

Abstract
Impaired function of the endoplasmic reticulum (ER stress) is a hallmark of many human diseases including stroke. To restore ER function in stressed cells, the unfolded protein response (UPR) is induced, which activates 3 ER stress sensor proteins including activating transcription factor 6 (ATF6). ATF6 is then cleaved by proteases to form the short-form ATF6 (sATF6), a transcription factor. To determine the extent to which activation of the ATF6 UPR branch defines the fate and function of neurons after stroke, we generated a conditional and tamoxifen-inducible sATF6 knock-in mouse. To express sATF6 in forebrain neurons, we crossed our sATF6 knock-in mouse line with Emx1-Cre mice to generate ATF6-KI mice. After the ATF6 branch was activated in ATF6-KI mice with tamoxifen, mice were subjected to transient middle cerebral artery occlusion. Forced activation of the ATF6 UPR branch reduced infarct volume and improved functional outcome at 24 h after stroke. Increased autophagic activity at early reperfusion time after stroke may contribute to the ATF6-mediated neuroprotection. We concluded that the ATF6 UPR branch is crucial to ischemic stroke outcome. Therefore, boosting UPR pro-survival pathways may be a promising therapeutic strategy for stroke.
Keywords: Brain ischemia, endoplasmic reticulum stress, unfolded protein response, neuroprotection, transgenic mice
Introduction
Brain ischemia/stroke activates a complex pathological process involving regulation of many signaling pathways.1–3 Two strategies could be envisioned to improve stroke outcome: interfering with pathological processes triggered by ischemia, an approach that has yielded disappointing results in clinical trials,4 or boosting pro-survival pathways and thereby improving the self-healing capacity of the affected tissue. This strategy has not yet received much attention. Here, the unfolded protein response (UPR) could be a promising target, a complex of adaptive processes that enable cells to cope with stress conditions that impair the functioning of the endoplasmic reticulum (ER), a state termed ER stress. The key function of the ER is the folding and processing of newly synthesized membrane and secretory proteins, reactions that are sensitive to many stress conditions. Restoration of these processes is critical for cell survival after stress.5
A variety of diseases of major clinical significance are associated with ER stress including neurodegenerative diseases, myocardial ischemia/heart failure, and brain ischemia/stroke.6–8 The UPR has three response branches that are controlled by stress sensors in the ER membrane – activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and protein kinase RNA-like ER kinase (PERK). These UPR branches activate adaptive stress responses to resolve ER stress, including shutdown of translation to reduce the load of newly synthesized proteins that need to be folded (PERK branch), activation of a genetic program to increase folding capacity (ATF6 and IRE1 branches), and activation of ER-associated protein degradation (ERAD; ATF6 and IRE1 branches) to clear accumulated unfolded/misfolded proteins from the ER. The UPR also modulates other pro-survival pathways, which include autophagy and O-linked β-N-acetylglucosamine modification of proteins (O-GlcNAcylation; IRE1 branch).9,10 However, if ER stress persists and cellular homeostasis fails to be restored, the UPR triggers apoptosis, which is mediated primarily through the PERK branch,11 and involves B-cell lymphoma-2 (Bcl-2) family members, such as Bcl-2 and Bax.12
Considering the UPR as a potentially promising target for neuroprotection, we first need to know which of the UPR pro-survival pathways is critical for stroke outcome. It is well established that brain ischemia/stroke impairs ER function and activates the UPR.6,8 However, a systematic analysis of the role of each UPR branch in the fate and function of post-ischemic neurons has not yet been performed. In contrast, novel mouse models that target individual UPR branches specifically in cardiomyocytes have been generated, and there is now evidence that activation of the ATF6 and IRE1 UPR branches significantly improves outcome after myocardial ischemia.9,13
As a first attempt to explore the potential of boosting UPR pro-survival pathways as strategy for neuroprotection, we investigated the role of the ATF6 UPR branch in stroke. When activated under ER stress conditions, ATF6 translocates to the Golgi where it is cleaved by S1P and S2P proteases to form the short-form of ATF6 (sATF6), an active transcription factor.14,15 sATF6 then translocates to the nucleus and binds to the ER stress element sequence in the promoter of target genes and thereby activates their expression.16 To avoid the potential problem that ATF6 does not translocate to the Golgi after brain ischemia,17 we decided to express the active form of ATF6, sATF6. Here, we generated a conditional and inducible sATF6 knock-in mouse (Rosa26-sATF6-MER; Emx1-Cre, hereafter referred to as ATF6-KI) in which sATF6 is expressed predominantly in the neurons of the hippocampus and cortex, and its activation is controlled by tamoxifen. Using this new ATF6-KI mouse model, we demonstrated that forced activation of the ATF6 UPR branch improved functional outcome and reduced infarct volume after stroke.
Materials and methods
All animal experiments conformed to protocols approved by the Duke University Animal Care and Use Committee and complied with National Institutes of Health guidelines and ARRIVE guidelines.
