

Abstract
Perinatal asphyxia in preterm infants remains a significant contributor to abnormal long-term neurodevelopmental outcomes. Recombinant human erythropoietin has potent non-haematopoietic neuroprotective properties, but there is limited evidence for protection in the preterm brain. Preterm (0.7 gestation) fetal sheep received sham asphyxia (sham occlusion) or asphyxia induced by umbilical cord occlusion for 25 min, followed by an intravenous infusion of vehicle (occlusion-vehicle) or recombinant human erythropoietin (occlusion-Epo, 5000 international units by slow push, then 832.5 IU/h), starting 30 min after asphyxia and continued until 72 h. Recombinant human erythropoietin reduced neuronal loss and numbers of caspase-3-positive cells in the striatal caudate nucleus, CA3 and dentate gyrus of the hippocampus, and thalamic medial nucleus (P < 0.05 vs. occlusion-vehicle). In the white matter tracts, recombinant human erythropoietin increased total, but not immature/mature oligodendrocytes (P < 0.05 vs. occlusion-vehicle), with increased cell proliferation and reduced induction of activated caspase-3, microglia and astrocytes (P < 0.05). Finally, occlusion-Epo reduced seizure burden, with more rapid recovery of electroencephalogram power, spectral edge frequency, and carotid blood flow. In summary, prolonged infusion of recombinant human erythropoietin after severe asphyxia in preterm fetal sheep was partially neuroprotective and improved electrophysiological and cerebrovascular recovery, in association with reduced apoptosis and inflammation.
Keywords: Asphyxia, erythropoietin, neuroprotection, preterm, white matter
Introduction
Preterm infants continue to have a very high rate of neurodevelopmental disability.1 The etiology of preterm brain injury is clearly multifactorial;2 however, one contributing factor is the increased risk of acute hypoxic-ischemic encephalopathy (HIE) in moderately preterm infants compared to term.3 Therapeutic hypothermia improves outcomes in term infants with HIE but has not been shown to be safe in preterm infants.4 Thus, there is a considerable, unmet need for safe neuroprotective treatments for moderately preterm infants with acute HIE.
There is evidence that the haematopoietic cytokine, erythropoietin (Epo), can improve histological and behavioural outcomes after ischemia and hypoxia-ischemia (HI) in adult and neonatal rodents, as recently reviewed.5 Although there is limited information in the preterm-equivalent brain,6,7 brain maturity of postnatal day (P)7 rat pups is broadly consistent with that of the late preterm infant.8 In P7 rats, neuroprotection with peripheral injection of recombinant human erythropoietin (rEpo) immediately after HI is reported with a wide range of doses, from 1000 to 30,000 international units (IU)/kg.5
Critically, repeated injection with 5000 IU/kg rEpo daily for three days, started immediately after HI, was more neuroprotective after seven days recovery, than either a single dose of 5000 IU/kg or three injections of 30,000 IU/kg,9 but neuroprotection was largely lost when treatment was delayed for 1 to 3 h after HI.10 This finding parallels the experience with mild induced hypothermia that optimal neuroprotection after acute HI injury is seen when cooling is started within the first 6 h and continued until secondary events such as seizures have resolved, after approximately three days.11 This suggests that continued exposure to rEpo during this critical phase will be needed for optimal outcomes after acute HI.
Further, small clinical studies report that rEpo injections of 3000 IU/kg started within 3 h of birth, then at 12–18 h and 36–42 h, reduced the risk of white matter injury,12 and improved white matter development at term-equivalent.13 However, functionally, meta-analysis of small randomized trials of early, repeated treatment of preterm infants with rEpo suggests that while it was associated with a small improvement in neurodevelopment, there was no effect on severe disability.14 These studies used intermittent injections based on the relatively long half-life (up to ∼ 18 h) of high-dose rEpo.15 However, intermittent injections must be associated with significant variation in concentration of Epo. Thus, it is possible that a sustained infusion which provides more stable levels may be neuroprotective even after delayed initiation of treatment.
In the present study, we examined the hypothesis that continuous infusion with rEpo, started 30 min after severe asphyxia induced by umbilical cord occlusion and continued until 72 h, would improve electrophysiological recovery and alleviate white and grey matter damage in the immature 0.7 gestation fetal sheep. The focus of this study was on potential for acute neuroprotection through suppression of apoptosis and inflammation; we also quantified proliferation in the white matter tracts as rEpo has longer term effects on oligodendrogenesis that may promote neurorestoration.5 Brain development at this fetal age is broadly similar to 28 to 32 weeks in the human infant.16
Materials and methods
Experimental preparation
All animal procedures were approved by the Animal Ethics Committee of the University of Auckland under the provisions of the New Zealand Animal Welfare Act, and the Code of Ethical Conduct for animals in research established by the Ministry of Primary Industries, Government of New Zealand. This manuscript complies with the ARRIVE guidelines for reporting animal research.17 Twenty-six time-mated Romney/Suffolk fetal sheep were instrumented at 98 to 99 days gestation (term = 147 days). Food, but not water was withdrawn 18 h before surgery. Ewes were given long-acting oxytetracycline (20 mg/kg, Phoenix Pharm., Auckland, New Zealand) intramuscularly for prophylaxis 30 min before the start of surgery. Anaesthesia was induced by intravenous (i.v.) injection of propofol (5 mg/kg, AstraZeneca Limited, Auckland, New Zealand), and after intubation, anaesthesia was maintained with 2%–3% isoflurane (Medsource, Ashburton, New Zealand) in O2. The depth of anaesthesia, maternal heart rate, and respiration were monitored by trained anaesthetic staff during surgery. Ewes received a constant isotonic saline drip (250 mL/h) to maintain fluid balance.
All surgical procedures were performed under aseptic conditions.18 Following a maternal midline abdominal incision, the fetus was exposed and polyvinyl catheters were inserted into the fetal right brachial vein for drug infusion, and both brachial arteries for pre-ductal blood sampling and mean arterial blood pressure (MAP) monitoring. An amniotic catheter was secured to the fetal shoulder. Electrocardiogram (ECG) electrodes (AS633-3SSF, Cooner Wire Co., Chatsworth, CA, USA) were placed subcutaneously over the right shoulder and the fetal chest at the apex to record fetal heart rate (FHR). Two pairs of electroencephalogram (EEG) electrodes (AS633-5SSF, Cooner Wire Co.) were placed on the dura over the parasagittal parietal cortex (5 mm and 10 mm anterior to bregma and 5 mm lateral), with a reference electrode sewn over the occiput. One thermistor (IncuTemp-1, Mallinckrodt Pharmaceuticals, Chesterfield, UK) was placed over the parasagittal dura 20 mm anterior to bregma to measure extradural temperature. All burr holes were sealed and the fetal scalp secured with cyanoacrylate glue. An ultrasonic flow probe (3S type, Transonic Systems Inc., Ithaca, NY, USA) was placed around the right carotid artery to provide an index of global cephalic blood flow.19,20 Finally, a silicone vascular occluder was fitted around the umbilical cord of all fetuses (In Vivo Metric, Healdsburg, CA, USA).
The uterus was closed, and antibiotics (80 mg gentamicin sulfate, Pfizer, Auckland, New Zealand) were administered into the amniotic sac. Any amniotic fluid lost during surgery was replaced with isotonic saline warmed to 37℃. The maternal laparotomy incision was repaired and the skin infiltrated with 10 mL of 0.25% bupivacaine plus adrenaline (AstraZeneca Limited). The maternal long saphenous vein was catheterized for post-operative care, and all fetal leads were exteriorized through the maternal flank.
Post-operative care
Sheep were housed together in separate metabolic cages with access to water and concentrate pellet feed (Dunstan Nutrition, Hamilton, New Zealand) ad libitum. The animal housing facility was temperature controlled (16 ± 1℃, humidity 50 ± 10%), and operated on a 12-h light/dark cycle (6 a.m. on, 6 p.m. off). During the post-operative period before experiments, ewes were given daily i.v. antibiotics, including gentamicin (40 mg for two days, Pfizer) and benzylpenicillin sodium (600 mg for four days, Onelink, Auckland, New Zealand). Vascular catheters were maintained patent by continuous infusion of heparinized saline (20 IU/mL at 0.15 mL/h), and the maternal catheter was maintained by daily flushing.
