

Abstract
Cerebrovascular insufficiency appears years prior to clinical symptoms in Alzheimer’s disease. The soluble, highly toxic amyloid-β species, generated from the amyloidogenic processing of amyloid precursor protein, are known instigators of the chronic cerebrovascular insufficiency observed in both Alzheimer’s disease patients and transgenic mouse models. We have previously demonstrated that pioglitazone potently reverses cerebrovascular impairments in a mouse model of Alzheimer’s disease overexpressing amyloid-β. In this study, we sought to characterize the effects of amyloid-β overproduction on the cerebrovascular proteome; determine how pioglitazone treatment affected the altered proteome; and analyze the relationship between normalized protein levels and recovery of cerebrovascular function. Three-month-old wildtype and amyloid precursor protein mice were treated with pioglitazone- (20 mg/kg/day, 14 weeks) or control-diet. Cerebral arteries were surgically isolated, and extracted proteins analyzed by gel-free and gel-based mass spectrometry. 193 cerebrovascular proteins were abnormally expressed in amyloid precursor protein mice. Pioglitazone treatment rescued a third of these proteins, mainly those associated with oxidative stress, promotion of cerebrovascular vasocontractile tone, and vascular compliance. Our results demonstrate that amyloid-β overproduction perturbs the cerebrovascular proteome. Recovery of cerebrovascular function with pioglitazone is associated with normalized levels of key proteins in brain vessel function, suggesting that pioglitazone-responsive cerebrovascular proteins could be early biomarkers of Alzheimer’s disease.
Keywords: Amyloid peptide, cerebral artery, oxidative stress, proliferator-activated receptor gamma, vascular biomarkers
Introduction
Cognitive decline in Alzheimer’s disease (AD) is the end result of chronic brain synaptic and neuronal alterations, as well as early changes in the cerebrovasculature.1–3 Cerebral blood vessels in AD exhibit alterations in both structure and function.4,5 Chronic cerebral hypoperfusion typically appears prior to measurable mnemonic impairment and persists well into the latest stages of the disease.1,3 Recommended as a robust preclinical biomarker of AD,3 cerebrovascular hypoperfusion has also been correlated with negative treatment outcome in AD.2 Similarly, reduced cerebral blood flow (CBF) is an early feature of AD transgenic mouse models, including transgenic mice expressing a mutated form of the human amyloid precursor protein (APP mice), and precedes Aβ plaque deposition and cognitive deficits.6–8
Increased levels of Aβ peptide, especially soluble Aβ species, are the primary instigator of cerebrovascular dysfunction in APP mice. Young APP mice lacking Aβ plaque deposition, but harboring increased levels of soluble Aβ, exhibit oxidative stress in their brain vasculature,9,10 a major contributor of vascular abnormalities, including reductions in endothelial-mediated dilatory function9,10 and CBF.11 Oxidative stress and reductions in CBF are also observed in the brain vasculature of wildtype (WT) mice exposed to Aβ.9 Consistent with its detrimental effects on mouse cerebrovasculature, excess Aβ has also been shown to disrupt cerebral hemodynamics in isolated human cerebral arteries.12 We previously demonstrated that the peroxisome proliferator-activated receptor gamma (PPARγ) agonist pioglitazone, which belongs to the thiazolidinedione (TZD) class of anti-diabetic drugs, fully reversed Aβ-induced cerebrovascular dysfunction in APP mice.13,8 Pioglitazone exerts these benefits likely through vascular transcription factor PPARγ, which is capable of activating or trans-repressing a multitude of genes.14
While a previous study using proteomics on microvessel-enriched fractions of whole-brain homogenate in an AD mouse model found age-related changes,15 to date, no study has characterized the differential protein expression patterns in surgically extracted and individually isolated cerebral arteries between WT and APP mice, nor tested the impact of effective pharmacotherapy on cerebrovascular function. Addressing this gap in knowledge, our aims were to (a) characterize the effect of Aβ overproduction on the cerebrovascular proteome of APP mice; (b) determine the extent to which pioglitazone rescues the Aβ-altered cerebrovascular proteome; and (c) determine the link between normalized protein levels by pioglitazone and cerebrovascular function recovery. Our study is timely considering pioglitazone has recently entered a Phase III trial for delay of onset of AD in high risk, cognitively normal participants.16
Material and methods
Mice
Six-month-old male APP mice (line J20, n = 18) and their wildtype (WT, n = 18) littermates (C57BL/6J) were used. The APP/J20 line carries one copy of the familial early onset AD Swedish (670/671KM→NL) and Indiana (717V→F) APP mutations on the C57BL/6J background,17 which leads to increased production of APP-derived Aβ peptides. Mice were housed under a 12-h light-dark cycle, in a room with controlled temperature (23℃) and humidity (50%). All experiments were approved by the Animal Ethics Committee of the Montreal Neurological Institute and complied with the Canadian Council on Animal Care and the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines.
Drug treatment
A 14-week treatment regime was initiated in three-month-old mice, an age when soluble Aβ is present in APP mice, but not deposited forms of Aβ peptide.17 It should be noted that vascular Aβ deposits or cerebral amyloid angiopathy (CAA) is clearly detectable in J20 APP mice at 12-months of age in the distal segments and small branches of the arteries of the circle of Willis.18 The treated WT and APP groups (n = 9 each) received a pioglitazone-containing-diet [20 mg/kg/day (Takeda Pharmaceuticals)], while the untreated groups (n = 9 each) received non-medicated control-diet. This dose of pioglitazone was selected since it previously showed efficacy in restoring cerebrovascular function in APP mice.13 Both diets were mixed in Teklad Rodent chow (Research Diets Inc., New Brunswick, NJ, USA). Mice had access to tap water and food ad libitum.
Surgical extraction of cerebral arteries
The circle of Willis and major cerebral arteries along with their main branches were surgically removed from mice and individually stripped from the attached pia matter to obtain a clean preparation of vascular tissue, as previously described.19 Arteries extracted from three mice were combined to constitute one biological replicate and stored at −80℃. Three biological replicates (B1, B2, B3) were prepared for each of the four groups. Such a preparation of readily accessible cerebral blood vessels that are bathed in cerebrospinal fluid offers a reliable window to the brain microvasculature since both vascular beds are exposed to increased levels of soluble Aβ proteins, and previous proteomic analyses have revealed a very high level of similarities between these two segments of the brain circulation.19
Cerebrovascular proteomics workflow
Protein extraction from mouse cerebral arteries was performed using our recently published and validated protocol.19 A brief description of the procedure is provided in Supplemental information.
Generation of statistically significant protein lists
For generation of protein lists, statistical analyses were performed on proteins with ≥2 peptides, peptide score ≥35, and fold-change ≥1.5. In addition, proteins detected using both gel and gel-free approaches were retained only if the change in protein expression level was in the same direction. Visual validation of average peptide intensity distribution per biological and technical replicate was performed on all statistically identified proteins.
Proteins differentially expressed between WT and APP cerebral arteries were identified through parametric Student t and non-parametric Mann–Whitney U tests, with p < 0.05 considered statistically significant. Similarly, to identify differentially expressed proteins in the cerebral vasculature of APP mice that were affected by pioglitazone, we performed (a) parametric Student t-test, (b) non-parametric Mann–Whitney U test between APP and pioglitazone-treated APP mice, and (c) a two-way ANOVA taking transgene and treatment as the two factors (MATLAB, http://www.mathworks.com/products/matlab/). Proteins were considered fully rescued by pioglitazone treatment if they did not significantly differ from WT protein levels. Significance was considered to be p < 0.05.
