

Abstract
Hepatic encephalopathy has traditionally been considered a reversible disorder. However, recent studies suggested that repeated episodes of hepatic encephalopathy cause persistent impairment leading to neuronal loss. The aims of our study were the development of a new animal model that reproduces the course of episodic hepatic encephalopathy and the identification of neurodegeneration evidences. Rats with portacaval anastomosis underwent simulated episodes of hepatic encephalopathy, triggered by the regular administration of ammonium acetate, and/or lipopolysaccharide. The neurological status was assessed and neuronal loss stereologically quantified in motor areas. During the simulated episodes, ammonia induced reversible motor impairment in portacaval anastomosis rats. In cerebellum, stereology showed a reduction in Purkinje cell population in portacaval anastomosis and PCA+NH3 groups and morphological changes. An increase in astrocyte size in PCA+NH3 group and activated microglia in groups treated with ammonium acetate and/or lipopolysaccharide was observed. A modulation of neurodegeneration-related genes and the presence of apoptosis in Bergmann glia were observed. This new animal model reproduces the clinical course of episodic hepatic encephalopathy when ammonia is the precipitant factor and demonstrates the existence of neuronal loss in cerebellum. The persistence of over-activated microglia and reactive astrocytes could participate in the apoptosis of Bergmann glia and therefore Purkinje cell degeneration.
Keywords: Apoptosis, astrocytes, inflammation, microglia, neurodegeneration
Introduction
Hepatic encephalopathy (HE) is considered a reversible neurophysiological syndrome that causes brain disturbances, ranging from behavioral alterations as confusion or depressed consciousness to ataxia and coma.1 It is characterized by astrocyte swelling, secondary to cytotoxic edema, probably induced by the deleterious effects of ammonia and oxidative stress.
HE has classically been considered a metabolic disorder affecting astrocytes, but not neuronal architecture. However, the presence of neuronal loss in basal ganglia, thalamus and cerebellum is well documented in the most extreme form of HE: the acquired hepatocerebral degeneration.2,3 This disorder is characterized by chronic symptoms as ataxia, dysarthria, apraxia and parkinsonism, generally related to repeated or long episodes of HE. End-stage liver failure patients (alcoholic and nonalcoholic) show a high incidence of cerebellar lesions,4 in addition to a differential response of Bergman glia, a specialized cerebellar astrocyte, in comparison to other astrocytes.5,6
Several studies confirmed that HE episodes lead to persistent cognitive impairment,7 which perseveres even after hepatic transplantation.8 A decrease in the brain volume and in the N-acetyl-aspartate (neuronal marker) signal was observed by magnetic resonance imaging, suggesting that episodes of HE could induce neuronal loss.9 This loss could be more important in some areas, explaining some of the clinical manifestations. For example, it has been reported that cerebellum is especially susceptible to oxidative stress and together with basal ganglia, the regions with a greater supply of ammonia during HE.10,11
Experimental models are necessary to study the pathophysiology of HE and to test new drugs.12 These models induce diverse grades of liver failure and could be combined with portosystemic shunting and precipitating factors to simulate the different types of HE emulating the clinical manifestation.
Accordingly, the first aim of our study was the development of a new animal model that reproduces the clinical course of episodic HE. Starting from an established model of minimal HE in rats, we tested the periodical administration of common precipitating factors, ammonia and/or lipopolysaccharide (LPS), to assess the involvement of both factors and its synergic effects in the neurological outcome and pathophysiology. The second aim was to investigate whether the impact of repeated HE episodes in the brain, (hyperammonemia independently or in combination with peripheral inflammation) could lead to neurodegeneration in this new model of HE, and identify the main signaling pathways involved in this neurodegenerative process.
Materials and methods
Animal model
Adult male Sprague-Dawley rats (200–250 g; Charles River Laboratories France, L'Arbresle Cedex, France) were housed in polycarbonate cages under standard laboratory conditions. Standard food (A04, Panlab, Barcelona, Spain) and water were available ad libitum. All procedures were conducted following SECAL guidelines (http://secal.es/), in compliance with the ARRIVE guidelines for reporting animal experiments and approved by the Catalan Animal Research Committee at the facilities of Vall d'Hebron Research Institute (VHIR).
Portacaval anastomosis
Rats were anesthetized with isoflurane (Aerrane-Isoflurane, Baxter, Deerfield, IL, USA). Portacaval anastomosis (PCA) was performed following previous studies13 and after surgery, PCA rats recovered for four weeks before performing studies. Sham rats underwent a similar procedure in which portal vein was clamped for 15 min.
Episodic HE
PCA rats were regularly administered once every two weeks by continuous infusion for 3 h (20 µL/min; Harvard Apparatus, Holliston, Massachusetts), up to 10 infusions (five months) with ammonium acetate (55 µmol/kg/min; Sigma-Aldrich, St. Louis, USA), and/or LPS from Klebsiella pneumoniae (3 mg/kg; L4268; Sigma-Aldrich, St. Louis, USA), both prepared in saline (Fresenius Kabi, Sevres, France). Saline was also injected to a group of PCA and sham rats following the same protocol (supplementary data). Groups: Sham, PCA, PCA+NH3, PCA+LPS, PCA+NH3+LPS (n = 14). All infusions were administered through a permanent intraperitoneal catheter. Animal weight, blood ammonia, cytokines, and neurological manifestations were checked during the experiment.
Ammonia and cytokine levels in plasma
Ammonia levels were quantified by an enzymatic method (COBAS INTEGRA ammonia Kit, Roche, Basel, Switzerland) and three different cytokines (interleukin-6, IL-6; interleukin-1β, IL-1β; and tumor necrosis factor-α, TNF-α) were measured by immunoassay (Quantikine ELISA kit, R&D Systems, Minneapolis, USA) before and after the infusions (n = 7).
