frangione B. 1986. Amyloid fibrils in hereditary cerebral hemorrhage with amyloidosis of Icelandic type is a variant of gamma‐trace basic protein (cystatinC). Proc Natl Acad Sci USA 83:2974–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilal S, Saini M, Tan CS, Catindig JA, Koay WI, Niessen WJ, Vrooman HA, Wong TY, Chen C, Ikram MK, Venketasubramanian N. 2014. Cerebral microbleeds and cognition: The epidemiology of dementia in Singapore study. Alzheimer Dis Assoc Disord 28:106–112. [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G. 2014. ApoE and Abeta in Alzheimer's disease: Accidental encounters or partners? Neuron 81:740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Samii A, Bertalanffy H. 2015. PTEN/PI3 K/Akt/VEGF signaling and the cross talk to KRIT1, CCM2, and PDCD10 proteins in cerebral cavernous malformations. Neurosurg Rev 38:229–236. [DOI] [PubMed] [Google Scholar]
- Krut JJ, Zetterberg H, Blennow K, Cinque P, Hagberg L, Price RW, Studahl M, Gisslen M. 2013. Cerebrospinal fluid Alzheimer's biomarker profiles in CNS infections. J Neurol 260:620–626. [DOI] [PubMed] [Google Scholar]
- Laberge‐le CS, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier‐Lasserve E. 1999. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 23:189–193. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L, Marchuk DA. 2003. Mutations in a gene encoding a novel protein containing a phosphotyrosine‐binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 73:1459–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tan L, Wang HF, Liu Y, Hao XK, Tan CC, Jiang T, Liu B, Zhang DQ, Yu JT. 2015. Multiple effect of APOE genotype on clinical and neuroimaging biomarkers across Alzheimer's disease spectrum. Mol Neurobiol. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. 1984. Clinical diagnosis of Alzheimer's disease: Report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34:939–944. [DOI] [PubMed] [Google Scholar]
- Morrison L, Akers A. 2003. Cerebral cavernous malformation, familial In: Pagon RA, Adam MP, Ardinger HH, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington; http://www.ncbi.nlm.nih.gov/books/NBK1293/ [Google Scholar]
- Nochlin D, Bird TD, Nemens EJ, Ball MJ, Sumi SM. 1998. Amyloid angiopathy in a Volga German family with Alzheimer's disease and a presenilin‐2 mutation (N141I). Ann Neurol 43:131–135. [DOI] [PubMed] [Google Scholar]
- Otto M, Esselmann H, Schulz‐Shaeffer W, Neumann M, Schroter A, Ratzka P, Cepek L, Zerr I, Steinacker P, Windl O, Kornhuber J, Kretzschmar HA, Poser S, Wiltfang J. 2000. Decreased beta‐amyloid1‐42 in cerebrospinal fluid of patients with Creutzfeldt‐Jakob disease. Neurology 54:1099–1102. [DOI] [PubMed] [Google Scholar]
- Pagenstecher A, Stahl S, Sure U, Felbor U. 2009. A two‐hit mechanism causes cerebral cavernous malformations: Complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet 18:911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC. 2004. Mild cognitive impairment as a diagnostic entity. J Intern Med 256:183–194. [DOI] [PubMed] [Google Scholar]
- Poels MM, Ikram MA, van der Lugt A, Hofman A, Niessen WJ, Krestin GP, Breteler MM, Vernooij MW. 2012. Cerebral microbleeds are associated with worse cognitive function: The Rotterdam Scan Study. Neurology 78:326–333. [DOI] [PubMed] [Google Scholar]
- Poels MM, Vernooij MW, Ikram MA, Hofman A, Krestin GP, van der Lugt A, Breteler MM. 2010. Prevalence and risk factors of cerebral microbleeds: An update of the Rotterdam scan study. Stroke 41:S103–S106. [DOI] [PubMed] [Google Scholar]
- Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee‐Lin SQ, Kosofsky B, Kurth JH, Louis DN, Mettler G, Morrison L, Gil‐Nagel A, Rich SS, Zabramski JM, Boguski MS, Green ED, Marchuk DA. 1999. Mutations in the gene encoding KRIT1, a Krev‐1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet 8:2325–2333. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Valle R, Llado A, Ezquerra M, Rey MJ, Rami L, Molinuevo JL. 2007. A novel mutation in the PSEN1 gene (L286P) associated with familial early‐onset dementia of Alzheimer type and lobar haematomas. Eur J Neurol 14:1409–1412. [DOI] [PubMed] [Google Scholar]
- Shams S, Martola J, Granberg T, Li X, Shams M, Fereshtehnejad SM, Cavallin L, Aspelin P, Kristoffersen‐Wiberg M, Wahlund LO. 2015. Cerebral microbleeds: Different prevalence, topography, and risk factors depending on dementia diagnosis—The Karolinska Imaging Dementia Study. AJNR Am J Neuroradiol 36:661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small BJ, Rosnick CB, Fratiglioni L, Backman L. 2004. Apolipoprotein E and cognitive performance: A meta‐analysis. Psychol Aging 19:592–600. [DOI] [PubMed] [Google Scholar]
- Sveinbjornsdottir S, Sigurdsson S, Aspelund T, Kjartansson O, Eiriksdottir G, Valtysdottir B, Lopez OL, van Buchem MA, Jonsson PV, Gudnason V, Launer LJ. 2008. Cerebral microbleeds in the population based AGES‐Reykjavik study: Prevalence and location. J Neurol Neurosurg Psychiatry 79:1002–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier WM, Pijnenburg YA, Prins N, Lemstra AW, Bouwman FH, Teunissen CE, van Berckel BN, Stam CJ, Barkhof F, Visser PJ, van EE, Scheltens P. 2014. Optimizing patient care and research: The Amsterdam Dementia Cohort. J Alzheimers Dis 41:313–327. [DOI] [PubMed] [Google Scholar]
- van der Flier WM, Schoonenboom SNM, Pijnenburg YAL, Fox NC, Scheltens P. 2006. The effect of APOE genotype on clinical phenotype in Alzheimer disease. Neurology 67:526–527. [DOI] [PubMed] [Google Scholar]
- Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J. 1999. A stop‐codon mutation in the BRI gene associated with familial British dementia. Nature 399:776–781. [DOI] [PubMed] [Google Scholar]
- Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, Bojsen‐Moller M, Braendgaard H, Plant G, Ghiso J, Frangione B. 2000. A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci USA 97:4920–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O'Brien JT, Barkhof F, Benavente OR, Black SE, Brayne C, Breteler M, Chabriat H, DeCarli C, de Leeuw FE, Doubal F, Duering M, Fox NC, Greenberg S, Hachinski V, Kilimann I, Mok V, Oostenbrugge R, Pantoni L, Speck O, Stephan BC, Teipel S, Viswanathan A, Werring D, Chen C, Smith C, van BM, Norrving B, Gorelick PB, Dichgans M. 2013. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates PA, Desmond PM, Phal PM, Steward C, Szoeke C, Salvado O, Ellis KA, Martins RN, Masters CL, Ames D, Villemagne VL, Rowe CC. 2014. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 82:1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, Drayer BP, Brown B, Rigamonti D, Brown G. 1994. The natural history of familial cavernous malformations: Results of an ongoing study. J Neurosurg 80:422–432. [DOI] [PubMed] [Google Scholar]