Characterization of proteins
APP peptide alignment was performed using ClustalW2, a sequence alignment program for DNA or proteins (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Proteins known to directly or indirectly (secondary and tertiary interactions only) interact with APP and/or with each other were identified by querying PubMed, the STRING database that contains known and predicted protein interactions (http://string-db.org, version 9.1, confidence: medium to high), the IntAct database (http://www.ebi.ac.uk/intact/), and an in-house database that is a compendium of several databases, including BIND, BioGRID, HPRD, HIMAP, and EcoCyc. Species selected for the analyses were Mus musculus and/or Homo sapiens. APP-associated proteins, i.e. proteins co-mentioned with APP or amyloid-β peptides in the literature that may or may not be a direct interacting partner, were identified by querying the PubMed reference library. Proteins were indicated as associated with increased AD risk if present in AlzGene (http://alzgene.org), a database of AD genetic association studies, and/or by querying PubMed. Subcellular localizations of proteins were determined using data from Gene Ontology (http://geneontology.org/) and the Universal Protein Resource (www.uniprot.org) databases combined. The PANTHER Classification System (http://www.pantherdb.org/pathway/) was used to classify proteins by ‘molecular function’. Genes (of proteins) harboring peroxisome proliferator responsive element (PPRE) and PACM (PPAR-associated conserved motif) were identified using a published database.20 Proteins associated with extracellular microvesicles (or exosomes) released from brain endothelial cells (BECs) into the blood were identified using the BEC Exosome database.21 These proteins are a rich source of brain vasculature-specific biomarkers, as well as receptors known for delivering molecules across the blood–brain barrier.
Results
Proteins differentially expressed in brain vessels of APP and WT mice
We identified APP in both gel-free and gel-based proteomic approaches from one peptide (LVFFAEDVGSNK, Figure 1(a)) with protein scores of 93 and 71, respectively. Compared to WT mice, APP protein levels were significantly (p < 0.01) increased in the cerebral arteries of APP mice (Figure 1(b) and (c)). We identified an additional 192 proteins, corresponding to 3% of total proteins detected, differentially expressed between WT and APP mice cerebral arteries (Supplementary Table 1). The majority (131 proteins, 68.2%) of these proteins were identified using both parametric Student’s t and non-parametric Mann–Whitney U distributions (p < 0.05), while 50 (26.1%) and 11 (5.7%) proteins were detected using only parametric and non-parametric analyses, respectively (Figure 2(a)). Moreover, 78 (40.6%) of the 192 proteins were also identified using two-way ANOVA (Supplementary Table 1). The majority (62.5%) of proteins in the APP cerebral vasculature were upregulated with fold-changes ranging from ±1.5 to ±5.0 (Figure 2(b), Supplementary Table 1). As much as 122 (63.2%) of the differentially expressed proteins were found to interact with each other, and 32 (16.7%) were also present in the BEC Exosome database (Supplementary Table 1).
Figure 1.

Quantification of APP protein levels in cerebral arteries. (a) Alignment of the detected APP peptide (LVFFAEDVGSNK) in relation to the full-length APP protein. Also, indicated are the cleavage sites for the α, β, and γ secretases, as well as the AD-associated Swedish (KM) and Indiana (V) mutations. (b) Peptide intensity distribution of WT and APP mice as visualized using MSight. For both genotypes, biological replicate 1, each consisting of vessels extracted from three different mice is shown. The shaded grey area and/or the purple cross hair represent APP protein level. (c) As expected, APP mice (blue) display significantly elevated cerebrovascular APP protein levels relative to WT controls (green). Pioglitazone therapy moderately rescued APP protein levels in treated APP (red), but had no effect on WT (grey) mice. Error bars denote standard error or SEM and ⋆⋆ p < 0.01.
PIO: pioglitazone.
Figure 2.

Proteins differentially expressed in brain vessels of APP mice. (a) Distribution (in percentage) of proteins identified with either parametric or non-parametric Student t-test or both statistical analyses. (b) Fold-change distribution of differentially expressed proteins along with the percentage of upregulated or downregulated proteins in the APP vasculature. (c) Protein interactors of APP or proteins known to directly (black line) or indirectly (secondary: purple line and tertiary: grey line interactions only) interact with APP were identified using the STRING, IntAct, and in-house, and/or PubMed reference library. Note that only a few indirectly interacting proteins have been shown. Proteins associated with increased AD risk in the AlzGene database and/or by querying the PubMed reference library are identified in red and underlined (e.g. APOE). Average (d) APOE, and (e) HSP90B1, AQP1, and ACTN4 peptide/s intensities for WT (green), WT(pio) (grey), APP(blue), and APP(pio) (red). Error bars denote standard error or SEM, ⋆ p < 0.05 and ⋆⋆⋆ p < 0.001 denote significant genotype effect using two-way ANOVA. PIO: pioglitazone.
Proteins interacting or associated with APP
We found that over a third (68 proteins) of the differentially expressed proteins were either (a) protein interactors of APP (60 proteins) and/or (b) associated with APP (eight unique proteins, namely AIFM1, ATP5F1, BSN, CLOCK, DCN, PLA2G4A, PPP2CB, and SRF; protein names and additional information in Table 1). Of the established interactors, the 40 identified to interact directly with APP are as follows: ACTN4, ALDH7A1, APBB3, APOE, AQP1, ASPH, CCNG1, CYFIP, DEPDC6, GABRA4, GAK, GAS2, HAVCR2, HSP90AA1, HSP90BI, HSPA5, HTR2C, KIF2C, MAPRE1, MYH9, NAPA, NFASC, PCBP1, PGM1, PRDX5, RAB5C, RNF151, SENP1, SENP2, SERBP1, SHOC2, SOD1, STX12, TCP11L2, TMCC2, TPI1, TRIM41, USP5, VIM, and VTN (Table 1, Figure 2(c)). Of these, all except AQP1, CYFIP1, DEPDC6, HAVCR2, HTR2C, MAPRE1, NAPA, PGM1, SENP2, STX12, and TRIM41 were upregulated in APP mice (Figure 2(c)). Representative increased peptide levels of APOE, ACTN4, and HSP90B1 and decreased levels of AQP1 are illustrated in Figure 2(d) and (e). Nineteen established interactors of APP, 14 directly interacting (ACTN4, ASPH, CYFIP1, HSP90AA1, HSP90B1, HSPA5, MAPRE1, MYH9, PCBP1, RAB5C, SERBP1, SHOC2, TPI1, and VIM) and 5 indirectly interacting (CCT2, DES, HSPB1, RDX, RPLP0) were also present in the BEC Exosome database (Table 1).
Table 1.
Cerebral arterial proteins differentially expressed between WT and APP mice.