Experimental recordings
Experiments were conducted at 103–104 days gestation. Fetal MAP, corrected for maternal movement by subtraction of amniotic pressure (MDX-MX866; Medex Inc., Hilliard, OH, USA), ECG, EEG, carotid blood flow (CaBF) and extradural temperature were recorded continuously from 24 h before until 72 h after occlusion or sham occlusion. The CaBF signal was low-pass filtered with a second-order Butterworth filter at 10 Hz, then digitized at 512 Hz. Similarly, the pressure signal was low-pass filtered with a Butterworth filter, with the −3 decibel (dB) point set to a cut-off frequency of 20 Hz, then digitized at 512 Hz.
The analogue EEG signal was filtered with a first-order high-pass filter at 1.6 Hz and a sixth-order low-pass Butterworth anti-aliasing filter, with the −3 dB point set at a cut-off frequency of 50 Hz, then digitized at 512 Hz. EEG power and spectral edge frequency (SEF, Hz) were extracted between 1 and 20 Hz from four second(s) epochs. The EEG power signal was log transformed (dB, 20 × log (power)).21 SEF was defined as the frequency below which 90% of the EEG power lies. Data from left and right EEG electrodes were averaged and normalized with respect to the 24 h baseline period. All experimental data were collected for offline analysis using custom software (LabVIEW for Windows, National Instruments, Auckland, New Zealand).
Experimental protocols
Fetuses were randomly assigned to either sham occlusion (n = 9), occlusion-vehicle (n = 9), or occlusion-Epo (n = 8). Fetal asphyxia was induced for 25 min by rapid inflation of the umbilical cord occluder with sterile saline of a defined volume known to completely occlude the umbilical blood flow.22 Occlusion was confirmed by rapid onset of bradycardia, hypertension, and changes in blood gases. This duration represents an acute, near-terminal asphyxial insult that is associated with diffuse white matter lesions and moderate subcortical neural loss,23 similarly to the pattern seen in preterm infants.24 The umbilical cord occluder was not inflated in the sham occlusion animals. All occlusions were performed at 0900 h.
Fetuses received either sterile isotonic saline with 0.1% bovine serum albumin (BSA: vehicle) or rEpo (Janssen-Cilag Ltd, Auckland, New Zealand) in vehicle solution, started from 30 min after asphyxia and continued for 72 h. Catheters were pre-infused with vehicle solution. rEpo or the same volume of vehicle was injected as a single loading bolus of 5000 IU by slow push over 5 min, followed by continuous i.v. infusion of 832.5 IU/h (2500 IU/mL at 0.333 mL/h).
Fetal arterial blood samples were taken 60 min before occlusion, at 5 and 17 min during occlusion, and 10 min, 1, 2, 4, 6, 24, 48, and 72 h after occlusion for pre-ductal pH, blood gas, glucose, and lactate determination (ABL800 Flex analyzer, Radiometer, Auckland, New Zealand). All fetuses had normal biochemical variables for their gestational age.18 Fetal blood samples were collected on ice in EDTA vacutainers (Onelink, Auckland, New Zealand) at the same times as above, for Epo measurement. Samples were immediately centrifuged at 3000 r/min for 10 min at 4℃ (Heraeus Megafuge 8R, Thermo Fisher Scientific Ltd., Auckland, New Zealand), and plasma was collected and stored at −80℃.
Three days after occlusion, the ewes and fetuses were killed by overdose of sodium pentobarbitone (9 g, i.v. to the ewe; Pentobarb 300, Provet, Auckland, New Zealand). The fetuses were removed by hysterectomy, and the fetus and selected organs were weighed. The brains were perfusion fixed in situ with saline followed by 500 mL of 10% phosphate buffered formalin. Brain tissue was post-fixed in 10% formalin solution for five to six days and then processed and embedded using a standard paraffin preparation.
Immunohistochemistry
Coronal slices (10 µm thick) at the level of the mid-striatum and dorsal hippocampus were cut using a microtome (Leica Jung RM2035, Leica Microsystems Ltd, Albany, New Zealand), and mounted on chrome alum-coated slides. Slides were then dewaxed in xylene, rehydrated in decreasing concentrations of ethanol and washed in 0.1 mol/L phosphate buffered saline (PBS). Antigen retrieval was performed for 2 h in 0.01 mol/L citrate buffer using an antigen retrieval system (2100 Retriever, Aptum Biologics Ltd, Southampton, UK), followed by endogenous peroxidase quenching for 30 min through incubation in 1% H2O2 in methanol for neuronal nuclei (NeuN), cysteine-aspartic acid protease 3 (activated caspase-3), ionized calcium-binding adapter molecule 1 (Iba1), glial fibrillary acidic protein (GFAP), 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), and the proliferation marker Ki67, and PBS for oligodendrocyte transcription factor 2 (Olig2, which labels oligodendrocytes at all stages of the lineage).25
Blocking was performed in 3% normal goat serum (NGS; Life Technologies Limited, Auckland, New Zealand) in PBS for 1 h at room temperature. Sections were labelled with 1:200 rabbit anti-NeuN (AB177487, Abcam, Melbourne, Australia), 1:200 rabbit cleaved caspase-3 (CTE9661S, Thermo Fisher Scientific, Auckland, New Zealand), 1:200 rabbit anti-Iba1 (AB178680, Abcam), 1:200 rabbit anti-GFAP (AB68428, Abcam), 1:200 mouse anti-CNPase (AB6319, Abcam), 1:200 mouse anti-Ki67 (M7240, Dako Limited, Sydney, Australia), and 1:200 rabbit anti-Olig2 (AB109186, Abcam) in 3% NGS-PBS, overnight at 4℃. Sections were labelled with 1:200 goat anti-rabbit (NeuN, Caspase-3, Iba1, GFAP, and Olig2) or goat anti-mouse biotin-conjugated IgG (CNPase and Ki67; In Vitro Technologies, Auckland, New Zealand) in 3% NGS-PBS, overnight at 4℃. Slides were then incubated in 1:200 ExtrAvidin-Peroxidase (Sigma-Aldrich, Auckland, New Zealand) in 3% NGS-PBS for 2 h at room temperature, and reacted in diaminobenzidine (Sigma-Aldrich). The reaction was stopped by washing in PBS, and slides dehydrated and mounted with DPX (Scharlab, Barcelona, Spain).
Brain regions used for analysis included the mid-striatum (including the caudate nucleus and putamen), and the parasagittal intragyral white matter (IGWM) and periventricular white matter (PVWM) tracts on sections taken 23 mm anterior to stereotaxic zero. The cornu ammonis (CA) regions of the dorsal horn of the anterior hippocampus (CA1/2, CA3, CA4), the dentate gyrus (DG) and thalamus were assessed on sections taken 17 mm anterior to stereotaxic zero. Digital images were obtained from labelled sections by light microscopy at ×20 magnification on a Nikon 80i microscope equipped with a DS-Fi1-U3 camera and NIS Elements Br 4.0 imaging software (Nikon Instruments, Melville, NY, USA), using four fields in the striatum (two each in the caudate nucleus and putamen), three fields in the white matter (two parasagittal intragyral, one periventricular), two fields in the thalamus (one medial geniculate nucleus, one medial nucleus), and one field in each of the hippocampal divisions.
Numbers of surviving neurons (NeuN) and activated caspase-3-positive cells, total oligodendrocytes (Olig2) and immature and mature oligodendrocytes (CNPase), astrocytes (GFAP), microglia (Iba1), and proliferating cells (Ki67) were quantified using ImageJ software (National Institutes of Health, USA) by a single assessor masked to the treatment groups through independent coding of the slides (GW). Average scores across both hemispheres from two sections were calculated for each region. The area fraction of GFAP was quantified using the isodata threshold filter (ImageJ). NeuN-positive neurons were identified morphologically by the presence of normal appearing nuclei; pyknotic cells showing condensed nuclei or fragmented appearance were not counted. Microglia with either amoeboid or ramified morphology were both included.
Erythropoietin measurements
Fetal plasma concentrations of Epo were measured in duplicate using the commercially available Quantikine human erythropoietin enzyme-linked immunosorbent assay (DEP00, R&D Systems, Minneapolis, MN, USA), validated for fetal ovine plasma. All results were measured spectrophotometrically at 450 nm with the reference wavelength set at 570 nm. Samples were evaluated between 1:100 and 1:800 dilution, or undiluted as required; samples below the reported lower detection limit of the assay were assigned the value < 2.5 mIU/mL. Epo serum controls (CEP01 and 03, R&D Systems) were included to validate the assay. The average correlation coefficient for the four parameter logistic fitted standard curve (R2, 2.5–200 mIU/mL) was 1; the intra-assay and inter-assay coefficients of variation were 1.7 and 6.2%, respectively.