Neurological examination
Reflex score: A reflex test was performed similarly as previously described.14 Twelve different reflexes (including flexion, righting, grasping, placing reaction, equilibrium, corneal, auditory startle, head hacking) were checked in each animal before and after a simulated HE episode. The presence of the appropriate reaction was scored with one point, thus, a normal rat had a reflex score of 12 points (n = 14).
Memory examination: One week after the last HE episode, rats underwent a one-trial object recognition test to evaluate short-term and long-term memory, performed following Bevins and Besheer's protocol.15 In short, rats were exposed to two identical objects (a) as training. One hour later, short-term memory was assessed by the exposition to a novel object (b) and the familiar object (a). After 24 h, rats were exposed to a second new object (c) and the familiar one (a) to evaluate long-term memory. The test was videotaped and scored measuring the time invested exploring each object. The results were expressed as discrimination ratio, calculated as interaction with novel object divided by total interaction time (n = 6).
Brain analysis
Quantitative morphology
Animals were perfused with 1% heparinized saline solution and then with 4% paraformaldehyde solution (PFA; Merk, New York, USA). Brains were post-fixed 24 h in PFA, cryoprotected in sucrose and finally stored at −80℃. Continuous coronal sections of 30 µm thick from entire brain were obtained with a cryostat (Leica CM3050S, Leica Microsystems, Wetzlar, Germany).
The dopaminergic neurons of Substantia Nigra pars compacta (SNpc) were assessed after free-floating inmunohistochemistry (anti-tyroxine hydroxylase, TH; 1/2000; Calbiochem, Merk milipore Billerica, USA) and Purkinje neurons of cerebellum after Nissl staining. The stereological study (supplementary data) was realized in entire SNpc following a periodicity of in one every six sections (n = 8) and in cerebellum one every 24 sections (n = 3) using stereo investigator software (version 7.00; MSF Bioscience-Micro Bright Field, Williston, USA). All sections were blind counted based on nucleus identification to estimate number of neurons. Striatal TH innervation (n = 8) was evaluated by optical densitometry (OD) using Image J software (version 1.45s; NIH, Bethesda; USA), as previously described.16
For neuroglia analysis, four evenly spaced sections of the entire cerebellum were immunostained using primary antibody against microglia (anti-Iba-1; 1/1000; Wako Chemicals, Osaka, Japan) or astrocytes (anti-GFAP; 1/1000; Sigma-Aldrich, San Luis, MO, USA). Apoptosis was assessed using cleaved-caspase-3 antibody (anti-casp3 (Asp175); 1/600, Cell Signalling, Danvers, MA, USA). Eight representative images (20 × objectives) for microglia analysis, two for astrocytes (40 × objectives) and four for Bergmann glia (20 × objectives) were blind taken in every section (n = 3–4). Images were quantified using Image J.
Molecular mechanisms
For RNA analysis, brain samples were maintained 24 h in RNA later (Life technologies, Carlsbad, USA), frozen in liquid nitrogen, and stored at −80℃. Total RNA was extracted from cerebellum using Rneasy micro kit (QIAGEN, Venlo, Nederland) and RNA quality was checked by Bioanalyzer (2100, Agilent Technologies, Santa Clara, CA, USA). Gene expression profile of cerebellum was assessed using Rat Gene 1.1 ST 24-array plate (16,500 genes, Affimetrix, Santa Clara, USA) and genes differentially expressed by >2-fold and involved in neurological processes in accordance with Ingenuity® Pathway Software (IPA®, Qiagen, Redwood city, USA; n = 5) were verified by RT-PCR (TaqMan gene expression assay, Life Technologies, Carlsbad, USA). The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus17 and are accessible through GEO Series accession number GSE71425 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71425).
For Western blot, brains were frozen in methylbutane for storage at −80℃; 30 µg of lysated protein were loaded in SDS-polyacrylamide gel (12%, BioRad, Hercules, USA), transferred to a polyvinylidenedifluoride membrane (BioRad, Hercules, USA), and incubated overnight at 4℃ with primary antibody transthyretin (Ttr; 1/2000, Thermo Fisher, Waltham, USA). β-actin was used as loading control (1/25,000; Sigma-Aldrich, San Luis, MO, USA). Membrane was developed using ECL kit (GE Healthcase; Little Chalfont, UK), and quantified using Image J (n = 6). The results were an average of three replicates.
Statistical analysis
The differences between intergroup data (sham, PCA, PCA+NH3, PCA+LPS, and PCA+NH3+LPS) were verified with one-way ANOVA followed by Holm-Sidak. Data from blood ammonia and blood cytokines were analyzed by two-way ANOVA. The results obtained from reflexes were summarized in a contingency table and analyzed by McNemar test followed by Fisher method. Discrimination index was analyzed by one-way ANOVA. Neuronal counts were compared by one-way ANOVA followed by Holm-Sidak. Differences between groups in microglia and astrocyte reactivity and apoptosis were analyzed by one-way ANOVA followed by Holm-Sidak. Data were expressed as mean ± standard deviation and P values < 0.05 were considered statistically significant. All the statistical analyses were performed with Sigma Stat package (version 3.00 for windows, SPSS Inc Chicago, USA).
Results
Animal weight was monitored during the development of the model and no evidence of malnutrition was found in any group. The weight increased in all groups during the infusion period (Supplementary data).