| Protein ID | Protein Symbol | Protein Name | Protein Interactors of APP | Associated with APP or AD (*) or both (bold) in Pubmed (PMID provided) | Present in AlzGene Db | Linked to APP and AD | Direction of change (APP) |
|---|---|---|---|---|---|---|---|
|
Q8QZT1 |
ACAT1
|
Acetyl-CoA acetyltransferase, mitochondrial |
inD |
20398792 24587158 |
y |
b |
↑ |
|
P57780 |
ACTN4
|
Alpha-actinin-4 |
D |
23055000* |
– |
b |
↑ |
|
Q9Z0X1 |
AIFM1
|
Apoptosis-inducing factor 1, mitochondrial |
– |
14767566 |
– |
– |
↓ |
|
Q9DBF1 |
ALDH7A1
|
Alpha-aminoadipic semialdehyde dehydrogenase |
D |
21832049 |
– |
– |
↑ |
|
O70423 |
AOC3
|
Membrane primary amine oxidase |
– |
11461168* |
– |
– |
↑ |
|
Q8R1C9 |
APBB3
|
Amyloid-beta A4 precursor protein-binding family B member 3 |
D |
14527950 16973241
|
– |
b |
↑ |
|
P08226 |
APOE
|
Apolipoprotein E |
D |
multiple
|
y |
b |
↑ |
|
Q02013 |
AQP1
|
Aquaporin-1 |
D |
16871401* 17123487* 18509662 21107133* 21955788* 22805778* 19687153 |
– |
b |
↓ |
|
Q8BSY0 |
ASPH
|
Aspartyl/asparaginyl beta-hydroxylase |
D |
21832049 |
– |
– |
↑ |
|
Q9CQQ7 |
ATP5F1
|
ATP synthase subunit b, mitochondrial |
– |
24587158 |
– |
– |
↑ |
|
P50516 |
ATP6V1A
|
V-type proton ATPase catalytic subunit A |
inD |
24587158 |
– |
– |
↓ |
|
O88737 |
BSN
|
Protein bassoon |
– |
24587158 |
– |
– |
↓ |
|
P51945 |
CCNG1
|
Cyclin-G1 |
D |
21832049 12901840* 17006763* |
– |
b |
↑ |
|
P80314 |
CCT2
|
T-complex protein 1 subunit beta |
inD |
11441917* |
– |
b |
↑ |
|
P53566 |
CEBPA
|
CCAAT/enhancer-binding protein alpha |
– |
18405661* |
– |
– |
↑ |
|
O08785 |
CLOCK
|
Circadian locomoter output cycles protein kaput |
– |
22634208 23781009* |
– |
b |
↑ |
|
Q9CPQ1 |
COX6C
|
Cytochrome c oxidase subunit 6C |
inD |
– |
– |
– |
↑ |
| Q7TMB8 | CYFIP1 | Cytoplasmic FMR1- interacting protein 1 | D | – | – | – | ↓ |
|
P28654 |
DCN
|
Decorin |
– |
1370306* 7793988* 17321057* 17660861* 21310214 |
– |
b |
↓ |
|
Q570Y9 |
DEPDC6
|
DEP domain-containing mTOR-interacting protein |
D |
21832049 |
– |
– |
↓ |
|
P31001 |
DES
|
Desmin |
inD |
– |
– |
– |
↑ |
|
O08749 |
DLD
|
Dihydrolipoyl dehydrogenase, mitochondrial |
inD |
– |
y |
b |
↑ |
|
Q9D832 |
DNAJB4
|
DnaJ homolog subfamily B member 4 |
inD |
– |
– |
– |
↑ |
|
P70372 |
ELAVL1
|
ELAV-like protein 1 |
– |
19221430* |
– |
– |
↓ |
|
Q8VHX6 |
FLNC
|
Filamin-C |
– |
9437013* 20847418* 22815492* |
– |
– |
↑ |
|
Q9D6F4 |
GABRA4
|
Gamma-aminobutyric acid receptor subunit alpha-4 |
D |
21832049 |
– |
– |
↑ |
|
Q99KY4 |
GAK
|
Cyclin G-associated kinase |
D |
21832049 |
– |
– |
↑ |
|
P11862 |
GAS2
|
Growth arrest-specific protein 2 |
D |
21832049 |
– |
– |
↑ |
|
Q01097 |
GRIN2B
|
Glutamate [NMDA] receptor subunit epsilon-2 |
inD |
– |
y |
b |
↓ |
|
Q8BH60 |
GOPC
|
Golgi-associated PDZ and coiled-coil motif-containing protein |
– |
– |
– |
– |
↑ |
|
Q8BMS1 |
HADHA
|
Trifunctional enzyme subunit alpha, mitochondrial |
– |
20538040* |
– |
– |
↑ |
|
Q8VIM0 |
HAVCR2
|
Hepatitis A virus cellular receptor 2 homolog |
D |
21832049 |
– |
– |
↓ |
|
P07901 |
HSP90AA1
|
Heat shock protein HSP 90-alpha |
D |
19449461 16049941 |
– |
b |
↑ |
|
P08113 |
HSP90B1
|
Endoplasmin |
D |
17934213 |
– |
– |
↑ |
|
P20029 |
HSPA5
|
78 kDa glucose-regulated protein |
D |
9748217 17132139 17934213 |
y |
b |
↑ |
|
P14602 |
HSPB1
|
Heat shock protein beta-1 |
inD |
16635482* 21670152* |
– |
b |
↑ |
| P34968 | HTR2C | 5-hydroxytryptamine receptor 2C | D | – | y | b | ↓ |
|
Q9Z0Y7 |
IRS4
|
Insulin receptor substrate 4 |
inD |
24274089* |
– |
b |
↓ |
|
A2ARA8 |
ITGA8
|
Integrin alpha-8 |
inD |
– |
– |
– |
↑ |
|
Q9Z0R4 |
ITSN1
|
Intersectin-1 |
– |
24573290 16442855* 21876463* |
– |
b |
↓ |
|
Q922S8 |
KIF2C
|
Kinesin-like protein KIF2C |
D |
21832049 |
– |
– |
↑ |
|
Q6PFD6 |
KIF18B
|
Kinesin-like protein KIF18B |
– |
– |
y |
– |
↑ |
|
Q80U28 |
MADD
|
MAP kinase-activating death domain protein |
– |
15007167* 16253995* 22678883* |
– |
– |
↓ |
|
Q61166 |
MAPRE1
|
Microtubule-associated protein RP/EB family member 1 |
D |
21832049 |
– |
– |
↓ |
|
O70583 |
MID1
|
Midline-1 |
– |
21098287* 22613722* |
– |
– |
↓ |
|
Q3V1L6 |
MTMR11
|
Myotubularin-related protein 11 |
– |
– |
– |
– |
↑ |
|
Q8VDD5 |
MYH9
|
Myosin-9 |
D |
16049941
|
– |
b |
↑ |
|
Q9DB05 |
NAPA
|
Alpha-soluble NSF attachment protein |
D |
21832049 24573290 |
– |
– |
↓ |
|
Q810U3 |
NFASC
|
Neurofascin |
D |
17934213 |
– |
– |
↑ |
|
P60335 |
PCBP1
|
Poly(rC)-binding protein 1 |
D |
21832049 |
– |
– |
↑ |
|
P08003 |
PDIA4
|
Protein disulfide-isomerase A4 |
inD |
– |
– |
– |
↑ |
|
Q9D0F9 |
PGM1
|
Phosphoglucomutase-1 |
D |
21832049 17522440* |
– |
b |
↓ |
|
P47713 |
PLA2G4A
|
Cytosolic phospholipase A2 |
– |
17713604 22188721 multiple* |
– |
b |
↑ |
|
Q3UYC0 |
PPM1H
|
Protein phosphatase 1H |
– |
– |
y |
– |
↓ |
|
Q6R891 |
PPP1R9B
|
Neurabin-2 |
– |
– |
– |
– |
↑ |
|
P62715 |
PPP2CB
|
Serine/threonine-protein phosphatase 2A catalytic subunit beta isoform |
– |
23020770 multiple* |
– |
b |
↑ |
|
P99029 |
PRDX5
|
Peroxiredoxin-5, mitochondrial |
D |
21832049 24573290 |
– |
– |
↑ |
|
P15331 |
PRPH
|
Peripherin |
inD |
– |
– |
– |
↑ |
| P35278 | RAB5C | Ras-related protein Rab-5C | D | 21832049 | – | – | ↑ |