Data analysis and statistics
All physiological analyses were performed using custom analysis programs (LabVIEW for Windows), by an investigator blinded to the treatment groups through coding of all experimental animals. Electroencephalographic seizure activity was identified visually and defined as the concurrent appearance of sudden, repetitive, evolving stereotypic waveforms in the EEG signal, lasting more than 10 s and greater than 20 µV.26 Seizure burden was calculated as the cumulative duration (s/h) of seizures after asphyxia. Signal artefact prevented EEG analysis in two fetuses in the sham occlusion and occlusion-vehicle groups, and one fetus in the occlusion-Epo group. CaBF from one fetus in each occlusion group was also excluded due to poor signal quality.
The effects of occlusion and rEpo on fetal physiological and histological parameters were evaluated by analysis of variance, with time or region treated as repeated measures and baseline values as a covariate for time series analysis (SPSS v22, SPSS Inc., Chicago, IL, USA). Between-group comparisons were performed with univariate analysis and Sidak correction when a significant overall effect of group or an interaction between group and time was found. Seizure duration and fetal biological data were compared by non-parametric Kruskal–Wallis test. Data are presented as mean ± standard error of the mean (SEM). Statistical significance was accepted when P < 0.05.
Results
There were no significant baseline differences between groups for EEG, SEF, CaBF, carotid vascular conductance (CaVC), FHR, MAP, extradural temperature, pH, or arterial blood gases. Umbilical cord occlusion was associated with profound hypoxia and mixed metabolic and respiratory acidosis (P < 0.05, Table 1), immediate bradycardia and progressive, severe hypotension (occlusion-Epo 10.9 ± 0.6 vs. occlusion-vehicle 10.8 ± 1.2 mmHg, not significant (N.S.)) and carotid hypoperfusion (8.6 ± 1.4 vs. 9.9 ± 1.8 mL/min, N.S.). The sham occlusion group showed no significant change from baseline (mean MAP: 32.6 ±0.8 mmHg, and mean CaBF: 23.3 ± 1.7 mL/min, P < 0.05 compared with occlusion).
Table 1.
Fetal biochemical parameters before, during, and after umbilical cord occlusion for 25 min.
| Group | Baseline | 17 min | +10 min | +1 h | +2 h | +4 h | +6 h | +24 h | +48 h | +72 h |
|---|---|---|---|---|---|---|---|---|---|---|
| pH | ||||||||||
| SHAM | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.36 ± 0.01 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.37 ± 0.01 | 7.37 ± 0.00 | 7.37 ± 0.01 | 7.37 ± 0.01 |
| OCCL-VEH | 7.38 ± 0.01 | 6.85 ± 0.02* | 7.16 ± 0.02* | 7.29 ± 0.02* | 7.33 ± 0.02 | 7.39 ± 0.01 | 7.39 ± 0.01 | 7.36 ± 0.01 | 7.36 ± 0.01 | 7.35 ± 0.01 |
| OCCL-EPO | 7.37 ± 0.00 | 6.86 ± 0.01* | 7.17 ± 0.01* | 7.31 ± 0.01* | 7.36 ± 0.01 | 7.42 ± 0.00# | 7.40 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.37 ± 0.01 |
| PaCO2 (mmHg) | ||||||||||
| SHAM | 47.6 ± 0.7 | 45.5 ± 1.1 | 47.9 ± 1.1 | 48.1 ± 1.0 | 50.0 ± 0.9 | 49.3 ± 1.0 | 50.1 ± 0.9 | 50.3 ± 0.9 | 49.7 ± 1.4 | 49.4 ± 1.3 |
| OCCL-VEH | 49.3 ± 1.4 | 133.7 ± 4.2* | 54.6 ± 2.9 | 51.2 ± 3.3 | 51.5 ± 3.0 | 47.6 ± 1.9 | 49.4 ± 1.6 | 48.3 ± 1.2 | 46.3 ± 1.8 | 49.1 ± 1.7 |
| OCCL-EPO | 49.6 ± 0.8 | 132.6 ± 4.3* | 50.6 ± 1.0 | 46.4 ± 1.3 | 48.0 ± 1.2 | 47.1 ± 1.1 | 46.3 ± 1.6 | 47.5 ± 1.2 | 46.9 ± 1.1 | 48.4 ± 0.9 |
| PaO2 (mmHg) | ||||||||||
| SHAM | 25.0 ± 1.2 | 24.2 ± 0.8 | 24.8 ± 1.0 | 24.6 ± 0.7 | 23.5 ± 1.0 | 24.1 ± 1.0 | 24.9 ± 1.4 | 23.7 ± 1.1 | 24.3 ± 1.1 | 23.5 ± 1.4 |
| OCCL-VEH | 24.3 ± 1.3 | 11.1 ± 0.8* | 32.3 ± 1.6* | 28.5 ± 1.9 | 25.4 ± 1.4 | 23.0 ± 1.8 | 23.3 ± 1.8 | 25.9 ± 2.0 | 27.0 ± 2.0 | 27.0 ± 2.0 |
| OCCL-EPO | 25.2 ± 0.9 | 11.7 ± 0.9* | 33.2 ± 0.7* | 26.5 ± 1.1 | 24.9 ± 0.8 | 24.5 ± 0.7 | 25.5 ± 1.8 | 26.6 ± 1.5 | 26.5 ± 1.6 | 26.1 ± 1.7 |
| Base Excess (mmol/L) | ||||||||||
| SHAM | 1.5 ± 0.4 | 0.5 ± 0.5 | 0.7 ± 0.8 | 1.8 ± 0.4 | 2.8 ± 0.4 | 2.5 ± 0.6 | 2.9 ± 0.3 | 2.8 ± 0.5 | 2.4 ± 0.6 | 2.1 ± 0.4 |
| OCCL-VEH | 2.3 ± 1.0 | −11.6 ± 1.1* | −9.7 ± 1.1* | −2.9 ± 1.7* | 0.6 ± 1.8 | 2.8 ± 1.0 | 4.1 ± 1.1 | 1.2 ± 1.2 | 0.4 ± 1.1 | 0.7 ± 0.7 |
| OCCL-EPO | 2.2 ± 0.6 | −10.9 ± 0.8* | −10.5 ± 0.7* | −3.4 ± 1.0* | 1.2 ± 1.2 | 5.2 ± 0.6 | 3.2 ± 0.8 | 1.5 ± 0.6 | 2.0 ± 0.9 | 1.7 ± 0.7 |
| Lactate (mmol/L) | ||||||||||
| SHAM | 0.7 ± 0.1 | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.1 | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.0 | 0.8 ± 0.1 | 0.8 ± 0.1 |
| OCCL-VEH | 0.9 ± 0.1 | 7.1 ± 0.4* | 6.1 ± 0.4* | 4.4 ± 0.6* | 4.1 ± 0.8* | 2.9 ± 0.7* | 2.5 ± 0.6* | 1.1 ± 0.2 | 0.9 ± 0.1 | 0.8 ± 0.1 |
| OCCL-EPO | 0.9 ± 0.0 | 5.8 ± 0.7* | 5.6 ± 0.5* | 3.7 ± 0.9* | 3.4 ± 0.6 | 1.6 ± 0.2 | 1.8 ± 0.2 | 1.0 ± 0.1 | 0.7 ± 0.1 | 0.7 ± 0.1 |
| Glucose (mmol/L) | ||||||||||
| SHAM | 1.0 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 |
| OCCL-VEH | 1.0 ± 0.1 | 0.7 ± 0.1 | 1.6 ± 0.1 | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.4 ± 0.1 | 1.4 ± 0.1 | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 |