Generation and breeding of ATF6 knock-in mice
The scheme of the targeting strategy is depicted in Figure 1(a). The FLAG-sATF6-MER fragment was assembled previously.13 The targeting vector was constructed and verified by restriction analysis and partial sequencing, and then linearized and electroporated into mouse embryonic stem (ES) cells derived from the 129 Sv strain at the Duke Neurotransgenic Laboratory. Positive ES clones were identified by PCR screening, and confirmed by Southern blot analysis. ES clones were microinjected into blastocysts derived from C57BL/6 mice to produce chimeric mice. After the germline transmission was confirmed, heterozygous mice (Rosa26-sATF6-MER) were backcrossed with C57BL/6. C57BL/6 mice were purchased from The Jackson Laboratory (Maine). The 5th or 6th generation of Rosa26-sATF6-MER mice was used for the present study. To express transgene FLAG-sATF6-MER in forebrain neurons, Emx1Cre/Cre mice (JAX stock #005628; C57BL/6 background) were crossbred with Rosa26-sATF6-MER mice to generate Rosa26-sATF6-MER; Emx1-Cre mice (ATF6-KI). Primers for genotyping are listed in Table S1.
Figure 1.

Generation of conditional and inducible ATF6 knock-in mice. (a) Scheme of the targeting strategy for generating Rosa26-sATF6-MER mice. FLAG-tagged ATF6 (39-373 aa; FLAG-sATF6) was fused to mutated estrogen receptor (MER) in-frame. A floxed STOP cassette was placed upstream of FLAG-sATF6-MER. After Cre-mediated deletion of the STOP cassette, the fusion protein FLAG-sTAF6-MER was expressed under control of the cytomegalovirus early enhancer/chickenβ−actin (CAG) promoter. The transgene was knocked in at the Rosa26 locus. (b) and (c) Rosa26-sATF6-MER mice were crossed with Emx1-Cre to generate Rosa26-sATF6-MER;Emx1-Cre mice (ATF6-KI). ATF6-KI mice were given corn oil (vehicle) or tamoxifen for five days, and their brains were then analyzed by immunohistochemistry. (b) Immunostaining with anti-FLAG and anti-ATF6 antibodies confirmed expression of FLAG-sATF6-MER and its nuclear translocation in the hippocampus and cortex after tamoxifen treatment. (c) FLAG-sATF6-MER was expressed predominately in neurons, as demonstrated by co-localization of FLAG staining with MAP2 (neuronal marker) staining. Scale bars: 50 µm.
Tamoxifen treatment
Tamoxifen (Cayman; 20 mg/mL) was suspended in corn oil (Sigma). Animals were treated with 100 mg/kg tamoxifen or corn oil (vehicle) by oral gavage once daily for five days.
Animal surgery
The surgeon was blind to the genotype of the animals. Transient middle cerebral artery occlusion (MCAO) was performed as described previously with minor modifications.18 Briefly, male mice (2–3 months old) were anesthetized with 5% isoflurane in 30% O2 balanced with N2O. Mice were then endotracheally intubated and mechanically ventilated. Isoflurane was decreased to 1.5%–1.8%. During the surgical procedure, the rectal temperature was maintained at 37℃ ± 0.2℃ using a heating pad and a heating lamp. Transient focal ischemia was induced by inserting a nylon monofilament (Doccol) into the right internal carotid artery via the external carotid artery and temporary ligation of the right common carotid artery. Laser-Doppler flowmetry (Moor) was used to monitor regional cerebral blood flow (rCBF) in the MCA cortex. After 45 min MCAO, mice were administrated 0.5 mL saline subcutaneously and were then transferred to a temperature-controlled incubator for 4 h before being returned to their home cages. Animals with brain hemorrhage, and those that did not show a reduction in rCBF > 80% during MCAO and recovery of rCBF > 80% after 5 min reperfusion, were excluded.
Acute neurologic assessment
Neurologic assessment was performed at 24 h post MCAO using a 48-point scoring system as recently detailed.19 Before evaluation, an observer who was blind to the genotype of the animals completed the video-assisted training and certification program in order to standardize the scoring process. This comprehensive scoring system evaluates general status (spontaneous activity, body symmetry, gait; 0–12), simple motor deficit (forelimb asymmetry, circling, hind limb placement; 0–14), complex motor deficit (vertical screen climbing, beam walking; 0–8), and sensory deficit (hind limb, trunk, vibrissae, and face touch; 0–14). After completion of testing, the score given to each animal was the sum of the four individual scores, with 0 = no deficit and 48 = maximal deficit.
Infarct volume
After evaluating neurologic deficits, mice were euthanized, and the brains were rapidly removed to measure infarct volume. Briefly, brains were coronally cut into 1-mm sections in a mouse brain matrix (ASI instruments) on ice. Brain sections were then incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) in 0.9% saline for 10 min. After fixing in 10% formaldehyde, the sections were digitally scanned, and the digital pictures were analyzed using the ImageJ software (NIH) by an observer blinded to the genotype of the animals. The infarct volume was measured indirectly to minimize any artifacts caused by post-ischemic edema. The infarct volume was calculated by subtracting the non-infarcted area in the ipsilateral hemisphere from the total area of the contralateral hemisphere.