|
P60764 |
RAC3
|
Ras-related C3 botulinum toxin substrate 3 |
inD |
– |
– |
– |
↓ |
|
P26043 |
RDX
|
Radixin |
inD |
– |
– |
– |
↓ |
|
Q9CQ29 |
RNF151
|
RING finger protein 151 |
D |
21832049 |
– |
– |
↑ |
|
P14869 |
RPLP0
|
60S acidic ribosomal protein P0 |
inD |
– |
– |
– |
↑ |
|
P59110 |
SENP1
|
Sentrin-specific protease 1 |
D |
21832049 24778618* |
– |
b |
↑ |
|
Q91ZX6 |
SENP2
|
Sentrin-specific protease 2 |
D |
21832049 |
– |
– |
↓ |
|
Q9CY58 |
SERBP1
|
Plasminogen activator inhibitor 1 RNA-binding protein |
D |
21832049 |
– |
– |
↑ |
|
Q8R0X7 |
SGPL1
|
Sphingosine-1-phosphate lyase 1 |
– |
– |
y |
– |
↑ |
|
O88520 |
SHOC2
|
Leucine-rich repeat protein SHOC-2 |
D |
21832049 |
– |
– |
↑ |
|
P08228 |
SOD1
|
Superoxide dismutase [Cu-Zn] |
D |
10195200 22790929 multiple* |
– |
b |
↑ |
|
Q9JM73 |
SRF
|
Serum response factor |
– |
22912719 17215356* 19098903* 22850315* |
– |
b |
↑ |
|
Q9ER00 |
STX12
|
Syntaxin-12 (alias Syntaxin-13) |
D |
20925061 |
– |
– |
↓ |
|
Q8JZP2 |
SYN3
|
Synapsin-3 |
– |
– |
y |
– |
↑ |
|
Q8K1H7 |
TCP11L2
|
T-complex protein 11-like protein 2 |
D |
21832049 |
– |
– |
↑ |
|
P39447 |
TJP1
|
Tight junction protein ZO-1 |
inD |
22745485* |
– |
b |
↑ |
|
Q80W04 |
TMCC2
|
Transmembrane and coiled-coil domains protein 2 |
D |
21832049 23409049 |
– |
b |
↑ |
|
Q3UHK8 |
TNRC6A
|
Trinucleotide repeat- containing gene 6A protein |
– |
– |
– |
– |
↓ |
|
P17751 |
TPI1
|
Triosephosphate isomerase |
D |
21832049 |
– |
– |
↑ |
|
Q5NCC3 |
TRIM41
|
E3 ubiquitin-protein ligase TRIM41 |
D |
21832049 |
– |
– |
↓ |
|
O08747 |
UNC5C
|
Netrin receptor UNC5C |
– |
24866402* |
– |
– |
↑ |
| P56399 | USP5 | Ubiquitin carboxyl-terminal hydrolase 5 | D | 21832049 24573290 | – | – | ↑ |
|
P20152 |
VIM
|
Vimentin |
D |
21832049 23038755 |
– |
– |
↑ |
|
P29788 |
VTN
|
Vitronectin |
D |
21832049 |
– |
– |
↑ |
|
Q60989 |
XIAP
|
Baculoviral IAP repeat- containing protein 4 |
inD |
16599295* 17292615* |
– |
b |
↑ |
| O88967 | YME1L1 | ATP-dependent metalloprotease YME1L1 | inD | – | – | – | ↓ |
Note: Only proteins mentioned in the manuscript are listed, while the complete list of proteins differentially expressed between APP and WT mice is provided in Supplementary Table 1. All proteins were identified with ≥2 peptides, exhibiting peptide scores ≥35. Protein IDs used follow the UniProt assignment (www.uniprot.org). In COLUMN 4 (Protein Interactors of APP), proteins known to directly interact with APP are indicated as ‘D’ and those known to indirectly (secondary or tertiary only) interact with APP are indicated as ‘inD’. These protein interactors of APP were identified using STRING, IntAct, an in-house database, and by querying PubMed. D indicates that the protein was listed as a direct interactor (medium to high confidence; accessed March 14, 2016) in the STRING database. COLUMN 5 provides PubMed identifiers or PMIDs of proteins/genes associated with APP only (PMID or multiple), AD only (PMID* or multiple*), or both (PMID or multiple*). For each of the three categories, PMIDs are listed in numerical order. COLUMN 7 (Linked to APP and AD) identifies proteins (‘b’) that interact/or are associated with APP and are associated with AD. AD: Alzheimer’s disease. Symbols: ↑, up; ↓, down; y, protein present in the AlzGene Db.
Proteins associated with AD
We identified 39 (20.3%) AD-associated proteins in our differentially expressed list (Table 1). Ten, namely, ACAT1, APOE, DLD, GRIN2B, HSPA5, HTR2C, KIF18B, PPM1H, SGPL1, and SYN3, are products of genes present in the AlzGene database and linked with increased AD risk (Table 1). Of these, APOE, HSPA5, and HTR2C are known to directly interact with APP (Figure 2(c)), while ACAT1, DLD, and GRIN2B demonstrate indirect interaction. Overall, we found that more than half (27/39 proteins, 69.2%) of the proteins identified as “associated with AD” were also known to “interact or associate with APP” (refer to subsection “Proteins interacting or associated with APP” and Table 1).
Molecular functions disrupted by mutant APP overexpression
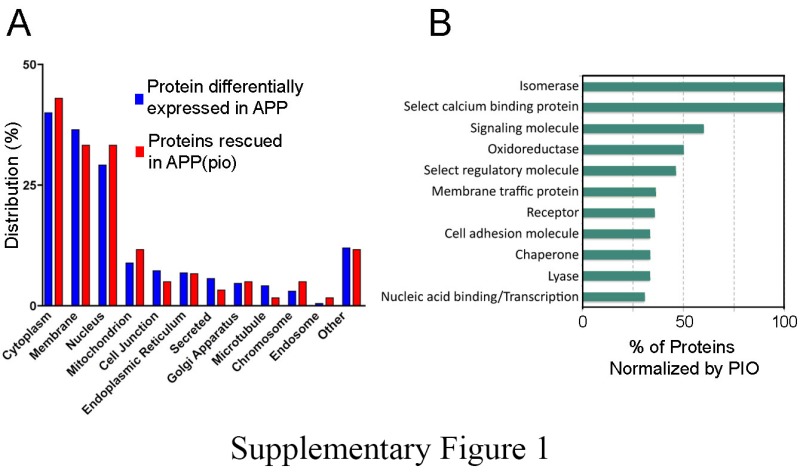
The top five locations of differentially expressed proteins were cytoplasm (40.0%), membrane (36.5%), nucleus (29.2%), mitochondrion (8.9%), and cell junction (7.3%) (Supplemental Figure 1(a)). 143 (75%) of these proteins also had known molecular functions in the PANTHER database, which corresponded to (i) proteins binding to nucleic acids (27 proteins), (ii) cytoskeletal filaments (14 proteins), (iii) receptors (14 proteins), (iv) regulatory molecules (13 proteins), and (v) oxidoreductases (12 proteins) (Supplementary Table 2).
Differentially expressed proteins rescued by pioglitazone in APP mice
We found that pioglitazone induced a near-significant reduction of APP levels (p = 0.07) in APP mice, but had no detectable effect on WT mice (Figure 1(c)). Pioglitazone further rescued 60 of the 192 (31%) differentially expressed proteins, with 45 (75%) proteins demonstrating full normalization, and 15 (25%) exhibiting partial albeit significant recovery to WT levels (Table 2).
Table 2.