| OCCL-EPO | 0.9 ± 0.0 | 0.7 ± 0.2 | 1.8 ± 0.4* | 1.0 ± 0.2 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.3 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 0.8 ± 0.1 |
| Haemoglobin (g/dL) | ||||||||||
| SHAM | 8.6 ± 0.1 | 8.6 ± 0.2 | 8.7 ± 0.3 | 8.5 ± 0.2 | 8.5 ± 0.2 | 8.5 ± 0.1 | 8.6 ± 0.1 | 8.7 ± 0.2 | 8.7 ± 0.3 | 8.5 ± 0.2 |
| OCCL-VEH | 9.3 ± 0.5 | 9.5 ± 0.5 | 9.6 ± 0.6 | 9.9 ± 0.5 | 9.7 ± 0.5 | 9.2 ± 0.5 | 9.3 ± 0.7 | 9.1 ± 0.5 | 8.5 ± 0.4 | 8.7 ± 0.4 |
| OCCL-EPO | 9.3 ± 0.5 | 9.7 ± 0.4 | 10.0 ± 0.7 | 9.7 ± 0.6 | 9.7 ± 0.6 | 9.4 ± 0.5 | 9.4 ± 0.5 | 9.5 ± 0.6 | 9.8 ± 0.7 | 10.2 ± 1.0 |
| Haematocrit (%) | ||||||||||
| SHAM | 25.3 ± 0.4 | 25.1 ± 0.7 | 25.6 ± 0.9 | 24.9 ± 0.7 | 25.2 ± 0.4 | 24.8 ± 0.4 | 25.0 ± 0.4 | 25.1 ± 0.5 | 25.1 ± 0.7 | 25.0 ± 0.6 |
| OCCL-VEH | 27.4 ± 1.6 | 27.9 ± 1.4 | 28.2 ± 1.7 | 29.2 ± 1.6 | 28.6 ± 1.6 | 26.9 ± 1.5 | 27.3 ± 2.1 | 26.8 ± 1.5 | 25.0 ± 1.3 | 25.7 ± 1.2 |
| OCCL-EPO | 26.8 ± 1.4 | 27.8 ± 1.4 | 29.4 ± 2.2 | 28.8 ± 1.8 | 28.4 ± 1.8 | 27.8 ± 1.6 | 27.5 ± 1.6 | 28.1 ± 1.7 | 28.9 ± 2.2 | 29.8 ± 2.9 |
| CtO2 (mmol/L) | ||||||||||
| SHAM | 3.9 ± 0.1 | 3.8 ± 0.1 | 3.8 ± 0.2 | 3.7 ± 0.2 | 3.6 ± 0.2 | 3.7 ± 0.1 | 3.9 ± 0.2 | 3.6 ± 0.1 | 3.7 ± 0.2 | 3.7 ± 0.2 |
| OCCL-VEH | 3.5 ± 0.1 | 0.5 ± 0.0* | 4.0 ± 0.2 | 4.1 ± 0.2 | 3.7 ± 0.2 | 3.4 ± 0.3 | 3.5 ± 0.3 | 3.7 ± 0.2 | 3.6 ± 0.2 | 3.7 ± 0.2 |
| OCCL-EPO | 3.7 ± 0.2 | 0.5 ± 0.0* | 4.3 ± 0.3 | 4.0 ± 0.2 | 4.0 ± 0.2 | 3.9 ± 0.2 | 3.9 ± 0.3 | 4.0 ± 0.2 | 4.0 ± 0.3 | 3.9 ± 0.4 |
| DO2 (µmol/min) | ||||||||||
| SHAM | 102.8 ± 10.6 | 114.0 ± 12.5 | 110.2 ± 13.2 | 106.7 ± 11.4 | 105.2 ± 11.5 | 112.6 ± 15.1 | 122.6 ± 16.7 | 121.6 ± 15.0 | 133.6 ± 16.2 | 140.1 ± 18.7 |
| OCCL-VEH | 101.6 ± 5.5 | 7.8 ± 1.1* | 152.9 ± 7.4 | 113.2 ± 7.4 | 62.1 ± 7.3* | 68.2 ± 6.2* | 73.4 ± 4.6* | 83.4 ± 8.7 | 83.0 ± 7.6* | 91.2 ± 8.9 |
| OCCL-EPO | 105.8 ± 10.0 | 5.9 ± 1.0* | 149.2 ± 21.1 | 117.9 ± 17.6 | 86.5 ± 13.1 | 74.8 ± 10.7 | 77.6 ± 12.4 | 94.4 ± 14.1 | 125.0 ± 10.2$ | 142.3 ± 9.6$ |
Fetal arterial blood samples were drawn from the sham occlusion (sham), occlusion-vehicle (occl-veh), and occlusion-erythropoietin (occl-Epo) animals at 60 min before the occlusion (baseline), 17 min during umbilical cord occlusion, and 10 min, 1, 2, 4, 6, 24, 48, and 72 h after reperfusion. The 5 min blood sample during cord occlusion was omitted for brevity. PaCO2: fetal arterial pressure of carbon dioxide; PaO2: fetal arterial pressure of oxygen; CtO2: fetal arterial oxygen content; DO2: cephalic oxygen delivery. Data are presented as mean ± SEM; between-group comparisons by one-way ANOVA with Sidak post-hoc correction. *P < 0.05 vs. sham occlusion group; $P < 0.05 vs. occlusion-vehicle group; #P < 0.05 vs. sham occlusion and occlusion-vehicle groups.
Blood composition
After release of occlusion PaCO2, PaO2, arterial oxygen content and cephalic oxygen delivery recovered to sham occlusion values within 10 min, and pH and base excess recovered within 1 h (Table 1). After reperfusion, in the occlusion-vehicle group cephalic oxygen delivery fell from 2 to 6 h and 48 to 72 h, compared with sham occlusion (P < 0.05). In contrast, occlusion-Epo was associated with increased cephalic oxygen delivery compared to occlusion-vehicle from 48 to 72 h (P < 0.05). rEpo did not affect haemoglobin or haematocrit values (N.S.).
Fetal plasma Epo levels remained below the detection limit of the immunoassay (<2.5 mIU/mL) in the occlusion-vehicle group during the study. rEpo infusion was associated with a rise in plasma Epo levels to a maximum of 32,639 ± 2886 mIU/mL after 30 min (Supplementary Figure 1) and was maintained at 25,061 ± 608 mIU/mL from 2 to 72 h.
EEG power and SEF
Severe asphyxia was associated with immediate, rapid suppression of EEG power and SEF, compared to sham occlusion (P < 0.05 vs. occlusion groups). After occlusion, EEG power and SEF remained significantly suppressed in the occlusion-vehicle group from 0 to 72 h (P < 0.05 vs. sham occlusion; Figure 1). In contrast, rEpo infusion was associated with more rapid recovery of EEG power and SEF after asphyxia, such that EEG power was increased from 24 to 48 h (P < 0.05 vs. occlusion-vehicle), and SEF from 54 h until the end of the experiment (P < 0.05 vs. occlusion-vehicle).
Figure 1.
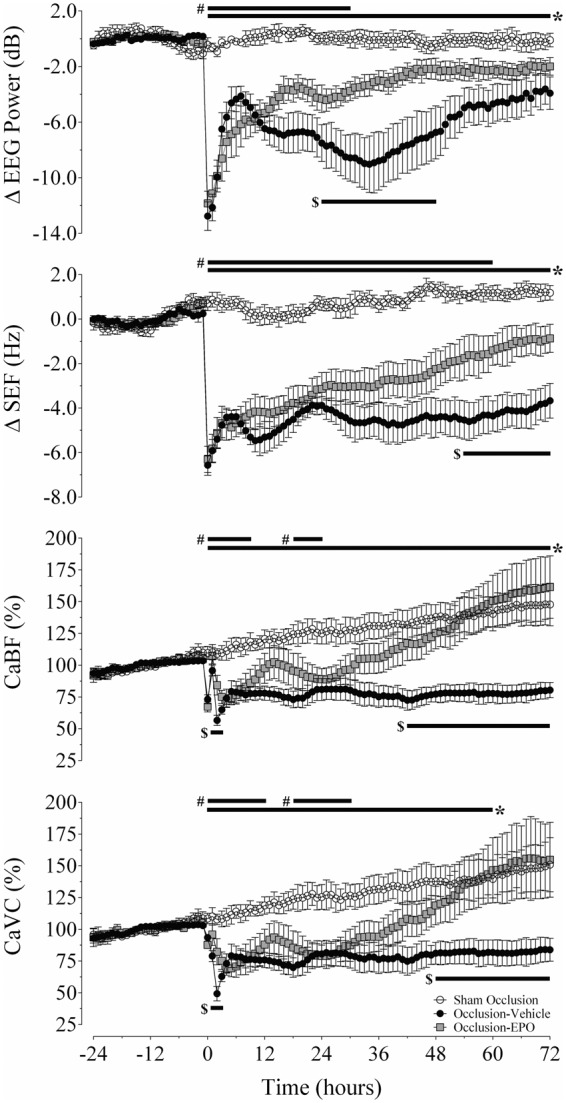
Changes in electroencephalographic power (ΔEEG, dB), spectral edge frequency (ΔSEF, Hz), carotid blood flow (CaBF, % baseline), and carotid vascular conductance (CaVC, % baseline) in the sham occlusion (open circles), occlusion-vehicle (closed circles), and occlusion-Epo animals (closed squares). Time point zero denotes the period (25 min) of umbilical cord occlusion. Data are hourly averages and presented as mean ± SEM. *P < 0.05, occlusion-vehicle vs. sham occlusion; #P < 0.05, occlusion-Epo vs. sham occlusion; $P < 0.05, occlusion-vehicle vs. occlusion-Epo.