Immunohistochemistry and microscopy
Immunohistochemistry was performed as described previously.20 In short, paraffin-embedded brain sections (5 µm) were deparaffinized, and then incubated with primary antibodies at 4℃ overnight. After extensive washing, the sections were incubated with fluorescent secondary antibodies for 1 h at room temperature. The primary antibodies used are listed in Table S2. Confocal images were captured on a Leica SP5 confocal microscope (Leica Microsystems).
Reverse-transcription PCR and quantitative PCR
Reverse-transcription PCR (RT-PCR) and quantitative PCR (qPCR) were performed as previously described.21 In short, total RNA was extracted from brain cortex samples using the TRIzol reagent (Invitrogen). One microgram of total RNA was reverse transcribed into cDNA (Invitrogen). To evaluate Xbp1 splicing, RT-PCR was performed. PCR products of spliced Xbp1 (Xbp1s) and unspliced Xbp1 (Xbp1u) were analyzed by agarose gel electrophoresis. qPCR was performed in a Lightcycler 2.0 (Roche). All primers used in this study are listed in Table S1.
Western blotting
Western blot analysis was performed essentially as described previously.21 Briefly, brain cortex samples were homogenized with sonication using 2% SDS lysis buffer. Protein samples were separated on pre-cast SDS-PAGE gels (Bio-Rad), and transferred to PVDF membranes. Membranes were blocked with TBST containing 5% milk or 5% BSA, and incubated with a primary antibody overnight at 4℃. After extensive washing, membranes were incubated with a secondary antibody for 1 h at room temperature. Proteins were then visualized using the enhanced chemiluminescence analysis system (GE Healthcare). After exposure, membranes were stripped and re-probed for β-actin as a loading control. Image analysis was performed using the ImageJ program (NIH). All primary antibodies used in this study are listed in Table S2.
Statistical analysis
All data analyses were performed with Prism 6 (GraphPad Software). Data are presented as mean ± SD or mean ± SEM. Statistical analysis was assessed by unpaired Student’s t-test (mRNA and protein levels, and infarct volumes) or Mann–Whitney U test (neurologic scores). The level of significance was set at p < 0.05.
Results
Generation and characterization of ATF6-KI mice
The targeting strategy to generate conditional and inducible sATF6 knock-in mice is illustrated in Figure 1(a). FLAG-tagged sATF6 (39-373 aa) was fused to the mutated estrogen receptor (MER). MER retains FLAG-sATF6-MER in the cytosol. When tamoxifen is introduced, it binds to MER, triggering translocation of FLAG-sATF6-MER into the nucleus, which activates expression of ATF6-dependent genes.13 In this targeting vector, FLAG-sATF6-MER was placed downstream of a loxP-flanked STOP cassette, and then cloned into a Rosa26 targeting vector. The cytomegalovirus early enhancer/chicken β-actin (CAG) promoter was used to achieve robust transgene expression. Using this targeting vector, we generated a new sATF6 knock-in mouse line, which we call Rosa26-sATF6-MER. To express transgene FLAG-sATF6-MER in forebrain neurons,22 we crossed Rosa26-sATF6-MER mice with Emx1-Cre mice to generate Rosa26-sATF6-MER;Emx1-Cre (ATF6-KI) mice. ATF6-KI mice are healthy and show no overt phenotype.
To induce nuclear translocation of FLAG-sATF6-MER and confirm its expression in neurons, ATF6-KI mice were treated with tamoxifen or corn oil (vehicle) for five days by oral gavage. As expected, in the brain of vehicle-treated ATF6-KI mice, FLAG-tagged sATF6-MER was retained in the cytoplasm, and tamoxifen exposure triggered its nuclear translocation, both in the cortex and hippocampus (Figure 1(b)). In ATF6-KI mouse brains, we found that FLAG-sATF6-MER was expressed predominately in neurons, as shown by its co-localization with the neuronal marker MAP2 (Figure 1(c); Figure S1), but not with the astrocyte marker GFAP (Figure S2(a)), or the endothelial cell marker CD31 (Figure S2(b)).
Next, we investigated whether tamoxifen-induced nuclear translocation of FLAG-sATF6-MER resulted in activation of ATF6-dependent genes. The mRNA levels of 4 ATF6-dependent genes that code for glucose-regulated proteins 78 and 94 (GRP78 and GRP94), protein disulfide isomerase (PDI), and ER stress protein 72 (ERp72), were analyzed by qPCR. On day 1 after the last dose of tamoxifen, the mRNA levels of all four genes were significantly increased in the brains of tamoxifen-treated ATF6-KI but not wild-type mice (Figure 2(a)). To detect the protein levels of ATF6-dependent genes, we collected brain samples on day 1 or 7 after the last dose of tamoxifen. On post-treatment day 1, GRP78 and PDI protein levels were markedly increased in the brains of tamoxifen-treated ATF6-KI but not wild-type mice (Figure 2(b), left panel). To clarify the subcellular localization of ER proteins activated in an ATF6-dependent manner in tamoxifen-treated ATF6-KI mice, we performed double immunofluorescence staining using antibodies against ATF6, and KDEL that cross-reacts with many ER-resident proteins, including GRP78, GRP94, and PDI. The KDEL signal intensity increased markedly after tamoxifen treatment and showed the extranuclear reticular pattern expected for ER-resident proteins (Figure S3). Finally, we evaluated the stability of proteins activated by sATF6. Levels of GRP78 and PDI were still increased seven days after the last tamoxifen dose (Figure 2(b), right panel) when FLAG-sATF6-MER immunoreactivity was present in the cytoplasm but no longer in the nucleus (Figure 2(c)), which suggested that tamoxifen was cleared from the brain. Notably, levels of FLAG-sATF6-MER were clearly decreased in the samples from day 1 but not day 7 post tamoxifen treatment (Figure 2(b)), an observation that confirmed decreased ATF6 stability after nuclear translocation reported earlier.13,23 Together, these findings confirmed that our new ATF6-KI mouse model is functional.