Cerebral arterial proteins normalized by pioglitazone.
| Protein ID | Protein symbol | Protein name | Direction of change (APP) | Normalization by Pioglitazone | Two-way ANOVA Interaction Term |
|---|---|---|---|---|---|
| Q8VCH0 | ACAA1B | 3-ketoacyl-CoA thiolase B, peroxisomal | ↓ | full | – |
| Q8JZV7 | AMDHD2 | Putative N-acetylglucosamine-6- phosphate deacetylase | ↓ | full | – |
| Q9CZK6 | ANKS3 | Ankyrin repeat and SAM domain-containing protein 3 | ↑ | full | – |
| Q99PT1 | ARHGDIA | Rho GDP-dissociation inhibitor 1 | ↑ | full | – |
| Q8C9S8 | ATG4A | Cysteine protease ATG4A | ↓ | full | – |
| Q9D4K7 | CCDC105 | Coiled-coil domain-containing protein 105 | ↑ | full | – |
| Q99PF4 | CDH23 | Cadherin-23 | ↑ | partial | – |
| Q69ZA1 | CDK13 | Cell division protein kinase 13 | ↓ | full | – |
| P53566 | CEBPA | CCAAT/enhancer-binding protein alpha | ↑ | full | – |
| Q9R0M0 | CELSR2 | Cadherin EGF LAG seven-pass G-type receptor 2 | ↓ | full | – |
| Q6PG95 | CRAMP1L | Protein cramped-like | ↓ | partial | – |
| P56395 | CYB5A | Cytochrome b5 | ↑ | full | – |
| O08749 | DLD | Dihydrolipoyl dehydrogenase, mitochondrial | ↑ | full | – |
| Q6XUX1 | DSTYK | Dual serine/threonine and tyrosine protein kinase | ↑ | full | y |
| P70372 | ELAVL1 | ELAV-like protein 1 | ↓ | full | – |
| Q99JW1 | FAM108A | Abhydrolase domain-containing protein FAM108A | ↑ | partial | – |
| Q8VHX6 | FLNC | Filamin-C | ↑ | full | – |
| Q9CYL5 | GLIPR2 | Golgi-associated plant pathogenesis-related protein 1 | ↑ | full | – |
| Q8BH60 | GOPC | Golgi-associated PDZ and coiled-coil motif-containing protein | ↑ | full | – |
| Q8BMS1 | HADHA | Trifunctional enzyme subunit alpha, mitochondrial | ↑ | full | – |
| P62806 | HIST1H4A | Histone H4 | ↑ | full | – |
| Q64523 | HIST2H2AC | Histone H2A type 2-C | ↓ | full | – |
| P20029 | HSPA5 | 78 kDa glucose-regulated protein | ↑ | full | – |
| P14602 | HSPB1 | Heat shock protein beta-1 | ↑ | full | – |
| Q9Z0R4 | ITSN1 | Intersectin-1 | ↓ | full | – |
| Q80V70 | MEGF6 | Multiple epidermal growth factor-like domains protein 6 | ↑ | full | – |
| Q791V5 | MTCH2 | Mitochondrial carrier homolog 2 | ↓ | full | – |
| Q3V1L6 | MTMR11 | Myotubularin-related protein 11 | ↑ | partial | – |
| Q8VDD5 | MYH9 | Myosin-9 | ↑ | full | – |
| Q7TSZ8 | NACC1 | Nucleus accumbens-associated protein 1 | ↑ | full | – |
| Q9DB05 | NAPA | Alpha-soluble NSF attachment protein | ↓ | partial | – |
| Q9Z1P6 | NDUFA7 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 7 | ↓ | partial | – |
| P29341 | PABPC1 | Polyadenylate-binding protein 1 | ↑ | partial | y |
| Q8CEE6 | PASK | PAS domain-containing serine/ threonine-protein kinase | ↓ | partial | – |
| P08003 | PDIA4 | Protein disulfide-isomerase A4 | ↑ | full | – |
| Q9D0F9 | PGM1 | Phosphoglucomutase-1 | ↓ | full | – |
| Q6R891 | PPP1R9B | Neurabin-2 | ↑ | partial | y |
| P99029 | PRDX5 | Peroxiredoxin-5, mitochondrial | ↑ | full | y |
| P15331 | PRPH | Peripherin | ↑ | full | – |
| P35278 | RAB5C | Ras-related protein Rab-5C | ↑ | partial | y |
| Q8BIV3 | RANBP6 | Ran-binding protein 6 | ↑ | full | – |
| P26043 | RDX | Radixin | ↓ | partial | – |
| P50543 | S100A11 | Protein S100-A11 | ↑ | full | – |
| Q8R2U0 | SEH1L | Nucleoporin SEH1 | ↓ | full | – |
| Q91ZX6 | SENP2 | Sentrin-specific protease 2 | ↓ | full | – |
| O88520 | SHOC2 | Leucine-rich repeat protein SHOC-2 | ↑ | partial | – |
| Q924W5 | SMC6 | Structural maintenance of chromosomes protein 6 | ↑ | full | – |
| Q91ZR2 | SNX18 | Sorting nexin-18 | ↓ | partial | – |
| P08228 | SOD1 | Superoxide dismutase [Cu-Zn] | ↑ | full | – |
| Q9D489 | SOHLH2 | Spermatogenesis- and oogenesis- specific basic helix-loop- helix-containing protein 2 | ↑ | partial | – |
| Q9DC40 | TELO2 | Telomere length regulation protein TEL2 homolog | ↑ | full | – |
| Q80W04 | TMCC2 | Transmembrane and coiled-coil domains protein 2 | ↑ | full | – |
| Q3UHK8 | TNRC6A | Trinucleotide repeat-containing gene 6A protein | ↓ | full | – |
| P17751 | TPI1 | Triosephosphate isomerase | ↑ | full | y |
| O08747 | UNC5C | Netrin receptor UNC5C | ↑ | full | – |
| P20152 | VIM | Vimentin | ↑ | full | – |
| Q8BX70 | VPS13C | Vacuolar protein sorting-associated protein 13C | ↑ | full | – |
| Q60989 | XIAP | Baculoviral IAP repeat-containing protein 4 | ↑ | full | y |
| P23607 | ZFA | Zinc finger autosomal protein | ↑ | full | y |
| Q5DU37 | ZFYVE26 | Zinc finger FYVE domain-containing protein 26 | ↑ | partial | – |
Note: Listed are cerebral arterial proteins significantly rescued (p < 0.05, t-test) by pioglitazone in APP mice compared to untreated APP mice. Only proteins identified with ≥ 2 peptides and exhibiting peptide scores ≥35 are included. Protein IDs used are those assigned by UniProt (www.uniprot.org). In COLUMN 2, Protein Symbol, Protein Symbol, and Protein Symbol, denotes associated with Alzheimer’s disease, interacting with and/or associated with APP, or both respectively. COLUMN 4 (two-way ANOVA) identifies (‘y’) proteins that are rescued by pioglitazone when analyzed using a two-way ANOVA taking genotype and treatment as factors. Shaded rows identify proteins present in the brain endothelial cell Exosome database and normalized by pioglitazone. Symbols: ↑, up; ↓, down.
Of the pioglitazone-rescued proteins, 8 (13%) had known PPARγ-PPRE/PACM binding sites (Figure 3(a), Table 1), 16 (27%) were found in circulating BEC microvesicles (Table 2, Figure 4(a)), and 4, namely FLNC, RAB5C, TNRC6A, and TPI1 exhibited both features. Of the aforementioned APP binding proteins, levels of primary interactors HSPA5, MYH9, PGM1, PRDX5, SENP2, SOD1, TMCC2, TPI1, and VIM, and secondary interactors DLD, HSPB1, PDIA4, PRPH, and XIAP were normalized (Figure 3(a), Table 1). Levels of AD-associated AlzGene proteins DLD and HSPA5 were also rescued by pioglitazone treatment (Figure 3(a), Table 1). Figure 3(b) demonstrates peptide levels of five representative proteins, namely SOD1, XIAP, TPI1, RAB5C, and PPP1R9B, normalized by pioglitazone.
Figure 3.

Pioglitazone-recovered proteins in the APP vasculature. (a) Pioglitazone-rescued protein that (i) directly or indirectly interact with APP, and/or (ii) harbor known PPARγ-PPRE/PACM binding sites in their genomic sequences. (b) Average SOD1, XIAP, TPI1, RAB5C, and PPP1R9B peptide intensities for WT (green), WT(pio) (grey), APP (blue), and APP(pio) (red) mice.