Ammonia and LPS infusions induce acute hyperammonemia and cytokine production respectively in PCA rats
Ammonia and cytokines were controlled before and after the infusions to check the acute increment of its levels in blood. As expected, the groups in which ammonia acted as precipitant factor showed significantly higher blood ammonia levels 3 h after infusion, in comparison with the other groups (P < 0.001, Table 1). The group PCA+NH3 also showed more elevated ammonia levels than the group PCA+NH3+LPS (P = 0.003). In the same way, plasma cytokine levels (IL-6, IL-1β and TNF-α) were strongly increased only after LPS injection in comparison with LPS-non-treated groups (P ≤ 0.001, Table 1). Unexpectedly, the PCA+NH3+LPS group showed significantly lower levels of IL1-β (P = 0.003) and TNF-α (P < 0.001), than the PCA+LPS.
Table 1.
Animal model features: Plasma ammonia, cytokine levels and neurological examination.
| Baseline | 3 h Post-infusion | Intragroup differences | |
|---|---|---|---|
| Plasma ammonia (nM) | |||
| Sham | 114 ± 40 | 159 ± 141♦ | |
| PCA | 329 ± 112 * | 219 ± 47 ♦ | |
| PCA+NH3 | 368 ± 116 * | 714 ± 414* | P < 0.001 |
| PCA+LPS | 292 ± 99 * | 171 ± 43 ♦ | |
| PCA+NH3+LPS | 242 ± 115 | 427 ± 96*♦ | P = 0.042 |
| Plasma cytokines (pg/ml) | |||
| IL-6 | |||
| Sham | 119 ± 16 | 163 ± 82 | |
| PCA | 238 ± 11 | 662 ± 325 | |
| PCA+NH3 | 255 ± 86 | 424 ± 208 | |
| PCA+LPS | 249 ± 51 | 3024 ± 4090* | P < 0.001 |
| PCA+NH3+LPS | 230 ± 19 | 4197 ± 4182* | P < 0.001 |
| IL-1β | |||
| Sham | 16 ± 11 | 56 ± 22 | |
| PCA | 27 ± 21 | 64 ± 37 | |
| PCA+NH3 | 17 ± 10 | 28 ± 27 | |
| PCA+LPS | 20 ± 22 | 255 ± 171* | P < 0.001 |
| PCA+NH3+LPS | 12 ± 12 | 168 ± 124*† | P < 0.001 |
| TNF-α | |||
| Sham | −2 ± 4 | −2 ± 5 | |
| PCA | −1 ± 4 | 21 ± 20 | |
| PCA+NH3 | −1 ± 4 | 16 ± 17 | |
| PCA+LPS | −1 ± 4 | 73 ± 16* | P < 0.001 |
| PCA+NH3+LPS | −1 ± 4 | 52 ± 6*† | P < 0.001 |
| Reflex score | |||
| PCA | 12 | 12 | |
| PCA+NH3 | 12 | 9 | P = 0.003 |
| PCA+LPS | 12 | 12 | |
| PCA+NH3+LPS | 12 | 11 | |
| Object recognition memory (discrimination ratio) | |||
| |
STM | LTM | |
| Sham | 0.54 ± 0.30 | 0.66 ± 0.16 | |
| PCA | 0.65 ± 0.15 | 0.61 ± 0.19 | |
| PCA+NH3 | 0.62 ± 0.32 | 0.50 ± 0.29 | |
| PCA+LPS | 0.61 ± 0.11 | 0.33 ± 0.26 | |
| PCA+NH3+LPS | 0.54 ± 0.29 | 0.51 ± 0.30 | |
PCA: portacaval anastomosis; LPS: lipopolysaccharide; IL-6: interleukin-6; IL-1β: interleukin 1-β; TNF-α: tumor necrosis factor- α; STM: short-term memory; LTM: long-term memory.
Note: Intergroup differences versus Sham: *P ≤ 0.05.
Intergroup differences versus PCA+NH3: ♦P ≤ 0.05.
Intergroup differences versus PCA+LPS: †P ≤ 0.05.
Intragroup differences: ANOVA/t paired baseline versus 3 h post-infusion.
Hyperammonemia induces loss of reflexes in PCA rats
Neurological evaluation revealed a significant loss of reflexes only in PCA+NH3 group between baseline stage and 3 h post-infusion (baseline=12 and post-infusion=9; P = 0.003, Table 1). Ammonia induced reversible impairment of motor capabilities in PCA rats; during the simulated episodes, ammonia-treated rats showed mild manifestation of encephalopathy.
Regarding memory examination, during the short-term memory session, rats exhibited a discrimination ratio above 0.5 in all cases without differences between groups (Table 1). This ratio means that animals were able to differentiate between objects. In long-term memory session, PCA+LPS group was not completely able to recognize the familiar object, it showed discrimination ratio of 0.32 ± 0.26, but without significant differences between groups.
Hyperammonemia induces Purkinje neurons degeneration probably mediated by Bergmann glia apoptosis
The evaluation of dopaminergic cell bodies in SNpc, assessed by stereological cell counting, did not show a reduction of the number of TH-reactive neurons compared to sham rats. The analysis of the axon terminal integrity of TH-positive fibers in striatum did not evidence degeneration, supporting the results obtained from SNpc cell counting (Figure 1(a)). The stereological evaluation of Purkinje cell layer in cerebellum revealed the presence of degenerating Purkinje neurons in all groups and these neurons exhibited shrinkage, dark cytoplasm, and nucleus and/or deformed nuclei (Figure 1(b) and (c)), as shown in other diseases.18 The stereological counting of normal and degenerative Purkinje neurons showed a significant reduction in the number of normal Purkinje cells in all treated groups in comparison to sham (P = 0.018; Figure 1(d)). In addition, a lower number of total neurons in PCA and PCA+NH3 groups was quantified compared to sham rats (P = 0.045). To elucidate the mechanism involved in Purkinje cell death, apoptosis was evaluated measuring cleaved-caspase-3 levels by immunohistochemistry. The immunohistochemical analysis revealed an increase in cleaved-caspase-3 staining in Bergmann glia cells (Figure 2(f)) of all hyperammonemic groups (PCA, PCA+NH3 and PCA+NH3+LPS), except in PCA+LPS compared to sham rats (P = 0.005; Figure 2).