Electrographic seizures
Stereotypical seizures developed from 12.5 ± 5.4 h in the occlusion-Epo group in 3/7 fetuses compared to 14.4 ± 3.9 h in 7/7 fetuses in the occlusion-vehicle group (N.S.). Occlusion-Epo was associated with a significant reduction in both the total number of seizures compared to occlusion-vehicle (16.3 ± 10.5 vs. 44.1 ± 9.0; P < 0.05) and the total seizure burden (13.3 ± 10.7 vs. 38.9 ± 8.3 min; P < 0.05, Supplementary Figure 2). There was no effect on duration of individual seizures (40 ± 10 vs. 54 ± 5 s; N.S.) or maximum seizure amplitude (183 ± 58 vs. 169 ± 13 µV; N.S.).
Extradural temperature
There were no significant differences in extradural temperature between groups, either before, during, or after asphyxia (Supplementary Figure 2).
FHR, MAP, CaBF, and CaVC
In both occlusion groups, FHR showed transient tachycardia followed by a small secondary reduction from 18 to 30 h compared with sham occlusion (P < 0.05, Supplementary Figure 2). Release of occlusion was associated with a transient increase in MAP above sham control values in both occlusion groups. In the occlusion-vehicle group, MAP remained at sham control values for the remainder of the experiment. By contrast, occlusion-Epo was associated with a small increase in MAP from 6 to 29 h after occlusion (P < 0.05 vs. occlusion-vehicle, Supplementary Figure 2).
Release of umbilical cord occlusion was associated with a prolonged secondary fall in CaBF and CaVC (Figure 1), from 0–72 h and 0–60 h, respectively, compared to sham occlusion (P < 0.05). The occlusion-Epo group showed progressive recovery of both CaBF and CaVC to sham control values, with values that were greater than occlusion-vehicle from 30 to 72 h and 48 to 72 h, respectively (P < 0.05; Figure 1).
Post-mortem
There was no significant difference in fetal body weight, heart or kidney weights, fetal sex or number of fetuses between experimental groups at post-mortem (Supplementary Table 1). Brain weight was reduced in the occlusion-vehicle group compared with sham occlusion (25.2 ± 0.7 g vs. 29.3 ± 0.9 g; P < 0.05); the occlusion-Epo group showed intermediate values between the other groups (26.4 ± 0.8 g, N.S.). Occlusion-Epo was associated with a significant increase in liver weight compared to sham occlusion and occlusion-vehicle (106.8 ± 6.9 g vs. 64.2 ± 3.6 g and 76.7 ± 3.0 g; P < 0.05).
Histopathology
Occlusion was associated with neuronal loss in the striatal caudate nucleus, CA1/2, CA3, CA4, DG of the hippocampus, and thalamic medial nucleus (P < 0.05, occlusion-vehicle vs. sham occlusion, Figures 2 and 3). In the occlusion-Epo group, neuronal survival was increased overall (P < 0.05 vs. occlusion-vehicle). Post-hoc analysis suggested significant improvement in the striatal caudate nucleus, CA3 and DG of the hippocampus, and thalamic medial nucleus. Analysis of the occlusion groups only showed an independent effect of rEpo (P < 0.001) but no significant interaction between rEpo and region, consistent with overall improvement. In parallel with changes in neuronal survival, occlusion-vehicle was associated with an overall increase in numbers of caspase-3-positive cells (P < 0.05 vs. sham occlusion, Figure 2 and Supplementary Figure 3), with an overall reduction of caspase-3 induction in the occlusion-Epo group (P < 0.05 vs. occlusion-vehicle).
Figure 2.
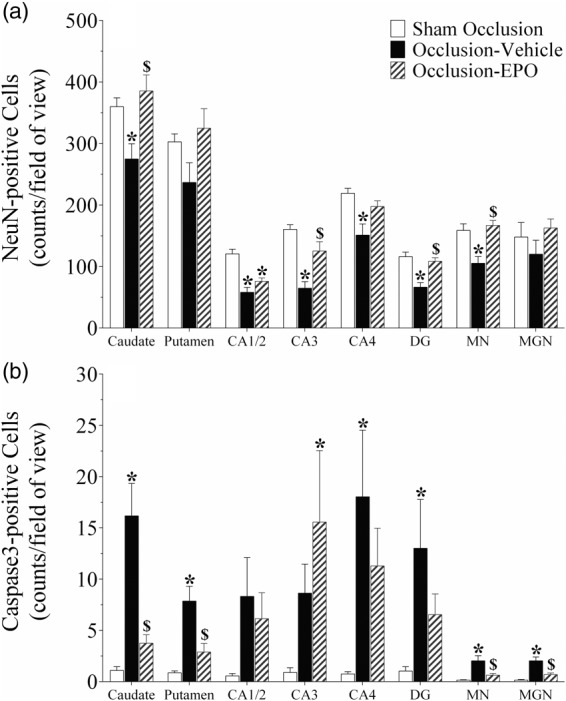
Neuronal (NeuN, panel a) and activated caspase-3 (panel b) -positive cell counts in the striatal caudate nucleus and putamen, CA1/2, CA3, CA4, and dentate gyrus (DG) of the hippocampus, thalamic medial nucleus (MN), and medial geniculate nucleus (MGN) in the sham occlusion, occlusion-vehicle, and occlusion-Epo groups three days after severe asphyxia. Epo: erythropoietin. Data are presented as mean ± SEM. *P < 0.05 vs. sham occlusion, $P < 0.05 vs. occlusion-vehicle.
Figure 3.
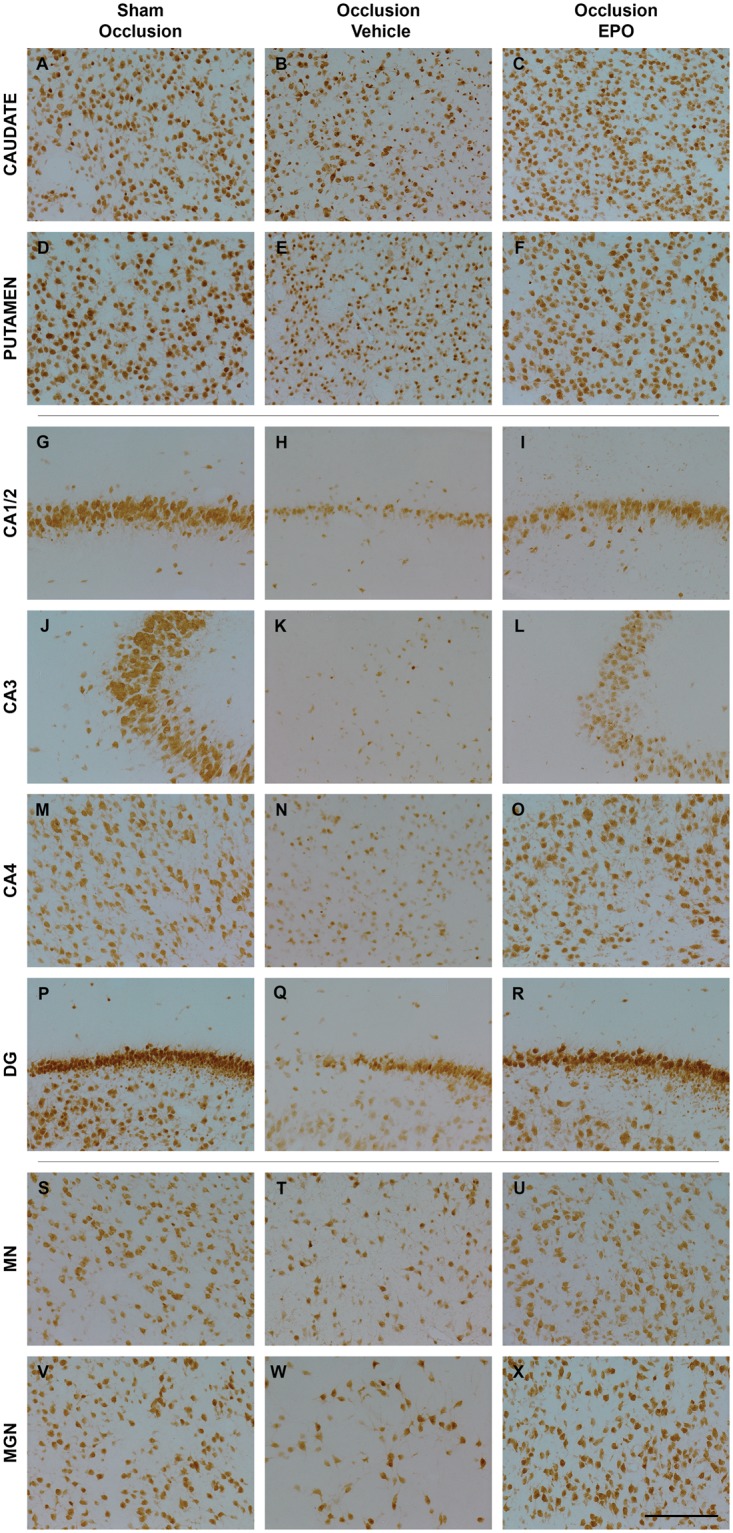
Photomicrographs of neurons (NeuN-positive cells) in the striatal caudate nucleus (caudate, panel A–C), putamen (panel D–F), CA1/2 (panel G–I), CA3 (panel J–L), CA4 (panel M–O), dentate gyrus (DG, panel P–R) of the hippocampus, thalamic medial nucleus (MN, panel S–U), and medial geniculate nucleus (MGN, panel V–X) from the sham occlusion (left column), occlusion-vehicle (middle column), and occlusion-Epo (right column) groups three days after severe asphyxia. Scale bar is 200 µm.