Figure 2.

Up-regulation of ATF6-dependent genes in ATF6-KI mouse brains. Wild-type (WT) and ATF6-KI mice were treated with corn oil (vehicle) or tamoxifen for five days. On day 1 or day 7 after the last dose of tamoxifen, mouse brain cortex samples were collected and analyzed by quantitative PCR (a) and Western blotting (b). (a) On day 1 after the last dose of tamoxifen, RNA samples were prepared from mouse brain cortex, and analyzed for mRNA levels of ATF6-dependent genes. All individual data were normalized to β-actin. To calculate fold change, the mean values of vehicle-treated WT mouse samples were set to 1.0. Data are presented as means ± SD (n = 3 per group), *p < 0.05. (b) On day 1 or day 7 after the last dose of tamoxifen, protein samples were prepared from mouse brain cortices, and analyzed for expression levels of FLAG-sATF6-MER and ATF6-dependent genes. (c) On day 7 after the last dose of tamoxifen, the brains of ATF6-KI were analyzed by immunohistochemistry with anti-FLAG. DAPI was used to stain nuclei (blue). FLAG-sATF6-MER was present in the cytoplasm, but not in the nucleus, in the forebrain neurons of ATF6-KI mice.
The ATF6 UPR branch is a pro-survival pathway in post-ischemic neurons after stroke
We first set out to confirm that transient brain ischemia activates the UPR branches in our MCAO model. After being activated, the ATF6 branch up-regulates many genes including GRP78, while IRE1 induces splicing of X-box binding protein-1 (Xbp1) mRNA, and PERK phosphorylates the alpha subunit of the eukaryotic initiation factor 2 (eIF2α).7 Indeed, we found that Xbp1 mRNA splicing and eIF2α phosphorylation, but not GRP78 expression, were increased after stroke (Figure S4), which is consistent with the current notion that the IRE1 and PERK branches, but not the ATF6 branch of UPR, are activated at early reperfusion time after brain ischemia/stroke.17,24,25 Recently, it was reported that mice with global deletion of Atf6 showed more pronounced spread of tissue damage from the ischemic core into the penumbra than wild-type mice in stroke.26 This implies that the ATF6 UPR branch is protective against ischemic brain damage. To determine whether activation of the ATF6 UPR branch in forebrain neurons is sufficient to improve stroke outcome, we subjected wild-type and ATF6-KI mice to 45 min MCAO seven days after dosing mice with tamoxifen. After 24 h reperfusion, animals were evaluated for neurologic deficits and infarct volumes. One animal in the ATF6-KI group was excluded because of brain hemorrhage. Representative TTC-stained brain slices and mean infarct volumes in brains of wild-type and ATF6-KI mice are depicted in Figure 3. Mean infarct volumes were 89.0 ± 19.6 mm3 versus 56.6 ± 32.1 mm3 in wild-type compared to ATF6-KI mice, respectively (Figure 3(b); p = 0.025). To extensively evaluate functional outcome at this early recovery interval after stroke, we used a 48-point scoring system that consists of 14 categories, and includes several behavioral tests.19 After testing, ATF6-KI mice showed better neurologic function (Figure 3(c); p = 0.0002). Together, our findings demonstrated that the ATF6 UPR branch plays an important role in protection of the brain from ischemic damage and in restoration of neurologic function impaired by ischemia.
Figure 3.
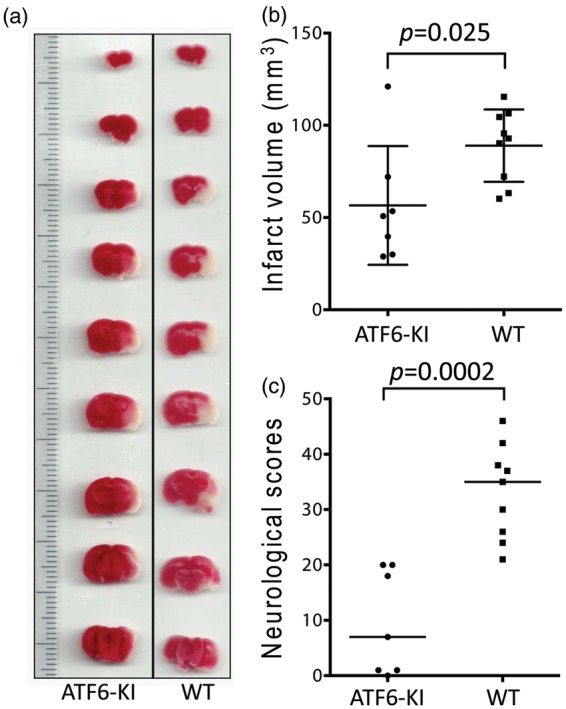
The effects of activation of the ATF6 UPR branch in neurons on infarct volume and neurologic function after stroke. Wild-type (WT) and ATF6-KI mice were treated with tamoxifen for five days. Seven days later, the mice (n = 9 or 7/group) were subjected to 45 min MCAO. After 24 h reperfusion, the animals were evaluated for neurologic deficits, and then sacrificed for measurement of infarct volumes by 2,3,5-triphenyltetrazolium chloride (TTC) staining. (a) Representative TTC-stained brain slices. (b) Total infarct volumes were presented as means ± SD. (c) Summary of neurologic scores. 0 = no deficit; horizontal bar = median values.