Figure 1.

Stereological analysis of dopaminergic neurons in SNpc and Purkinje neurons in cerebellum. (a) Relative number of dopaminergic neurons in SNpc (black bar) and the optical density of dopaminergic prolongations in striatum (grey bar). (b) Representative image of normal Purkinje neurons in PCA+NH3. (c) Representative image of Purkinje neurons with degenerative features (red arrows) in PCA+NH3. (d) Bar diagram represents the relative number of Purkinje neurons. Gray bar correspond to normal Purkinje neurons and black bar to degenerative cells (expressed in relation to the total number of Purkinje cells). *P ≤ 0.05 total number of Purkinje cells compared to sham. #P ≤ 0.05 normal Purkinje neurons compared to sham.
PCA: portocaval anastomosis; LPS: lipopolysaccharide; SNpc: substantia nigra pars compacta; TH: tyrosine hydroxylase.
Figure 2.
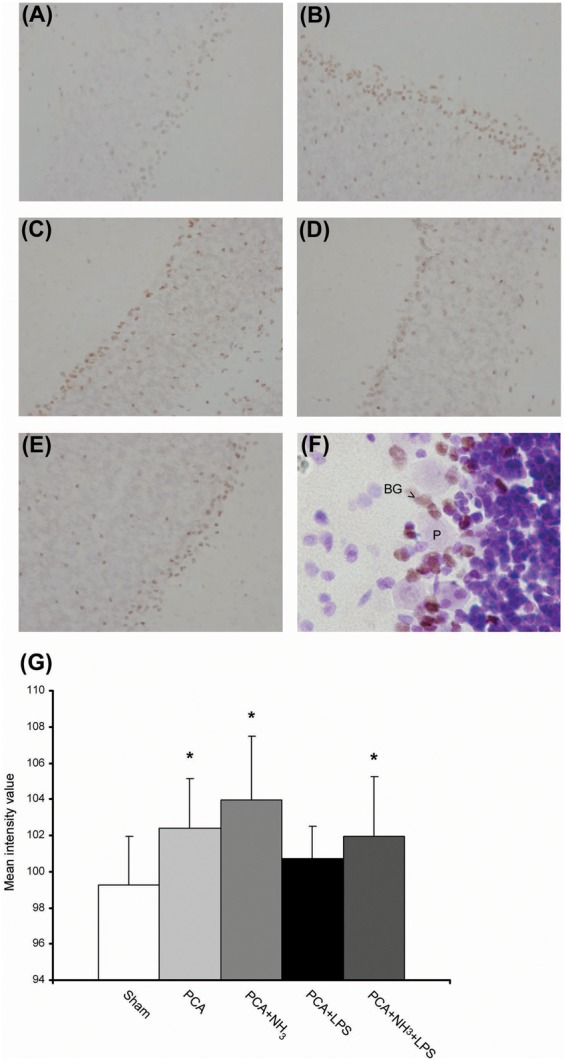
Bergmann glia apoptosis (a–g) representative images of cleaved-caspase-3 immunolabeling. Objective 20×. (a) Sham. (b) PCA. (c) PCA+NH3. (d) PCA+LPS. (e) PCA+NH3+LPS. (f) Representative image of cleaved-caspase-3 immunolabeling and Nissl counterstain, the double stain reveals cell death of Bergmann glia (BG; arrow) in Purkinje cell layer (P). Objective 60×. (g) Bar diagram represents mean intensity value of caspase-3 inmunostaining. *P ≤ 0.05 compared to sham.
PCA: portacaval anastomosis; LPS: lipopolysaccharide.
Ammonia infusion causes changes in astrocyte size
The differences in astrocyte size in cerebellum, estimated as the percentage of area immunostained with GFAP divided by the number of astrocytes, were statistically significant between sham (Figure 3(a)) and ammonia-treated group (P = 0.033, Figure 3(f)). In addition, the presence of astrocytes with extended processes was observed in all PCA groups (Figure 3(b) to (e)).
Figure 3.