Epo: erythropoietin.
Occlusion was associated with marked loss of total, Olig2-positive, oligodendrocytes (P < 0.05, occlusion-vehicle vs. sham occlusion, Figures 4 and 5), and CNPase-positive, immature to mature oligodendrocytes (P < 0.05, occlusion-vehicle vs. sham occlusion, Figures 4 and 5) in white matter. The occlusion-Epo group showed significantly greater total numbers of oligodendrocytes (P < 0.05 vs. occlusion-vehicle). In contrast, rEpo after occlusion had no effect on numbers of immature to mature oligodendrocytes in any white matter regions (N.S. vs. occlusion-vehicle). Occlusion was associated with a small increase in numbers of activated caspase-3-positive cells in the white matter tracts compared to sham occlusion (1.9 ± 0.2 vs. 1.0 ± 0.1 cells/field; P < 0.05), which was reduced after rEpo infusion (1.3 ± 0.2; P < 0.05 vs. occlusion-vehicle).
Figure 4.

Astrocyte cell counts and area fraction (GFAP, panel a and b), microglia (Iba1, panel c), total oligodendrocytes (Olig2, panel d), immature and mature oligodendrocytes (CNPase, panel e), and proliferating cell counts (Ki67, panel f) in the periventricular (PVWM), and first and second parasagittal white matter (IGWM 1–2) of the sham occlusion, occlusion-vehicle, and occlusion-Epo groups at three days after severe asphyxia. Data are presented as mean ± SEM. *P < 0.05 vs. sham occlusion, $P < 0.05 vs. occlusion-vehicle. #P < 0.05 vs. sham occlusion and occlusion-vehicle groups.
GFAP: glial fibrillary acidic protein; Epo: erythropoietin.
Figure 5.

Photomicrographs of total oligodendrocytes (Olig2-positive cells, panel A–F), immature and mature oligodendrocytes (CNPase-positive cells, panel G–L), and total proliferating cells (Ki67-positive cells, panel M–R) in the periventricular (PVWM) and intragyral white matter (IGWM) from sham occlusion (left column), occlusion-vehicle (middle column), and occlusion-Epo (right column) animals at three days after asphyxia. Representative images for IGWM were taken from the first parasagittal white matter (Olig2), and second parasagittal white matter (CNPase, Ki67). Scale bar is 200 µm.
Further, occlusion was associated with an overall increase in numbers and area fraction of GFAP-positive cells in the white matter tracts (P < 0.05, occlusion-vehicle vs. sham occlusion, Figures 4 and 6). rEpo-reduced numbers of GFAP-positive cells and GFAP-positive area fraction to sham control values (P < 0.05, occlusion-Epo vs. occlusion-vehicle). Similarly, occlusion was associated with marked induction of Iba1-positive microglia in the white matter tracts (P < 0.05 vs. sham occlusion, Figures 4 and 6), with attenuation of this increase in the occlusion-Epo group (P < 0.05 vs. occlusion-vehicle).
Figure 6.

Photomicrographs of the astroglial (GFAP-positive cells, panel A–F) and microglial cells (Iba1-positive cells, panel G–L) in the periventricular (PVWM) and intragyral white matter (IGWM) from sham occlusion (left column), occlusion-vehicle (middle column), and occlusion-Epo (right column) animals at three days after asphyxia. Representative images for IGWM were taken from the second parasagittal white matter. Scale bar is 200 µm.
Finally, occlusion-vehicle did not affect the number of Ki67-positive cells compared to sham occlusion (N.S.). In contrast, occlusion-Epo was associated with markedly increased numbers of Ki67-positive cells in the periventricular and parasagittal white matter, compared with sham occlusion and occlusion-vehicle (P < 0.05, Figures 4 and 5).
Discussion
This study demonstrates for the first time that continuous infusion with rEpo in preterm fetal sheep, started 30 min after severe asphyxia and continued for 72 h, was associated with partial neuroprotection in subcortical regions and markedly improved numbers of oligodendrocytes in the periventricular and parasagittal white matter tracts, after three days recovery. rEpo enhanced cell proliferation, reduced numbers of activated caspase-3-positive cells, and attenuated induction of astrocytes and microglia in the white matter tracts. Functionally, rEpo infusion markedly reduced the seizure burden, with faster recovery of electrocortical brain activity and cephalic perfusion. These data suggest that delayed treatment with rEpo, started within the early recovery phase and continued for three days to reflect a clinically plausible timing, can reduce hypoxic-ischemic white and subcortical grey matter injury in the immature brain, at least partially through anti-inflammatory and anti-apoptotic mechanisms.
Prolonged umbilical cord occlusion was associated with selective neuronal loss in the striatum, hippocampus and thalamus, with diffuse loss of total and immature to mature oligodendrocytes, and induction of astrocytes and microglia in the periventricular and parasagittal white matter, similarly to previous studies.27 Continuous i.v. infusion with rEpo started 30 min after occlusion was associated with sustained concentrations similar to levels achieved transiently after a single i.v. injection in healthy preterm fetal sheep, which led to potentially neuroprotective cerebrospinal fluid concentrations after 2.5 h,28 although the effect on recovery from HI was not tested. We now show that a three-day infusion of rEpo markedly reduced post-asphyxial neuronal loss in most subcortical regions in preterm fetal sheep, despite a 30-min delay after the end of asphyxia. Although this is the first study of rEpo after HI in a preterm-equivalent large animal, it is broadly consistent with the finding in P7 rats that optimal protection was seen with 3× repeated daily doses of 5000 IU/kg when the first injection was given immediately after HI.9 The present study did not evaluate the effect of intermittent boluses; further studies are needed to establish the optimal therapeutic regimen with rEpo for neuroprotection.