Increased level of GRP78 does not modulate activation of IRE and PERK UPR branches after stroke
ER stress is characterized by protein folding demand/capacity mismatch. Results from cell culture studies suggest that dissociation of GRP78 from ER stress sensors to increase folding capacity triggers activation of the UPR.27 If this model holds true in stroke, we would expect less activation of the IRE1 and PERK UPR branches in post-ischemic brains of ATF6-KI than wild-type mice because pre-ischemic GRP78 protein levels are considerably higher in ATF6-KI mice (Figure 2(b)). Surprisingly, ischemia-induced changes in Xbp1 mRNA splicing (IRE1 branch) and eIF2α phosphorylation (PERK branch) were similar in wild-type and ATF6-KI mice (Figure 4), an observation that seems not to support the GRP78 dissociation model in stroke. One possible explanation for this finding is that the potential mechanisms underlying UPR activation under a pathologic state in vivo are more complex than was originally thought based on earlier cell culture studies using potent pharmacologic ER stress inducers.27,28
Figure 4.

The UPR branches in ATF6-KI mouse brains after stroke. Wild-type (WT) and ATF6-KI mice were treated with tamoxifen for five days. Seven days later, mice (n = 3/group) were subjected to 45 min MCAO. After 1 h reperfusion, cortex samples from the contralateral (con) and ipsilateral (ips) hemispheres were collected and evaluated by Western blotting and reverse-transcription-PCR (RT-PCR) analysis. (a) Comparison of activation of the UPR branches in the post-ischemic brains of WT versus ATF6-KI mice. Xbp1 mRNA splicing was examined by RT-PCR to detect unspliced (Xbp1u) and spliced (Xbp1s) Xbp1 mRNAs. (b) Quantitative analysis of the Western blots shown in A. Intensities of each band were measured and normalized to β-actin. The mean values in contralateral cortex samples from WT mice were set to 1.0. Data were presented as means ± SEM. *p < 0.05.
Potential mechanisms underlying ATF6-mediated neuroprotection in stroke
ER stress-induced UPR is associated with pathways that could potentially impact cellular life/death decisions after stroke, including apoptosis and autophagy.12,29 To determine whether these processes may contribute to improved stroke outcome in ATF6-KI mice, we first analyzed levels of mediators involved in ER stress-induced apoptosis (Figure 5(a)) and autophagy signaling (Figure 5(b)) at 1 h reperfusion. Members of the Bcl-2 protein family, such as anti-apoptotic Bcl-2 and pro-apoptotic Bax, play a key role in ER stress-induced apoptosis.12 Compared to wild-type mice, ATF6-KI mice showed significantly higher Bcl-2 levels after stroke, while Bax appears increased in wild-type mice after stroke, although this increase did not reach statistical significance (Figure 5(a)). To investigate whether autophagy could be involved, we measured levels of microtubule-associated protein I light chain 3 (LC3)-II and p62. Increase in LC3-II or decrease in p62 is an indicator of autophagic activity. In support of a role for autophagy activation in ATF6-induced neuroprotection, we found a significant decrease in levels of p62 in ATF6-KI mice at 1 h reperfusion after stroke, while LC3-II levels were increased in both wild-type and ATF6-KI mice (Figure 5(b)). We then evaluated levels of the mammalian target of rapamycin (mTOR), which negatively regulates autophagy. Notably, activated mTOR (p-mTOR) levels were significantly decreased in ATF6-KI mice after stroke. To further clarify whether activation of the ATF6 UPR branch resulted in a more pronounced post-ischemic activation of autophagy or an earlier onset of activation, we compared levels of p62 and p-mTOR in wild-type and ATF6-KI mice at 6 h after stroke (Figure 5(c)). In agreement with a previous report, we found a significant decrease of p62 in the ipsilateral cortex of both wild-type and ATF6-KI mice (Figure 5(c)).30 Levels of p-mTOR continued to be significantly lower in ATF6-KI mice after stroke (Figure 5(c)). Although a similar trend of decreased p-mTOR levels after stroke appeared in wild-type mice, differences did not reach statistical significance at both reperfusion times. Together, these data implied that autophagy is activated at an earlier reperfusion time in ATF6-KI mice than wild-type mice after stroke. This earlier activation in ATF6-KI mice is possibly mediated through suppression of the mTOR pathway.