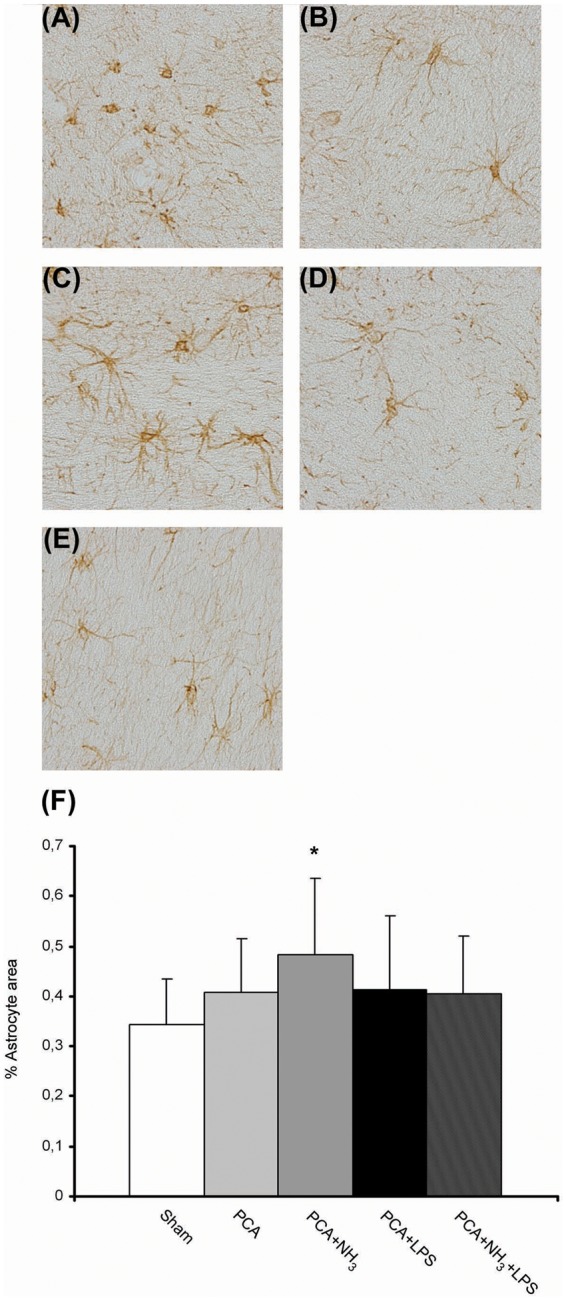
Reactive astrocytes in cerebellum of episodic HE model (a–e) Representative images of GFAP immunohistochemistry. Objective 40×. (a) Sham. (b) PCA. (c) PCA+NH3 activated phenotype (arrow). (d) PCA+LPS. (e) PCA+NH3+LPS. (f) Quantification of GFAP stained area divided by the number of astrocytes per image, along whole cerebellum. *P ≤ 0.05 compared to sham.
PCA: portacaval anastomosis; LPS: lipopolysaccharide.
Ammonia and/or LPS infusion induce microglia activation in PCA rats
Iba-1 stained area was measured to quantify the grade of microglia activation. An increase in microglia population was evident in cerebellar tissue following ammonia and or LPS injection in relation to sham (Figure 4(f)). Moreover, several of those cells presented activated phenotype, larger soma with shorter processes (P ≤ 0.001; Figure 4(c) to (e)). Microglia cells in sham rats possessed numerous and long ramifications characteristic of resting microglia (Figure 4(a)). The ammonia levels generated by PCA group appeared to be insufficient to produce substantial changes in microglia (Figure 4(b)).
Figure 4.
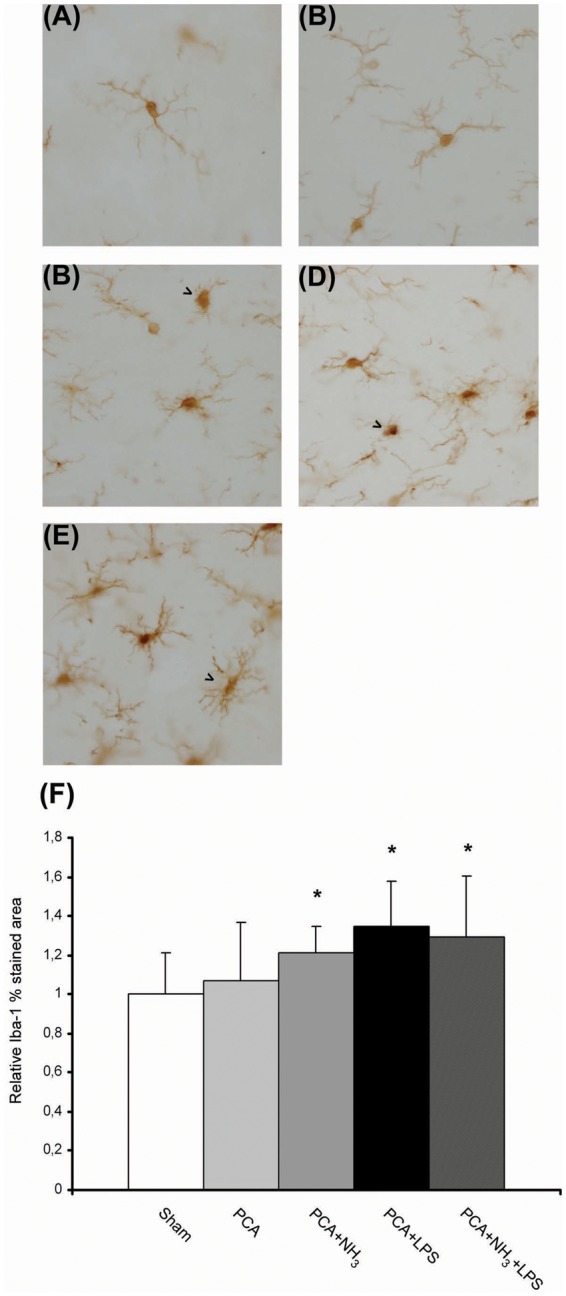
Microglial activation in cerebellum of episodic HE model (a–e) Representative images of Iba-1 immunohistochemistry. Objective 40×. (a) Sham. (b) PCA. (c) PCA+NH3 activated phenotype (arrow). (d) PCA+LPS activated phenotype (arrow). (e) PCA+NH3+LPS activated phenotype (arrow). (f) Quantification of Iba-1 stained area along whole cerebellum. *P ≤ 0.05 compared to sham.
PCA: portacaval anastomosis; LPS: lipopolysaccharide.