Erythropoietin promotes expression of anti-apoptotic relative to pro-apoptotic genes and inhibits caspase activation with DNA fragmentation after HI injury.29,30 Consistent with these effects, in the present study, rEpo improved neuronal survival with an overall reduction in numbers of caspase-3-positive cells. Interestingly, in post-hoc analysis, the CA1/2 region of the hippocampus did not appear to show any improvement in neuronal survival. The reason for this apparent selective lack of neuroprotection is unclear. The Epo receptor is highly expressed in the hippocampus,31 and there is no evidence for lack of penetration into the hippocampus.5 Thus, it is improbable that there was a regional difference in rEpo activity. We speculate that this lack of neuroprotection might reflect relatively rapid evolution of cell death, in a region that is highly vulnerable to HI.32
There was a significant loss of both total (Olig2 labelled) oligodendrocytes and CNPase-labelled immature/mature oligodendrocytes, in the periventricular and parasagittal white matter at three days after occlusion. The prolonged infusion of rEpo was associated with a striking restoration of the number of Olig2-labelled oligodendrocytes in the white matter tracts, but had no effect on loss of CNPase-labelled immature/mature oligodendrocytes after occlusion. The reported mechanisms of acute white matter protection with rEpo include anti-apoptotic and anti-inflammatory effects.33–35 Consistent with this, in the present study, there was an overall reduction in activated caspase-3-positive cells, microgliosis and astrogliosis in white matter after rEpo infusion. Post-mortem analysis of preterm brain injury in modern cohorts strongly suggests that astrocytosis in white matter is associated with arrest of maturation of oligodendroglia.36 Thus, reduced astrocytosis may contribute to improved long-term recovery. Of interest, within both the intragyral and periventricular WM tracts, rEpo was associated with a significant overall increase in proliferating cells compared with vehicle infusion. Collectively, these data strongly suggest that in addition to reducing acute oligodendroglial death, rEpo selectively stimulated proliferative regeneration of pre-oligodendrocytes, although it did not prevent asphyxia-induced death of immature/mature oligodendrocytes.
A limitation of this study is that, for technical reasons, we were not able to label pre-oligodendrocytes. However, this hypothesis is highly consistent with considerable in vitro and in vivo evidence that Epo promotes oligodendrogenesis,37,38 and survival and maturation of immature oligodendrocytes.39–41 Further, the selective lack of protection of immature and mature oligodendrocytes in the present study is consistent with a previous report in adult rats which showed that high-dose rEpo (5000 IU/kg) injection after stroke did not reduce loss of CNPase-positive oligodendrocytes in the striatum and corpus callosum at seven days, even though it improved long-term recovery of myelinating oligodendrocytes.41 The reasons for lack of protection of more mature oligodendrocytes are unknown, but may be related to maturational loss of Epo receptors.42,43
Severe asphyxia was associated with long-lasting suppression of EEG activity and SEF, with delayed onset of evolving seizures. These features are highly predictive of adverse outcomes in term and preterm infants.44,45 Infants who show recovery of EEG background activity within 48 h after hypoxic-ischemia have better long-term outcomes than those neonates that develop seizures and persistent suppression of background activity.44,46 Thus, the greatly reduced seizure burden and faster recovery of EEG power and SEF during rEpo infusion are highly consistent with reduced white and grey matter damage.
Faster recovery of brain activity with rEpo was also associated with gradual restoration of CaBF and cephalic oxygen delivery to sham occlusion values. This improvement was mediated by improved vascular conductance, with no change in fetal haematocrit. This combination of improved brain activity and blood flow strongly infers that increased CaBF was an autoregulatory adjustment to support improving cerebral metabolism rather than a direct effect of rEpo. This is highly consistent with evidence in near-term fetal sheep that greater post-ischemic histological neuroprotection with early initiation of cerebral cooling was associated with improved recovery of EEG activity and CaBF after rewarming.47
There is reassuring evidence that three doses of 2500 IU/kg did not increase the risk of complications of prematurity in extremely low birth weight infants.5 Similarly, in the present study, there were few systemic effects. The infusion of high-dose rEpo for three days was associated with a small increase in mean arterial blood pressure (of ∼3.6 mmHg). This was not related to changes in fetal haemoglobin or haematocrit. Given that heart rate was normal or modestly reduced after occlusion in the present study, the increase in arterial blood pressure during rEpo infusion must be related to either a small increase in peripheral vascular resistance or to enhanced stroke volume. rEpo increased contractility and reduced tissue injury after myocardial HI in adult mice and rats,48,49 suggesting that there would be value in assessing the effects of rEpo on the neonatal heart after perinatal asphyxia.
Further, in the present study, rEpo infusion was associated with increased liver size at post-mortem. Tissues were not kept for histology; however, potentially this may reflect stimulation of hepatic erythropoiesis. This finding is consistent with a previous report of increased liver size in male rats after repeated daily injections for the first five days of life with 5000 IU/kg rEpo.6 Finally, we found no difference in extradural temperature between experimental groups; thus excluding the possibility that rEpo-mediated neuroprotection was confounded by hypothermia.
Some potential limitations of this study should be considered. Post-asphyxial white matter and neural injury may continue to progress over days to weeks.11 Thus, in future studies, it will be important to assess whether the protection was maintained after the end of the rEpo infusion. Nevertheless, although the continued expression of apoptotic caspase-3-labelled cells is consistent with some ongoing injury, the absolute numbers of apoptotic cells was very low. Further, this study shows that despite rEpo infusion, after three days recovery there was loss of more mature oligodendrocytes. Thus, longer recovery times are also important to confirm whether rEpo improves the maturation of pre-oligodendrocytes after severe HI.36
Finally, it is important to appreciate that the present study targeted acute isolated asphyxial injury at a moderately preterm stage of brain development. Future studies are needed to examine the effects of rEpo on the normal brain, the impact of greater delay in starting treatment, and the potential to improve outcome after the rather complex inflammatory insults that are more common in extremely preterm infants.50 These issues will be particularly relevant for clinical trials of rEpo in extremely premature infants, where typically all infants within the selected gestational age range are treated.14
In summary, the present study demonstrates for the first time that infusion of high-dose rEpo for three days after severe asphyxia improves survival of subcortical neurons and total oligodendrocytes in white matter in the preterm fetal sheep with a corresponding improvement in recovery of electrocortical brain activity and CaBF. These improved outcomes were at least partially mediated through anti-apoptotic and anti-inflammatory mechanisms, while the recovery of total numbers of oligodendrocytes was also associated with increased white matter proliferation. These findings support rEpo as a potential therapeutic intervention for preterm infants after HI.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the Health Research Council of New Zealand. Mr Guido Wassink was supported by a University of Auckland Health Research Doctoral Scholarship. Dr Robert Galinsky was supported by a National Health and Medical Research Council of Australia CJ Martin Postdoctoral Fellowship (1090890).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
GW, JOD, LB, and AJG conceptualized and designed the study. GW, JOD, and RG undertook experiments. GW, RG, and SD undertook immunohistochemistry. GW and MF undertook immunoassays. GW undertook data analysis and cell quantification, preparation of tables and figures, statistics and drafted the manuscript. AJG provided overall oversight of the research. All authors reviewed the manuscript and approved the final manuscript as submitted, and agree to be accountable for the performed work.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Committee on Understanding Premature Birth and Assuring Healthy Outcomes, Behrman RE and Butler AS (eds) Preterm birth: causes, consequences, and prevention. Washington, DC: Institute of Medicine of the National Academies, 2007.