Figure 5.

Potential mechanisms underlying ATF6-mediated neuroprotection in stroke. Wild-type (WT) and ATF6-KI mice were treated with tamoxifen for five days. Seven days later, mice (n = 3/group) were subjected to 45 min MCAO. After 1 h or 6 h reperfusion, cortex samples from the contralateral (con) and ipsilateral (ips) hemispheres were collected and evaluated by Western blot analysis. (a) Levels of ER stress-related anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax at 1 h reperfusion. (b) and (c) Autophagy signaling at 1 h (b) or 6 h (c) reperfusion. Quantitative analysis of the Western blots is shown as bar graphs. Intensities of selected proteins were measured and normalized to β-actin or mTOR. The mean values in contralateral cortex samples from WT mice were set to 1.0. Data were presented as means ± SEM. *p < 0.05.
Discussion
Here, we have presented a novel UPR-related genetically modified mouse model (Rosa26-sATF6-MER), and performed the first experimental study to clarify the role of the UPR in stroke outcome using mice in which the ATF6 UPR branch was modulated predominantly in the neurons of the cortex and hippocampus. Since Rosa26-sATF6-MER is a Cre-loxP-based conditional mouse model, this mouse line can be used to activate the ATF6 UPR branch in a cell/tissue/organ-specific manner and therefore will be also of value to researchers who investigate mechanisms of other ER stress-associated diseases. In the present study, Rosa26-sATF6-MER mice were crossed with Emx1-Cre mice to modulate the ATF6 UPR branch in forebrain neurons. To avoid any potential negative side effects caused by a permanently active ATF6 UPR branch, we used a tamoxifen-inducible approach to activate expression of ATF6-dependent genes before performing ischemia experiments. Our results confirmed that this approach works in our ATF6-KI mice, as expected; sATF6 was constitutively expressed but retained in the cytoplasm, and after tamoxifen treatment, sATF6 translocated to the nucleus and activated expression of ATF6-dependent genes (Figure 1). Importantly, the levels of GRP78 and PDI were still markedly increased seven days after tamoxifen treatment when FLAG-sATF6-MER was present in the cytoplasm but no longer in the nucleus. This suggests that tamoxifen was cleared from the brain, and did not contribute to neuroprotection, in contrast to acute tamoxifen treatment.31 Together, the findings demonstrate that our novel mouse model with tamoxifen-induced activation of the ATF6 UPR branch in forebrain neurons is functional.
About 20 years ago, it was first hypothesized that brain ischemia impairs ER function causing ER stress, and that this impairment contributes to ischemic brain damage.32 This hypothesis triggered a large number of experimental studies on various aspects of ER stress-induced activation of the UPR in brain ischemia, summarized in many reviews.8,10,32–35 Despite this wealth of information, we still know little about the role of individual UPR branches in the fate and function of neurons after ischemic stroke because most of those experimental studies either were descriptive, used global brain ischemia or cell/tissue culture models, or modulated the UPR branches in the whole body. Manipulating UPR pathways in neurons for brain ischemia studies provides, however, an advantage over global approaches because neurons are the primary target in experimental stroke research. Indeed, neurons are particularly sensitive to even short periods of ischemia, and recovery of neurologic function ultimately defines quality of life for stroke patients. Moreover, global approaches affect the UPR branches in all organs, which could have unpredictable effects on pathologic processes in post-ischemic brains.
In search for UPR pro-survival pathways that have the potentials to be considered in future studies as targets for therapeutic intervention in stroke, we first focused on the ATF6 UPR branch, because some evidence is at hand that the ATF6 pathway is not or only slightly activated after both transient global and focal cerebral ischemia (Figure S4).17,24 It has therefore been concluded that the dysfunctional UPR activation after transient brain ischemia may play a significant role in neuronal cell death during reperfusion.17 We therefore expected improved stroke outcome by boosting the ATF6 pathway. Our findings presented here have indeed provided evidence that neurologic function was significantly improved in ATF6-KI mice (Figure 3), suggesting that restoration of ER protein homeostasis by activation of the ATF6 pathway could result in better stroke outcome.
Since ER chaperons and folding enzymes including GRP78, GRP94 and PDI are notably up-regulated in pre-ischemic ATF6-KI mice, it is likely that the increased folding capacity contributes to improved stroke outcome. It has been shown that up-regulation of GRP78 and PDI protects neurons from ischemic damage.36,37 Notably, Bcl-2, an anti-apoptotic protein that modulates ER stress-induced apoptosis, was increased in ATF6-KI compared to wild-type mice after stroke, suggesting less ER stress-induced apoptosis in the ATF6-KI mice (Figure 5(a)). Our results also imply modulation of autophagy to be a contributing factor. Recently, mounting evidence indicates autophagy activation is associated with neuroprotection in brain ischemia and crosstalk between the UPR and autophagy plays a critical role in stroke outcome.30,38–40 For example, in a rat model of ischemic pre-conditioning, blocking autophagy with 3-methyladenine (3-MA) inhibits GRP78 upregulation, exacerbates ER stress, increases apoptosis, and worsens stroke outcome.39 Using a rat stroke mode, a significant decrease of p62, indication of autophagy activation, was not observed until 6 h reperfusion.30 In contrast, in our ATF6-KI mice, we showed that activation of the ATF6 UPR branch significantly reduces the protein levels of p62 in the post-ischemic brain at 1 h reperfusion, suggesting earlier onset of increased autophagic activity (Figure 5(c)). This activation of autophagy was likely mediated through decreased levels of phosphorylated mTOR, a key inhibitory regulator of autophagy. Collectively, we reported here a correlation between ATF6 activation and changes in molecular markers of apoptosis and autophagy. Whether these changes contributed to the better stroke outcome observed in ATF6-KI mice needs to be evaluated in future studies.