Note: The function listed for RAB5C is probable, but not confirmed. Error bars denote standard error or SEM and ⋆ p < 0.05 denote significant interaction effect using two-way ANOVA. NO: nitric oxide; PIO: pioglitazone.
Figure 4.

Cerebrovascular functional recovery by pioglitazone in APP mice. (a) 16 cerebrovascular shed proteins (or proteins present in circulating BEC microvesicles) constituting 27% of pioglitazone-rescued proteins were identified. Fourteen of these shed proteins were upregulated (↑) in APP mice and normalized by pioglitazone: ARHGDIA, FLNC, GLIPR2, HIST1H4A, HSPA5, HSPB1, MYH9, PABPC1, RAB5C, S100A11, SHOC2, TPI1, VIM, and VPS13C, whereas two were downregulated (↓) in APP mice and normalized by treatment: RDX and TNRC6A. These proteins could serve as surrogates to indicate efficacy and/or select responders in pioglitazone clinical trials. (b) APP overexpression and consequent Aβ increase in APP mice alter levels of proteins involved in mediating (i) oxidative stress (both ROS generators and scavengers), (ii) vascular elasticity and compliance, and (iii) increased basal tone, which then promotes impaired vasodilation. Pioglitazone treatment normalizes levels of these proteins and recovers cerebrovascular function. Of the proteins shown, FLNC, HSPB1, MYH9, TPI1, and VIM are shed proteins. Symbols: ↑, up and ↓, down.
The top five locations of pioglitazone-rescued proteins were cytoplasm (43.0%), membrane (33.3%), nucleus (33.3%), mitochondrion (11.7%), and endoplasmic reticulum (6.7%) (Supplemental Figure 1(a)). The majority (>75%) of disturbed molecular functions were normalized to various degrees by pioglitazone, the top five categories being: isomerase activity, calcium binding, signaling, oxidation-reduction, and regulatory processes (Supplemental Figure 1(b)).
Discussion
Using two complementary and previously validated proteomic approaches,19 we found perturbations in the cerebrovascular proteome of transgenic mice overexpressing the AD-associated mutant-APP gene. We detected increase in the protein-level of APP, and alterations in the levels of an additional 192 proteins. Breakdown of the differentially expressed proteins by subcellular localization was in good agreement with the relative sizes of cellular compartments (e.g. membrane > mitochondria),19 demonstrating that our proteomic approach was unbiased towards any one cellular compartment. Our finding that 82 (43%) of these proteins are linked to the APP and/or AD literature strengthens our confidence that the protein list contains numerous novel cerebrovascular candidates affected by APP overexpression. We identified proteins involved in/or responding to APP overexpression associated with molecular pathogenic changes, such as RNA/DNA damage, vascular cytoskeleton alterations, and deregulation of the oxidoreductase system. We further demonstrated that approximately a third of the APP-altered cerebrovascular proteins were partially or fully normalized to WT levels following pioglitazone treatment. Since more than a quarter (27%) of pioglitazone-rescued proteins corresponded to BEC microvesicles,21 they could serve as markers of efficacy in pioglitazone clinical trials (Figure 4(a)). While prior study suffered from limited sensitivity due to suboptimal cerebral vascular tissue extraction technique from brain homogenates,15 our use of surgically extracted cerebral arteries, individually isolated and cleaned of the attached pial membrane,19 has allowed us to identify a large number of relevant vascular proteins that could be tested in functional assays to validate the identified pathways as key therapeutic targets.
We previously showed that pioglitazone is highly effective at reversing soluble Aβ-induced cerebrovascular dysfunction in APP mice, in particular, impaired vasodilation and reductions in neurovascular coupling.13,8 Since oxidative stress, alterations in vascular elasticity and increased basal tone can impair vascular dilation, we propose that rescue of these three categories of proteins by pioglitazone underlies the documented pioglitazone-mediated normalization of cerebrovascular function (Figure 4(b)). In the current study, we found that pioglitazone showed a near-significant ability to reduce APP protein levels, a result that was not observed in our previous study in older APP mice.13 The observed benefits of pioglitazone may thus be mediated both by reduction of cerebrovascular APP protein levels, and direct effects on the cerebrovasculature, as discussed below.
Mutant APP overexpression, oxidative stress, and pioglitazone
Pioglitazone reduces cerebrovascular APP/Aβ toxicity
The increase in cerebrovascular APP levels in APP mice was moderately reduced following early, long-term (three months) treatment with pioglitazone. Our finding is in line with a previous report that PPARγ activation by another TZD, rosiglitazone, increases APP protein ubiquination and degradation in cell cultures.22 Moreover, pioglitazone normalized the upregulated AD-susceptibility protein, HSPA5,23 a physiological binding partner of APP suggested to facilitate correct folding of APP and limit access of APP to β-/γ-secretases.24 Thus, the increase in cerebrovascular HSPA5 protein levels in APP mice might be a physiological attempt at reducing the amyloidogenic processing of APP and the ensuing Aβ toxicity. Normalized HSPA5 level in pioglitazone-treated APP mice likely results from reduced APP protein levels in cerebral vessels, and decrease in oxidative stress.
Pioglitazone normalizes altered levels of reactive oxygen species scavenging enzymes
We found upregulation of SOD1 and PRDX5 in the APP cerebrovasculature. Increase in these two reactive oxygen species (ROS)-scavenging enzymes signpost the presence of excess ROS. SOD1 reduces cytoplasmic ROS by catalyzing the dismutation of superoxide, and prevention of Aβ-induced cerebrovascular dysfunction has been demonstrated in APP transgenic mice co-overexpressing SOD1.25 Conversely, PRDX5 promotes decrease in mitochondrial ROS via reduction of hydrogen peroxide (H2O2) and other peroxide substrates.26 Our finding of increased mitochondrial ROS in the young APP vasculature is coherent with previous studies showing increased ROS production and concurrently elevated SOD2 protein levels10,13 in brain vessels from adult and aged APP mice. Furthermore, normalized SOD1 and PRDX5 protein levels in young APP mice by pioglitazone parallels the restored levels of SOD2 protein in pioglitazone-treated older mice,13 supporting pioglitazone as a potent restorer of antioxidant protection in the brain vasculature (Figure 4(b)).
Pioglitazone rescues altered levels of ROS promoting proteins
We identified two upregulated ROS promoting proteins, namely DLD and HADHA, in the APP brain vasculature that were normalized by pioglitazone (Figure 4(b)). DLD, an indirect protein interactor of APP, is a component of the pyruvate dehydrogenase complex (PDC) and oxidizes pyruvate to acetyl–coenzyme A.27 The enzymatic activity of DLD is coupled to the production of NADH,27 a major substrate for ROS generation by the mitochondrial electron transport chain and cytosolic NOX oxidases in arterial SMC.28 Increase in total NADH has also been associated with the initiation of SMC contraction in peripheral arteries.29 Since the increase in total NADH parallels the rise in PDC activity,29 it is possible that surplus DLD in APP arteries contributes to the destabilization of NADH homeostasis and, consequently, increased ROS. Previous findings implicating DLD as a source of ROS support our hypothesis.30 Single-nucleotide polymorphism variations in the DLD gene have been linked to increased AD risk in certain populations.31 In addition, upregulation of the inner mitochondrial membrane protein HADHA also contributes to increased NADH/NAD+ ratio in the APP vasculature.32 In contrast, reduced HADHA levels were reported in the tunica media of amyloid-laden pial arteries from AD patients,33 suggesting that HADHA upregulation in the cerebral arteries of our six-month-old APP mice may be a response to increased soluble Aβ peptide in the absence of detectable amyloid angiopathy at this age.