- 2.Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 2009; 8: 110–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards AD, Brocklehurst P, Gunn AJ, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 2010; 340: c363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higgins RD, Raju T, Edwards AD, et al. Hypothermia and other treatment options for neonatal encephalopathy: an executive summary of the Eunice Kennedy Shriver NICHD workshop. J Pediatr 2011; 159: 851–858.e851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rangarajan V, Juul SE. Erythropoietin: emerging role of erythropoietin in neonatal neuroprotection. Pediatr Neurol 2014; 51: 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McPherson RJ, Demers EJ, Juul SE. Safety of high-dose recombinant erythropoietin in a neonatal rat model. Neonatology 2007; 91: 36–43. [DOI] [PubMed] [Google Scholar]
- 7.van de Looij Y, Chatagner A, Quairiaux C, et al. Multi-modal assessment of long-term erythropoietin treatment after neonatal hypoxic-ischemic injury in rat brain. PloS One 2014; 9: e95643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rice JE, 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981; 9: 131–141. [DOI] [PubMed] [Google Scholar]
- 9.Kellert BA, McPherson RJ, Juul SE. A comparison of high-dose recombinant erythropoietin treatment regimens in brain-injured neonatal rats. Pediatr Res 2007; 61: 451–455. [DOI] [PubMed] [Google Scholar]
- 10.Alexander ML, Hill CA, Rosenkrantz TS, et al. Evaluation of the therapeutic benefit of delayed administration of erythropoietin following early hypoxic-ischemic injury in rodents. Dev Neurosci 2012; 34: 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wassink G, Gunn ER, Drury PP, et al. The mechanisms and treatment of asphyxial encephalopathy. Front Neurosci 2014; 8: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leuchter RH, Gui L, Poncet A, et al. Association between early administration of high-dose erythropoietin in preterm infants and brain MRI abnormality at term-equivalent age. JAMA 2014; 312: 817–824. [DOI] [PubMed] [Google Scholar]
- 13.O’Gorman RL, Bucher HU, Held U, et al. Tract-based spatial statistics to assess the neuroprotective effect of early erythropoietin on white matter development in preterm infants. Brain 2015; 138: 388–397. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Zhang L, Jin Y. A meta-analysis of the protective effect of recombinant human erythropoietin (rhEPO) for neurodevelopment in preterm infants. Cell Biochem Biophys 2015; 71: 795–802. [DOI] [PubMed] [Google Scholar]
- 15.Wu YW, Bauer LA, Ballard RA, et al. Erythropoietin for neuroprotection in neonatal encephalopathy: safety and pharmacokinetics. Pediatrics 2012; 130: 683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McIntosh GH, Baghurst KI, Potter BJ, et al. Foetal brain development in the sheep. Neuropathol Appl Neurobiol 1979; 5: 103–114. [DOI] [PubMed] [Google Scholar]
- 17.Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010; 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wassink G, Galinsky R, Drury PP, et al. Does maturity affect cephalic perfusion and T/QRS ratio during prolonged umbilical cord occlusion in fetal sheep? Obstet Gynecol Int 2014; 2014: 314159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Bel F, Roman C, Klautz RJ, et al. Relationship between brain blood flow and carotid arterial flow in the sheep fetus. Pediatr Res 1994; 35: 329–333. [DOI] [PubMed] [Google Scholar]
- 20.Covert RF, Schreiber MD, Torgerson LJ, et al. Prediction of cerebral blood flow in fetal lambs by carotid artery ultrasonic flow transducer. Reprod Fertil Dev 1996; 8: 157–162. [DOI] [PubMed] [Google Scholar]
- 21.Williams CE, Gluckman PD. Real-time spectral intensity analysis of the EEG on a common microcomputer. J Neurosci Meth 1990; 32: 9–13. [DOI] [PubMed] [Google Scholar]
- 22.Quaedackers JS, Roelfsema V, Heineman E, et al. The role of the sympathetic nervous system in post-asphyxial intestinal hypoperfusion in the preterm sheep fetus. J Physiol 2004; 557: 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennet L, Roelfsema V, George S, et al. The effect of cerebral hypothermia on white and grey matter injury induced by severe hypoxia in preterm fetal sheep. J Physiol 2007; 578: 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barkovich AJ, Truwit CL. Brain damage from perinatal asphyxia: correlation of MR findings with gestational age. AJNR Am J Neuroradiol 1990; 11: 1087–1096. [PMC free article] [PubMed] [Google Scholar]
- 25.Jakovcevski I, Filipovic R, Mo Z, et al. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat 2009; 3: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scher MS, Hamid MY, Steppe DA, et al. Ictal and interictal electrographic seizure durations in preterm and term neonates. Epilepsia 1993; 34: 284–288. [DOI] [PubMed] [Google Scholar]
- 27.Drury PP, Davidson JO, Bennet L, et al. Partial neural protection with prophylactic low-dose melatonin after asphyxia in preterm fetal sheep. J Cereb Blood Flow Metab 2014; 34: 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juul SE, McPherson RJ, Farrell FX, et al. Erytropoietin concentrations in cerebrospinal fluid of nonhuman primates and fetal sheep following high-dose recombinant erythropoietin. Biol Neonate 2004; 85: 138–144. [DOI] [PubMed] [Google Scholar]
- 29.Kumral A, Genc S, Ozer E, et al. Erythropoietin downregulates bax and DP5 proapoptotic gene expression in neonatal hypoxic-ischemic brain injury. Biol Neonate 2006; 89: 205–210. [DOI] [PubMed] [Google Scholar]
- 30.Schober ME, Requena DF, Block B, et al. Erythropoietin improved cognitive function and decreased hippocampal caspase activity in rat pups after traumatic brain injury. J Neurotrauma 2014; 31: 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ott C, Martens H, Hassouna I, et al. Widespread expression of erythropoietin receptor in brain and its induction by injury. Mol Med 2015; 21: 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keogh MJ, Drury PP, Bennet L, et al. Limited predictive value of early changes in EEG spectral power for neural injury after asphyxia in preterm fetal sheep. Pediatr Res 2012; 71: 345–353. [DOI] [PubMed] [Google Scholar]
- 33.Dzietko M, Felderhoff-Mueser U, Sifringer M, et al. Erythropoietin protects the developing brain against N-methyl-D-aspartate receptor antagonist neurotoxicity. Neurobiol Dis 2004; 15: 177–187. [DOI] [PubMed] [Google Scholar]
- 34.Villa P, van Beek J, Larsen AK, et al. Reduced functional deficits, neuroinflammation, and secondary tissue damage after treatment of stroke by nonerythropoietic erythropoietin derivatives. J Cereb Blood Flow Metab 2007; 27: 552–563. [DOI] [PubMed] [Google Scholar]
- 35.Matsushita H, Johnston MV, Lange MS, et al. Protective effect of erythropoietin in neonatal hypoxic ischemia in mice. Neuroreport 2003; 14: 1757–1761. [DOI] [PubMed] [Google Scholar]
- 36.Back SA, Miller SP. Brain injury in premature neonates: a primary cerebral dysmaturation disorder? Ann Neurol 2014; 75: 469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwai M, Stetler RA, Xing J, et al. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke 2010; 41: 1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez FF, Larpthaveesarp A, McQuillen P, et al. Erythropoietin increases neurogenesis and oligodendrogliosis of subventricular zone precursor cells after neonatal stroke. Stroke 2013; 44: 753–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jantzie LL, Miller RH, Robinson S. Erythropoietin signaling promotes oligodendrocyte development following prenatal systemic hypoxic-ischemic brain injury. Pediatr Res 2013; 74: 658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kako E, Kaneko N, Aoyama M, et al. Subventricular zone-derived oligodendrogenesis in injured neonatal white matter in mice enhanced by a nonerythropoietic erythropoietin derivative. Stem Cells 2012; 30: 2234–2247. [DOI] [PubMed] [Google Scholar]
- 41.Zhang L, Chopp M, Zhang RL, et al. Erythropoietin amplifies stroke-induced oligodendrogenesis in the rat. PLoS One 2010; 5: e11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugawa M, Sakurai Y, Ishikawa-Ieda Y, et al. Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci Res 2002; 44: 391–403. [DOI] [PubMed] [Google Scholar]
- 43.Nagai A, Nakagawa E, Choi HB, et al. Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J Neuropathol Exp Neurol 2001; 60: 386–392. [DOI] [PubMed] [Google Scholar]
- 44.Murray DM, Boylan GB, Ryan CA, et al. Early EEG findings in hypoxic-ischemic encephalopathy predict outcomes at 2 years. Pediatrics 2009; 124: e459–e467. [DOI] [PubMed] [Google Scholar]
- 45.Inder TE, Buckland L, Williams CE, et al. Lowered electroencephalographic spectral edge frequency predicts the presence of cerebral white matter injury in premature infants. Pediatrics 2003; 111: 27–33. [DOI] [PubMed] [Google Scholar]
- 46.van Rooij LG, Toet MC, Osredkar D, et al. Recovery of amplitude integrated electroencephalographic background patterns within 24 hours of perinatal asphyxia. Arch Dis Child Fetal Neonatal Ed 2005; 90: F245–F251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gunn AJ, Gunn TR, de Haan HH, et al. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest 1997; 99: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sterin-Borda L, Barcelo AC, Bozzini CE. Erythropoietin improves cardiac contractility in post-hypoxic mice. Br J Haematol 2003; 121: 180–186. [DOI] [PubMed] [Google Scholar]
- 49.Piuhola J, Kerkela R, Keenan JI, et al. Direct cardiac actions of erythropoietin (EPO): effects on cardiac contractility, BNP secretion and ischaemia/reperfusion injury. Clin Sci 2008; 114: 293–304. [DOI] [PubMed] [Google Scholar]
- 50.Mallard C, Davidson JO, Tan S, et al. Astrocytes and microglia in acute cerebral injury underlying cerebral palsy associated with preterm birth. Pediatr Res 2014; 75: 234–240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.