The PERK, IRE1, and ATF6 UPR branches fulfill different functions in cells under ER stress. These include activation of genetic programs that increase protein folding and processing capacity (ATF6 and IRE1 branches), shutdown of translation to reduce the ER load (PERK branch), and, if ER stress persists and protein homeostasis fails to be restored, induction of apoptosis to eliminate irreversibly damaged cells (PERK branch). Thus, the ATF6 and IRE1 UPR branches can be considered pro-survival pathways that are essential to restore key cellular functions impaired by stress and thereby, promote cell survival. Indeed, in the heart, both the ATF6 and IRE1 UPR branches have been reported to be pro-survival pathways that protect hearts from ischemic damage when activated in cardiomyocytes.9,13 IRE1-mediated splicing of Xbp1 mRNA triggers a frame-shift of the coding region, and formation of a new 54-kDa protein, the transcription factor XBP1s. In myocardial ischemia, the pro-survival effect of the IRE1 UPR branch is provided by XBP1s-induced activation of O-GlcNAcylation.9 A variety of studies reported a protective effect of increased O-GlcNAcylation in myocardial ischemia,41 and this post-translational modification is also activated after brain ischemia, implying similar protective function.21 Notably, ours is the first study to report that boosting the ATF6 UPR branch in neurons provided beneficial effects for neurologic function after stroke (Figure 3). Thus, results presented here and reported from myocardial ischemia studies suggest that UPR pathways could be considered for therapeutic intervention in stroke.
The findings reported here confirm our earlier hypothesis that recovery of ER function affects outcome after brain ischemia.32,35 In addition to brain ischemia/stroke, impaired ER function is associated with a variety of stress-related diseases. This suggests that ER-resident protein folding and processing reactions are particularly sensitive to stress conditions. Our results also support our hypothesis that boosting UPR pro-survival pathways could be a promising strategy to improve stroke outcome. In this study, we focused only on boosting the ATF6 UPR branch to improve stroke outcome. Results from myocardial ischemia experiments on wild-type and XBP1 loss-of-function and gain-of-function mice suggest that the IRE1/XBP1 branch of UPR is also a pro-survival pathway, and that the IRE1/XBP1/O-GlcNAc axis plays a major role in this process.9 We have recently reported that O-GlcNAcylation is activated after brain ischemia in young but not aged mice,21 but it still needs to be verified whether this activation is XBP1-dependent, and whether the IRE1/XBP1/O-GlcNAC axis is a pro-survival pathway after brain ischemia as it is after myocardial ischemia.
Supplementary Material
Acknowledgments
We thank Pei Miao for her excellent technical support, and Kathy Gage for her excellent editorial contributions.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by AHA Scientist Development Grant 12SDG11950003 (to WY), and by NIH R01 grant NS081299 and R03 grant NS078590 (to WP).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
ZY, HS, SL, SZ, CG, DW, WP, and WY performed the experiments and/or data analysis. WP and WY conceived the study and wrote the manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Neumar RW. Molecular mechanisms of ischemic neuronal injury. Ann Emerg Med 2000; 36: 483–506. [DOI] [PubMed] [Google Scholar]
- 2.Lo EH, Moskowitz MA, Jacobs TP. Exciting, radical, suicidal: how brain cells die after stroke. Stroke 2005; 36: 189–192. [DOI] [PubMed] [Google Scholar]
- 3.Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology 2008; 55: 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moretti A, Ferrari F, Villa RF. Neuroprotection for ischaemic stroke: current status and challenges. Pharmacol Ther 2015; 146: 23–34. [DOI] [PubMed] [Google Scholar]
- 5.Harding HP, Calfon M, Urano F, et al. Transcriptional and translational control in the Mammalian unfolded protein response. Annu Rev Cell Dev Biol 2002; 18: 575–599. [DOI] [PubMed] [Google Scholar]
- 6.Roussel BD, Kruppa AJ, Miranda E, et al. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol 2013; 12: 105–118. [DOI] [PubMed] [Google Scholar]
- 7.Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol 2013; 75: 49–67. [DOI] [PubMed] [Google Scholar]
- 8.Paschen W, Mengesdorf T. Cellular abnormalities linked to endoplasmic reticulum dysfunction in cerebrovascular disease – therapeutic potential. Pharmacol Ther 2005; 108: 362–375. [DOI] [PubMed] [Google Scholar]
- 9.Wang ZV, Deng Y, Gao N, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014; 156: 1179–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakka VP, Prakash-Babu P, Vemuganti R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: potential therapeutic targets for acute CNS injuries. Mol Neurobiol 2016; 53: 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318: 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szegezdi E, Logue SE, Gorman AM, et al. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 2006; 7: 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martindale JJ, Fernandez R, Thuerauf D, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res 2006; 98: 1186–1193. [DOI] [PubMed] [Google Scholar]