Pioglitazone rescues altered levels of proteins involved in regulating vascular tone and compliance
We identified three differentially expressed proteins in the APP cerebral vasculature, namely, HSPB1 (or HSP27), TPI1, and ELAVL1 (or HuR), known to promote increased vascular basal tone and, consequently, reduce vascular compliance (Figure 4(b)). The upregulated protein HSP27, a secondary interactor of APP, is known to mediate contraction in brain arteries upon phosphorylation,34 while increased levels of ROS induce HSP27 phosphorylation.35 Taken together, our findings suggest that in the milieu of excess, and potentially phosphorylated HSP27 in the APP vasculature, the response of vascular SMC to vasoconstrictors acting via the p38 mitogen-activated protein kinase (MAPK)/HSP27 pathway, such as angiotensin II,36 thrombin,37 and endothelin-1,34 is augmented. Since normal vascular tone requires balanced vasoconstrictor and vasodilator influences, selective augmentation of vasoconstrictor effects by HSP27 is likely to increase basal tone. The glycolytic enzyme TPI1, known to indirectly interact with APP, was also upregulated in APP mice. TPI1 is a component of the ATP-sensitive potassium (KATP) channel complex and plays a role in regulating channel function by altering ATP levels in the intracellular microenvironment.38 TPI1 activity and the subsequent increase in intracellular ATP reportedly inhibit activity at KATP channels,38 which are key mediators of vasodilatory responses in brain arteries.39 Notably, inhibition of KATP channel activity in SMC promotes increases in vascular basal tone and contraction.40 Protein level of HuR, a RNA-binding protein that regulates gene expression by modulating mRNA stability, was decreased in APP mice. In arterial SMC, binding of HuR to the β1-subunit mRNA of soluble guanylate cyclase (sGC) increases its stability, expression, and activity.41 Since sGC facilitates NO-dependent relaxation in cerebral arteries, decreased HuR levels in the APP vasculature and the accompanying reduction in sGC activity may also contribute to increased basal tone. In line with this hypothesis is the observation that NO-responsive sGC activity is reduced in AD brain.42 All three proteins normalized to WT levels in pioglitazone-treated APP mice.
Pioglitazone rescues altered levels of proteins reducing vascular elasticity
Increased cerebral arterial rigidity resulting in decreased dilatory capacity and compromised brain perfusion is present in AD.43 Impaired arterial elasticity most likely stems from soluble Aβ-triggered early onset vascular inflammation,44 as seen in AD transgenic mouse models.45 In our six-month old APP mice lacking vascular Aβ deposition, we identified four upregulated proteins potentially contributing to impaired arterial elasticity (Figure 4(b)), all of which were normalized by pioglitazone. Two were actin-cross-linking proteins with known PPARγ-PPRE/PACM binding sites.20 The muscle-specific FLNC that regulates cell membrane elasticity46 and PPP1R9B that inhibits protein phosphatase 1,47 a promoter of SMC relaxation, were both upregulated in APP brain vessels. Pioglitazone also normalized the intermediate filament protein VIM that promotes SMC rigidity and contractile state,48 and MYH9, an inherently contractile protein49 that acts through increased formation of the potent vasoconstrictor angiotensin II and degradation of the vasodilator bradykinin.50 MYH9 and VIM are known interactors of APP, and VIM has also been shown to co-localize with Aβ42 in brain tissues of AD patients and APP mice.51 Normalization of these upregulated proteins in pioglitazone-treated APP mice strongly supports the beneficial effects of TZD drugs in the reversal of decreased cell membrane elasticity via actin remodeling in the vasculature,52 and proper functioning of the perivascular drainage pathway within arterial and arteriolar basement membranes.53,54
Conclusions
In conclusion, our findings demonstrate that APP overexpression and increased soluble Aβ adversely impacts the cerebrovascular proteome. It should be noted that this detrimental effect occurs in the absence of detectable vascular Aβ deposits or CAA. We also show that pioglitazone reduces oxidative stress and normalizes the levels of several proteins involved in the reduced dilatory capacity of the APP brain vasculature. Moreover, pioglitazone-facilitated improvement in cerebral elasticity has the potential to diminish lymphatic congestion and improve brain Aβ clearance. Together, our findings indicate that pioglitazone significantly alleviates Aβ-induced cerebrovascular impairment and provides a link between protein rescue and cerebrovascular functional recovery by pioglitazone.
Supplementary Material
Supplementary Material
Acknowledgements
We thank Drs. A. Pshezhetsky and E. Kanshin (CHU Sainte-Justine, Research Center, Montréal, QC, Canada) for initial protein extraction and detection trials, Ms. C. Delaney and Mr. L. Tessier (IBS-National Research Council of Canada, Proteomics and Mass Spectrometry group, Ottawa, ON, Canada) for their technical assistance with protein isolation and mass spectrometry, and Dr. S. Narayanan (Montreal Neurological Institute, Montréal, QC, Canada) for helpful comments on the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by grants (E.H.) from the Canadian Institutes of Health Research (CIHR, MOP-84275 and MOP-126001) and Takeda Pharmaceuticals North America, Inc., and a CIHR Banting and Best Canada Graduate Scholarship (A.B.).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
APB designed the study, performed experiments, analyzed and interpreted the data, and wrote the manuscript; RB assisted in experiments; DS helped in the design of the study, data interpretation and preparation of the manuscript; ASH supervised data analysis and interpretation, and helped in the preparation of the manuscript; and EH designed the study, contributed to experiments, interpreted the data and contributed in the writing and revision of the manuscript. All co-authors meet conditions required for authorship credit as indicated on the JCBFM website, and all have approved the final manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Ruitenberg A, den Heijer T, Bakker SL, et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol 2005; 57: 789–794. [DOI] [PubMed] [Google Scholar]
- 2.Nicolakakis N, Hamel E. Neurovascular function in Alzheimer's disease patients and experimental models. J Cereb Blood Flow Metab 2011; 31: 1354–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wierenga CE, Hays CC, Zlatar ZZ. Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer's disease. J Alzheimers Dis 2014; 42: S411–S419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalaria RN. Cerebral vessels in ageing and Alzheimer's disease. Pharmacol Ther 1996; 72: 193–214. [DOI] [PubMed] [Google Scholar]
- 5.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol 2001; 64: 575–611. [DOI] [PubMed] [Google Scholar]
- 6.Niwa K, Younkin L, Ebeling C, et al. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A 2000; 97: 9735–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niwa K, Kazama K, Younkin SG, et al. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 2002; 9: 61–68. [DOI] [PubMed] [Google Scholar]
- 8.Badhwar A, Lerch JP, Hamel E, et al. Impaired structural correlates of memory in Alzheimer's disease mice. Neuroimage Clin 2013; 3: 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park L, Anrather J, Forster C, et al. Abeta-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab 2004; 24: 334–342. [DOI] [PubMed] [Google Scholar]
- 10.Tong XK, Nicolakakis N, Kocharyan A, et al. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J Neurosci 2005; 25: 11165–11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu X, Smith MA, Honda K, et al. Vascular oxidative stress in Alzheimer disease. J Neurol Sci 2007; 257: 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paris D, Humphrey J, Quadros A, et al. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer's disease: role of inflammation. Neurol Res 2003; 25: 642–651. [DOI] [PubMed] [Google Scholar]