Ammonia and/or LPS infusion induce changes in neurodegenerative-related genes in PCA rats
Gene expression array of cerebellum evidenced expression changes of several neurodegenerative-related genes in comparison to sham that were validated by RT-PCR (Table 2). Ingenuity pathway analysis software did not reveal any canonical pathway affected, even so a remarkable transthyretin (Ttr) up regulation was observed in ammonia group (fold change Ttr = 17.4). RT-PCR validation revealed a huge overexpression of Ttr in 50% of the animals (fold change Ttr = 200), while the others had similar levels to sham-controls. Those results were confirmed by Western blot. Overall, protein levels of Ttr were significantly higher in PCA and ammonia-treated group (Figure 5). In addition, those ammonia-treated animals overexpressing Ttr mRNA also revealed significantly higher levels of Ttr protein when compared with the other half of ammonia-treated animals that showed Ttr levels similar to sham (P = 0.025). Other genes showed appreciable modulation related to LPS injection, standing out lipocalin-2 (Lcn-2) and Chemokine ligand-2 (Ccl-2) (Table 2).
Table 2.
List of regulated genes in the rat cerebellum.
| Protein name | Gene symbol | Comparison versus sham (fold change) |
|||
|---|---|---|---|---|---|
| PCA | PCA+NH3 | PCA+LPS | PCA+NH3+LPS | ||
| Lipocalin-2 | Lcn-2 | 2.64 | 1.52 | 3.38 (8.04)a | 3.15 |
| Chemokine ligand-2 | Ccl-2 | 1.86 | 1.41 | 2.66 | 1.44 |
| OrthodenticleHomeobox 2 | Otx-2 | −1.19 | 1.93 | −2.74 (−1.84) | 1.00 |
| Transthyretin | Ttr | 2.65 | 17.45 (23.34) | 2.51 | 3.15 |
| 5-Hidroxythryptamine receptor 2C | Htr2C | −1.07 | 3.02 (6.08) | 2.67 | 1.71 |
RT-PCR: real time PCR; PCA: portacaval anastomosis; LPS: lipopolysaccharide.
Fold change by RT-PCR.
Figure 5.

Western blot of total TTR in cerebellum. Total Ttr was quantified as the addition of monomers (14 kDa), tetramers (56 kDa), and intermediate forms (28 kDa; 42 kDa) and normalized by β-actin. Liver protein extract is used as positive control. Bar diagram represents total protein levels of three replicates. *P ≤ 0.05 compared to sham.
PCA: portacaval anastomosis; LPS: lipopolysaccharide; TTR: transthyretin.
Discussion
Several experimental HE models have been described until now; however, none of them mimicked recurrent episodes of HE, the most common HE modality in cirrhotic patients. Our proposal is a new animal model that simulates the episodic course of HE, arising from PCA model and periodically sensitized with ammonia or LPS administration to trigger successive bouts of HE. The innate reflexes, along with the neuropathological analysis of the brain, including stereological cell counting for the first time in an HE model, showed signs of neurodegeneration at different levels in cerebellum.
Our results showed that ammonia, as precipitant factor of the HE episodes, provoked transitory neurological function impairment that was evidenced by the acute loss of innate reflexes in the animals, with subsequent recovery 24 h later.19 Although the correlation existing between ammonia levels in blood and the severity of HE is not well characterized, the increase of ammonia levels in plasma achieved in our animal model is compatible with levels found in other studies in EH patients.20,21 The neurological examination in rats allowed the evaluation of responses in front of stimuli and provided us an overall scale of central nervous system functions.22 This approach permitted perceiving fluctuations in the neurological state of the animals, suggesting that this animal model has the capacity to simulate transitory motor abnormalities similar to the ones occurring during an episode of HE in patients.
This work is focused on examining the presence of neurodegeneration in motor structures by stereological studies, which allow the quantification of the total number of neurons through an unbiased counting methodology.23 A decrease in total number of Purkinje neurons in cerebellum was discerned in PCA and PCA+NH3 groups as well as a reduction in the number of normal Purkinje neurons in all animal groups compared to sham groups. Those findings confirm the existence of neuronal loss, but also an increase in degenerating features of these neurons. Degenerative Purkinje cells exhibited shrinkage and dark cytoplasm, and deformed and dark nuclei similar to the degenerate structure found in spinocerebellar ataxia type 31 disease. The unexpected percentage of neurons with degenerative features found in sham group is attributable to a not uncommon artifact called “dark neuron.”24 In any case, this phenomenon would affect equally all groups and the results should remain the same.
It is assumed that the cerebellar degeneration, consisting on a significant loss of Purkinje cells and gliosis, is commonly present in alcoholic cirrhotic patients due to nutritional deficiency. However, cerebellar degeneration was observed with similar prevalence in end-stage cirrhotic patients in both alcoholic and nonalcoholic. Fact that suggests that mechanisms altered in liver disease, different from nutritional deficiencies, may also contribute to the cerebellar neurodegeneration.4
To elucidate the mechanism involved in this degenerating process, caspase-3 immunostaining was performed. It revealed an increase in apoptosis of Bergmann glia in hyperammonemic rats (PCA, PCA+NH3 and PCA+NH3+LPS) that could be responsible for the increase of degenerative neurons. Bergmann glia is a radial astrocyte in the cerebellar cortex located in Purkinje layer,25 it actively takes part in the processing and maintenance of synaptic information via glutamate transporters and participates in controlling cell-membrane potential in Purkinje neurons.26 Our results could suggest that the degenerative process of Purkinje neurons may be related to an increase in apoptosis of Bergman glia due to hyperammonemia or an abnormal glutamatergic transmission. Other studies also indicate significant differences in responses between Bergman glia and the astrocytes of other brain regions during end-stage liver disease. While astrocytes lose their immunereactivity to GFAP, Bergmann glia in PCA rats shows an increase in GFAP immunereactivity, but in EH patients changes in GFAP immunereactivity are not detected.5,6
Previous studies in six-month PCA rat model showed an increase in astrocyte swelling and a vacuolization in the cerebellum.27 Additionally, microglia activation and astrocyte dysfunction were found in acute liver failure models28 and bile duct ligated rats with hyperammonemic diet.29 For this reason, the glia of cerebellum was evaluated to identify activated microglia (via Iba-1 staining) and reactive astrocytes (by GFAP staining). Our results illustrate an over-activation of microglia, together with changes in astrocyte size, which could participate in cell death.
In addition, gene expression array was performed to identify the pathways involved in the neurodegenerative process. Although, no canonical pathways were altered, the changes in gene expression detected might be taken into account as putative mechanisms related to neuronal loss in cerebellum. The overexpression of Htr2c, a gene related to serotonin neurotransmission detected in ammonia-treated animals, could also be implicated in the abnormal neurotransmission suggested between Bergman glia and Purkinje cells. On the other hand, Lcn-2 is secreted by glial cells, mainly astrocytes, during diverse inflammatory conditions30 and sensitizes neurons with regard to cell death, motility, and morphological changes.31 Likewise, Ccl-2 has been related with microglia activation and neuronal decline in acute HE.32 The overexpression of both genes (Lcn-2 and Ccl-2) exacerbated by inflammatory conditions could illustrate the plausible role of microglia activation in cerebellum neuronal death. Meanwhile, a vast overexpression of Ttr in cerebellum, but not in other tissues (data not shown) was exhibited with hyperammonemia. Ttr is a homotetrameric protein that works as a carrier protein, and its monomers or small oligomers form cytotoxic amyloid depositions.33 A protective role for Ttr has been suggested in Alzheimer's disease by the interaction of Ttr with amyloidgenic β-amyloid peptide that could inhibit amyloid fibril formation.34 High levels of Ttr protein without Ttr protein aggregates (data not shown) were observed in our study, therefore Ttr may adopt a protective role as endogenous detoxifier to reduce hyperammonemia. Beyond, an increase in α-synuclein, the protein responsible of insoluble fibril formation in Parkinson disease, has been reported in the Purkinje layer of chronic hyperammonemic rats.35 This protein could also be one of the degeneration causing agents in our model. However, further experiments are necessary to elucidate the mechanism involved in this degeneration process of Purkinje neurons.
The study has some weaknesses that should be taken into consideration in the interpretation of the results. The lack of correlation between the grade of ammonia insult and the amount of Purkinje neuron death between hyperammonemic rats (PCA and PCA+NH3) that could be probably due to an excessive duration of the animal model. In addition, the damage in brain tissue caused by ammonia overdose could be produced at earlier stages and be masked by the powerful effect of the five-month PCA. Moreover, the group which combines hyperammonemia and inflammation did not show synergic effects of both factors aggravating the neurological status; it seems to be contradictory to what happens in patients. A plausible explanation for that could be that low dose of LPS up-regulated the capacity of urea synthesis in rats36 and thus reduced the ammonia levels in blood and its toxic effects in brain.
In conclusion, this new model of episodic HE achieved by the chronic administration of ammonia showed an intermittent loss of neurological skills, mimicking the intermittent neurological alterations in the clinical course of patients with HE. In addition, the results derived from this new model provide the first evidence of degeneration and death of Purkinje neurons, probably mediated by Bergmann glia apoptosis. This new model might be a valuable tool for the study of the neuropathological mechanisms behind the episodic HE.
Supplementary Material
Supplementary Material
Acknowledgment
The authors want to give a special thanks to their mentor Professor Juan Cordoba deceased in 2014. We will always remember him for his honesty, generosity and friendship. Thanks for showing us the best way to do science.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by grant from Instituto de Salud Carlos III (FIS PI11/0954) co-financed by the European Regional Development Fund (ERDF). CIBEREHD is supported by Instituto de Salud Carlos III. Jordi Bover is the recipient of grant CP11/00229 from Instituto de Salud Carlos III.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author's contribution
TG-L: Acquisition of data, analysis and interpretation of data, statistical analysis and drafting the manuscript.
MO: Study concept and design, acquisition of data, analysis and interpretation of data, critical revision of the manuscript.
JR-G: Acquisition of data, analysis and interpretation of data.
JB: Study supervision, critical revision of the manuscript.
MV: Critical revision of the manuscript.
JG: Study supervision, critical revision of the manuscript.
LC: Acquisition of data, analysis and interpretation of data, statistical analysis, study supervision, critical revision of the manuscript.
JC: Study concept and design, obtained funding, analysis and interpretation of data.
LC and JC: equal responsibility for all aspects of this publication.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data.
Accession number of repository for expression microarray data
References
- 1.Dharel N, Bajaj JS. Definition and nomenclature of hepatic encephalopathy. J Clin Exp Hepatol 2015; 5: S37–S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butterworth RF. Neuronal cell death in hepatic encephalopathy. Metab Brain Dis 2007; 22: 309–320. [DOI] [PubMed] [Google Scholar]
- 3.Soffer D, Sherman Y, Turkaspa R, et al. Acquired hepatocerebral degeneration in a liver-transplant recipient. Acta Neuropathol 1995; 90: 107–111. [DOI] [PubMed] [Google Scholar]
- 4.Kril JJ, Butterworth RF. Diencephalic and cerebellar pathology in alcoholic and nonalcoholic patients with end-stage liver disease. Hepatology 1997; 26: 837–841. [DOI] [PubMed] [Google Scholar]
- 5.Kril JJ, Flowers D, Butterworth RF. Distinctive pattern of Bergmann glial pathology in human hepatic encephalopathy. Mol Chem Neuropathol 1997; 31: 279–287. [DOI] [PubMed] [Google Scholar]
- 6.Suarez I, Bodega G, Arilla E, et al. Different response of astrocytes and Bergmann glial cells to portacaval shunt: an immunohistochemical study in the rat cerebellum. Glia 1992; 6: 172–179. [DOI] [PubMed] [Google Scholar]
- 7.Bajaj JS, Schubert CM, Heuman DM, et al. Persistence of cognitive impairment after resolution of overt hepatic encephalopathy. Gastroenterology 2010; 138: 2332–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sotil EU, Gottstein J, Ayala E, et al. Impact of preoperative overt hepatic encephalopathy on neurocognitive function after liver transplantation. Liver Transpl 2009; 15: 184–192. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Martinez R, Rovira A, Alonso J, et al. Hepatic encephalopathy is associated with posttransplant cognitive function and brain volume. Liver Transpl 2011; 17: 38–46. [DOI] [PubMed] [Google Scholar]
- 10.Ahl B, Weissenborn K, van den HJ, et al. Regional differences in cerebral blood flow and cerebral ammonia metabolism in patients with cirrhosis. Hepatology 2004; 40: 73–79. [DOI] [PubMed] [Google Scholar]
- 11.Singh S, Koiri RK, Trigun SK. Acute and chronic hyperammonemia modulate antioxidant enzymes differently in cerebral cortex and cerebellum. Neurochem Res 2008; 33: 103–113. [DOI] [PubMed] [Google Scholar]
- 12.Butterworth RF, Norenberg MD, Felipo V, et al. Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int 2009; 29: 783–788. [DOI] [PubMed] [Google Scholar]
- 13.Oria M, Raguer N, Chatauret N, et al. Functional abnormalities of the motor tract in the rat after portocaval anastomosis and after carbon tetrachloride induction of cirrhosis. Metab Brain Dis 2006; 21: 297–308. [DOI] [PubMed] [Google Scholar]
- 14.Oria M, Chatauret N, Chavarria L, et al. Motor-evoked potentials in awake rats are a valid method of assessing hepatic encephalopathy and of studying its pathogenesis. Hepatology 2010; 52: 2077–2085. [DOI] [PubMed] [Google Scholar]
- 15.Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 2006; 1: 1306–1311. [DOI] [PubMed] [Google Scholar]
- 16.Recasens A, Dehay B, Bove J, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014; 75: 351–362. [DOI] [PubMed] [Google Scholar]
- 17.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida K, Asakawa M, Suzuki-Kouyama E, et al. Distinctive features of degenerating Purkinje cells in spinocerebellar ataxia type 31. Neuropathology 2014; 34: 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmermann C, Ferenci P, Pifl C, et al. Hepatic-Encephalopathy in thioacetamide-induced acute liver-failure in rats – characterization of an improved model and study of amino acid-ergic neurotransmission. Hepatology 1989; 9: 594–601. [DOI] [PubMed] [Google Scholar]
- 20.Luo M, Li L, Yang EN, et al. Correlation between interleukin-6 and ammonia in patients with overt hepatic encephalopathy due to cirrhosis. Clin Res Hepatol Gastroenterol 2013; 37: 384–390. [DOI] [PubMed] [Google Scholar]
- 21.Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 2003; 114: 188–193. [DOI] [PubMed] [Google Scholar]
- 22.Tupper DE, Wallace RB. Utility of the neurological examination in rats. Acta Neurobiol Exp (Wars) 1980; 40: 999–1003. [PubMed] [Google Scholar]
- 23.Fiala JC, Harris KM. Extending unbiased stereology of brain ultrastructure to three-dimensional volumes. J Am Med Inform Assoc 2001; 8: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graeber MB, Moran LB. Mechanisms of cell death in neurodegenerative diseases: fashion, fiction, and facts. Brain Pathol 2002; 12: 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellamy TC. Interactions between Purkinje neurones and Bergmann glia. Cerebellum 2006; 5: 116–126. [DOI] [PubMed] [Google Scholar]
- 26.Wang F, Xu Q, Wang W, et al. Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K + uptake. Proc Natl Acad Sci U S A 2012; 109: 7911–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavanagh JB, Lewis PD, Blakemore WF, et al. Changes in the cerebellar cortex in rats after portocaval anastomosis. J Neurol Sci 1972; 15: 13–26. [DOI] [PubMed] [Google Scholar]
- 28.Jiang W, Desjardins P, Butterworth RF. Cerebral inflammation contributes to encephalopathy and brain edema in acute liver failure: protective effect of minocycline. J Neurochem 2009; 109: 485–493. [DOI] [PubMed] [Google Scholar]
- 29.Rodrigo R, Cauli O, Gomez-Pinedo U, et al. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology 2010; 139: 675–684. [DOI] [PubMed] [Google Scholar]
- 30.Bi F, Huang C, Tong J, et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A 2013; 110: 4069–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S, Lee WH, Lee MS, et al. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J Neurosci Res 2012; 90: 540–550. [DOI] [PubMed] [Google Scholar]
- 32.McMillin M, Frampton G, Thompson M, et al. Neuronal CCL2 is upregulated during hepatic encephalopathy and contributes to microglia activation and neurological decline. J Neuroinflammation 2014; 11: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reixach N, Deechongkit S, Jiang X, et al. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci U S A 2004; 101: 2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wisniewski T, Castano E, Ghiso J, et al. Cerebrospinal fluid inhibits Alzheimer beta-amyloid fibril formation in vitro. Ann Neurol 1993; 34: 631–633. [DOI] [PubMed] [Google Scholar]
- 35.Suarez I, Bodega G, Fernandez B. Upregulation of alpha-synuclein expression in the rat cerebellum in experimental hepatic encephalopathy. Neuropathol Appl Neurobiol 2010; 36: 422–435. [DOI] [PubMed] [Google Scholar]
- 36.Nielsen SS, Grofte T, Tygstrup N, et al. Effect of lipopolysaccharide on in vivo and genetic regulation of rat urea synthesis. Liver Int 2005; 25: 177–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.