- 14.Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 2000; 6: 1355–1364. [DOI] [PubMed] [Google Scholar]
- 15.Haze K, Yoshida H, Yanagi H, et al. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 1999; 10: 3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida H, Okada T, Haze K, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol 2000; 20: 6755–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar R, Krause GS, Yoshida H, et al. Dysfunction of the unfolded protein response during global brain ischemia and reperfusion. J Cereb Blood Flow Metab 2003; 23: 462–471. [DOI] [PubMed] [Google Scholar]
- 18.Chaparro RE, Izutsu M, Sasaki T, et al. Sustained functional improvement by hepatocyte growth factor-like small molecule BB3 after focal cerebral ischemia in rats and mice. J Cereb Blood Flow Metab 2015; 35: 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taninishi H, Pearlstein M, Sheng H, et al. Video training and certification program improves reliability of postischemic neurologic deficit measurement in the rat. J Cereb Blood Flow Metab.– Epub ahead of print 24 November 2015. DOI: 10.1177/0271678X15616980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W, Sheng H, Thompson JW, et al. Small ubiquitin-like modifier 3-modified proteome regulated by brain ischemia in novel small ubiquitin-like modifier transgenic mice: putative protective proteins/pathways. Stroke 2014; 45: 1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S, Sheng H, Yu Z, et al. O-linked beta-N-acetylglucosamine modification of proteins is activated in post-ischemic brains of young but not aged mice: implications for impaired functional recovery from ischemic stress. J Cereb Blood Flow Metab 2016; 36: 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan CH, Godinho LN, Thomaidou D, et al. Emx1 is a marker for pyramidal neurons of the cerebral cortex. Cereb Cortex 2001; 11: 1191–1198. [DOI] [PubMed] [Google Scholar]
- 23.Thuerauf DJ, Morrison LE, Hoover H, et al. Coordination of ATF6-mediated transcription and ATF6 degradation by a domain that is shared with the viral transcription factor, VP16. J Biol Chem 2002; 277: 20734–20739. [DOI] [PubMed] [Google Scholar]
- 24.Paschen W, Aufenberg C, Hotop S, et al. Transient cerebral ischemia activates processing of xbp1 messenger RNA indicative of endoplasmic reticulum stress. J Cereb Blood Flow Metab 2003; 23: 449–461. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto N, Oida Y, Shimazawa M, et al. Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience 2007; 147: 957–967. [DOI] [PubMed] [Google Scholar]
- 26.Yoshikawa A, Kamide T, Hashida K, et al. Deletion of Atf6alpha impairs astroglial activation and enhances neuronal death following brain ischemia in mice. J Neurochem 2015; 132: 342–353. [DOI] [PubMed] [Google Scholar]
- 27.Bertolotti A, Zhang Y, Hendershot LM, et al. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2: 326–332. [DOI] [PubMed] [Google Scholar]
- 28.Credle JJ, Finer-Moore JS, Papa FR, et al. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A 2005; 102: 18773–18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 2015; 40: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao L, Jiang T, Guo J, et al. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PloS One 2012; 7: e46092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimelberg HK, Feustel PJ, Jin Y, et al. Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport 2000; 11: 2675–2679. [DOI] [PubMed] [Google Scholar]
- 32.Paschen W. Disturbances of calcium homeostasis within the endoplasmic reticulum may contribute to the development of ischemic-cell damage. Med Hypotheses 1996; 47: 283–288. [DOI] [PubMed] [Google Scholar]
- 33.Paschen W. Shutdown of translation: lethal or protective? Unfolded protein response versus apoptosis. J Cereb Blood Flow Metab 2003; 23: 773–779. [DOI] [PubMed] [Google Scholar]
- 34.DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem 2004; 91: 1–8. [DOI] [PubMed] [Google Scholar]
- 35.Paschen W, Frandsen A. Endoplasmic reticulum dysfunction – a common denominator for cell injury in acute and degenerative diseases of the brain? J Neurochem 2001; 79: 719–725. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka S, Uehara T, Nomura Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J Biol Chem 2000; 275: 10388–10393. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi T, Saito A, Okuno S, et al. Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J Cereb Blood Flow Metab 2003; 23: 949–961. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Yuan Y, Jiang L, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy. Autophagy 2014; 10: 1801–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng R, Liu XQ, Zhang LS, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 2012; 8: 310–325. [DOI] [PubMed] [Google Scholar]
- 40.Papadakis M, Hadley G, Xilouri M, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med 2013; 19: 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marsh SA, Collins HE, Chatham JC. Protein O-GlcNAcylation and cardiovascular (patho)physiology. J Biol Chem 2014; 289: 34449–34456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.