- 13.Nicolakakis N, Aboulkassim T, Ongali B, et al. Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci 2008; 28: 9287–9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beyer AM, Baumbach GL, Halabi CM, et al. Interference with PPARgamma signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension 2008; 51: 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Searcy JL, Le Bihan T, Salvadores N, et al. Horsburgh. Impact of age on the cerebrovascular proteomes of wild-type and Tg-SwDI mice. PLoS One 2014; 9: e89970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crenshaw DG, Gottschalk WK, Lutz MW, et al. Using genetics to enable studies on the prevention of Alzheimer's disease. Clin Pharmacol Ther 2013; 93: 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mucke L, Masliah E, Yu GQ, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000; 20: 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tong XK, Nicolakakis N, Fernandes P, et al. Simvastatin improves cerebrovascular function and counters soluble amyloid-beta, inflammation and oxidative stress in aged APP mice. Neurobiol Dis 2009; 35: 406–414. [DOI] [PubMed] [Google Scholar]
- 19.Badhwar A, Stanimirovic DB, Hamel E, et al. The proteome of mouse cerebral arteries. J Cereb Blood Flow Metab 2014; 34: 1033–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemay DG, Hwang DH. Genome-wide identification of peroxisome proliferator response elements using integrated computational genomics. J Lipid Res 2006; 47: 1583–1587. [DOI] [PubMed] [Google Scholar]
- 21.Haqqani AS, Delaney CE, Tremblay TL, et al. Method for isolation and molecular characterization of extracellular microvesicles released from brain endothelial cells. Fluids Barriers CNS 2013; 10: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.d'Abramo C, Massone S, Zingg JM, et al. Role of peroxisome proliferator-activated receptor gamma in amyloid precursor protein processing and amyloid beta-mediated cell death. Biochem J 2005; 391: 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu WC, Wang HK, Lee LC, et al. Promoter polymorphisms modulating HSPA5 expression may increase susceptibility to Taiwanese Alzheimer's disease. J Neural Transm 2008; 115: 1537–1543. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem 1998; 273: 25552–25555. [DOI] [PubMed] [Google Scholar]
- 25.Iadecola C, Zhang F, Niwa K, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci 1999; 2: 157–161. [DOI] [PubMed] [Google Scholar]
- 26.Graves JA, Metukuri M, Scott D, et al. Regulation of reactive oxygen species homeostasis by peroxiredoxins and c-Myc. J Biol Chem 2009; 284: 6520–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan LJ, Thangthaeng N, Forster MJ. Changes in dihydrolipoamide dehydrogenase expression and activity during postnatal development and aging in the rat brain. Mech Ageing Dev 2008; 129: 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao Q, Wolin MS. Effects of hypoxia on relationships between cytosolic and mitochondrial NAD(P)H redox and superoxide generation in coronary arterial smooth muscle. Am J Physiol Heart Circ Physiol 2008; 295: H978–H989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barron JT, Gu L, Parrillo JE. Relation of NADH/NAD to contraction in vascular smooth muscle. Mol Cell Biochem 1999; 194: 283–290. [DOI] [PubMed] [Google Scholar]
- 30.Tahara EB, Barros MH, Oliveira GA, et al. Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. Faseb J 2007; 21: 274–283. [DOI] [PubMed] [Google Scholar]
- 31.Brown AM, Gordon D, Lee H, et al. Testing for linkage and association across the dihydrolipoyl dehydrogenase gene region with Alzheimer's disease in three sample populations. Neurochem Res 2007; 32: 857–869. [DOI] [PubMed] [Google Scholar]
- 32.Yao J, Hamilton RT, Cadenas E, et al. Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim Biophys Acta 2010; 1800: 1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frackowiak J, Mazur-Kolecka B, Kaczmarski W, et al. Deposition of Alzheimer's vascular amyloid-beta is associated with decreased expression of brain L-3-hydroxyacyl-coenzyme A dehydrogenase (ERAB). Brain Res 2001; 907: 44–53. [DOI] [PubMed] [Google Scholar]
- 34.Tong XK, Hamel E. Transforming growth factor-beta 1 impairs endothelin-1-mediated contraction of brain vessels by inducing mitogen-activated protein (MAP) kinase phosphatase-1 and inhibiting p38 MAP kinase. Mol Pharmacol 2007; 72: 1476–1483. [DOI] [PubMed] [Google Scholar]
- 35.Huot J, Houle F, Spitz DR, et al. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res 1996; 56: 273–279. [PubMed] [Google Scholar]
- 36.Meloche S, Landry J, Huot J, et al. Giasson. p38 MAP kinase pathway regulates angiotensin II-induced contraction of rat vascular smooth muscle. Am J Physiol Heart Circ Physiol 2000; 279: H741–H751. [DOI] [PubMed] [Google Scholar]
- 37.Hirade K, Kozawa O, Tanabe K, et al. Thrombin stimulates dissociation and induction of HSP27 via p38 MAPK in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 2002; 283: H941–H948. [DOI] [PubMed] [Google Scholar]
- 38.Dhar-Chowdhury P, Harrell MD, Han SY, et al. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the K(ATP) channel macromolecular complex and regulate its function. J Biol Chem 2005; 280: 38464–38470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koide M, Syed AU, Braas KM, et al. Pituitary adenylate cyclase activating polypeptide (PACAP) dilates cerebellar arteries through activation of large-conductance Ca(2+)-activated (BK) and ATP-sensitive (K ATP) K (+) channels. J Mol Neurosci 2014; 54: 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flagg TP, Enkvetchakul D, Koster JC, et al. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev 2010; 90: 799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin-Garrido A, Gonzalez-Ramos M, Griera M, et al. H2O2 regulation of vascular function through sGC mRNA stabilization by HuR. Arterioscler Thromb Vasc Biol 2011; 31: 567–573. [DOI] [PubMed] [Google Scholar]
- 42.Bonkale WL, Winblad B, Ravid R, et al. Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer's disease. Neurosci Lett 1995; 187: 5–8. [DOI] [PubMed] [Google Scholar]
- 43.Roher AE, Garami Z, Tyas SL, et al. Transcranial doppler ultrasound blood flow velocity and pulsatility index as systemic indicators for Alzheimer's disease. Alzheimers Dement 2011; 7: 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiffrin EL. Vascular stiffening and arterial compliance. Implications for systolic blood pressure. Am J Hypertens 2004; 17: 39S–48S. [DOI] [PubMed] [Google Scholar]
- 45.Yu D, Corbett B, Yan Y, et al. Early cerebrovascular inflammation in a transgenic mouse model of Alzheimer's disease. Neurobiol Aging 2012; 33: 2942–2947. [DOI] [PubMed]
- 46.Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr 2011; 5: 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh-Wilson LC, Allen PB, Watanabe T, et al. Characterization of the neuronal targeting protein spinophilin and its interactions with protein phosphatase-1. Biochemistry 1999; 38: 4365–4373. [DOI] [PubMed] [Google Scholar]
- 48.Tang DD. Intermediate filaments in smooth muscle. Am J Physiol Cell Physiol 2008; 294: C869–C878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Even-Ram S, Doyle AD, Conti MA, et al. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol 2007; 9: 299–309. [DOI] [PubMed] [Google Scholar]
- 50.Kohlstedt K, Kellner R, Busse R, et al. Signaling via the angiotensin-converting enzyme results in the phosphorylation of the nonmuscle myosin heavy chain IIA. Mol Pharmacol 2006; 69: 19–26. [DOI] [PubMed] [Google Scholar]
- 51.Levin EC, Acharya NK, Sedeyn JC, et al. Neuronal expression of vimentin in the Alzheimer's disease brain may be part of a generalized dendritic damage-response mechanism. Brain Res 2009; 1298: 194–207. [DOI] [PubMed] [Google Scholar]
- 52.Millioni R, Puricelli L, Iori E, et al. The effects of rosiglitazone and high glucose on protein expression in endothelial cells. J Proteome Res 2010; 9: 578–584. [DOI] [PubMed] [Google Scholar]
- 53.Saito S, Ihara M. New therapeutic approaches for Alzheimer's disease and cerebral amyloid angiopathy. Front Aging Neurosci 2014; 6: 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhat NR. Vasculoprotection as a convergent, multi-targeted mechanism of anti-AD therapeutics and interventions. J Alzheimers Dis 2015; 46: 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials