

Abstract
Cobalt-containing metal-on-metal hip replacements are associated with adverse reactions to metal debris (ARMD), including inflammatory pseudotumours, osteolysis, and aseptic implant loosening. The exact cellular and molecular mechanisms leading to these responses are unknown. Cobaltions (Co2+) activate human Toll-like receptor 4 (TLR4), an innate immune receptor responsible for inflammatory responses to Gram negative bacterial lipopolysaccharide (LPS).
We investigated the effect of Co2+-mediated TLR4 activation on human microvascular endothelial cells (HMEC-1), focusing on the secretion of key inflammatory cytokines and expression of adhesion molecules. We also studied the role of TLR4 in Co2+-mediated adhesion molecule expression in MonoMac 6 macrophages.
We show that Co2+ increases secretion of inflammatory cytokines, including IL-6 and IL-8, in HMEC-1. The effects are TLR4-dependent as they can be prevented with a small molecule TLR4 antagonist. Increased TLR4-dependent expression of intercellular adhesion molecule 1 (ICAM1) was also observed in endothelial cells and macrophages. Furthermore, we demonstrate for the first time that Co2+ activation of TLR4 upregulates secretion of a soluble adhesion molecule, sICAM-1, in both endothelial cells and macrophages. Although sICAM-1 can be generated through activity of matrix metalloproteinase-9 (MMP-9), we did not find any changes in MMP9 expression following Co2+ stimulation.
In summary we show that Co2+ can induce endothelial inflammation via activation of TLR4. We also identify a role for TLR4 in Co2+-mediated changes in adhesion molecule expression. Finally, sICAM-1 is a novel target for further investigation in ARMD studies.
Keywords: cobalt, TLR4, endothelium, inflammation, metal-on-metal, Immunology and Microbiology Section, Immune response, Immunity
INTRODUCTION
Metal-on-metal (MoM) hip replacements are associated with the development of adverse reactions to metal debris (ARMD), which includes inflammatory pseudotumours, soft tissue necrosis, osteolysis and resulting aseptic implant loosening. Peri-implant tissues are often infiltrated by monocytes, macrophages and lymphocytes (referred to as aseptic lymphocyte-dominated vasculitis-associated lesion, ALVAL) which is indicative of an inflammatory response. However the cellular and molecular mechanisms that underlie ARMD are not well-understood.
Co2+ from MoM implants activates human Toll-like receptor 4 (TLR4) [1-3], an innate immune receptor expressed on immune cells as well as endothelial and epithelial cells. The major ligand for TLR4 is lipopolysaccharide from Gram negative bacteria, and receptor activation causes adaptor protein recruitment and an intracellular signalling cascade that upregulates the activity of transcription factors including NFκB [3].
We have previously shown that activation of TLR4 by Co2+ increases the secretion of inflammatory cytokines, including interleukin-8 (IL-8) and chemokine (C-X-C motif) ligand 10 (CXCL10), in MonoMac 6 macrophages [4]. Previous studies investigating the inflammatory effects of Co2+ in endothelial cells have primarily focused on endothelial cells transfected with TLR4 and its co-receptor MD2 [3, 5], but few studies have investigated the effect of Co2+ on endogenous TLR4-expressing endothelial cell lines. Endothelial cells are exposed to Co2+ present in the blood of MoM hip replacement patients [6] and therefore understanding the cellular response is important in defining the causes of ARMD and identifying potential therapeutic targets for ARMD prevention.
In the present study we assessed the immune response of endothelial cells to Co2+, with a focus on the role of TLR4. We also investigated the effect of Co2+ on adhesion molecule expression by endothelial cells and macrophages because of their critical role in inflammatory process such as leukocyte binding and extravasation.
RESULTS
Co2+ activation of TLR4 increases IL-8 and IL-6 secretion
HMEC-1 cells were stimulated with 0.25-1mM cobalt chloride hexahydrate (Co2+) or 100ng/ml LPS for 24h and supernatant was collected for ELISA. IL-8 secretion was significantly increased by all concentrations of Co2+ (all p < 0.001 except 0.25mM where p = 0.026), peaking at 1300pg/ml with 1mM Co2+. The positive control LPS also increased IL-8 secretion (Figure 1A). IL-6 secretion was similarly upregulated by the agonists (all p < 0.001 except 0.25mM where p = 0.011) (Figure 1B).
Figure 1. Effect of Co2+ ions and TLR4 activation on cytokine secretion by HMEC-1.
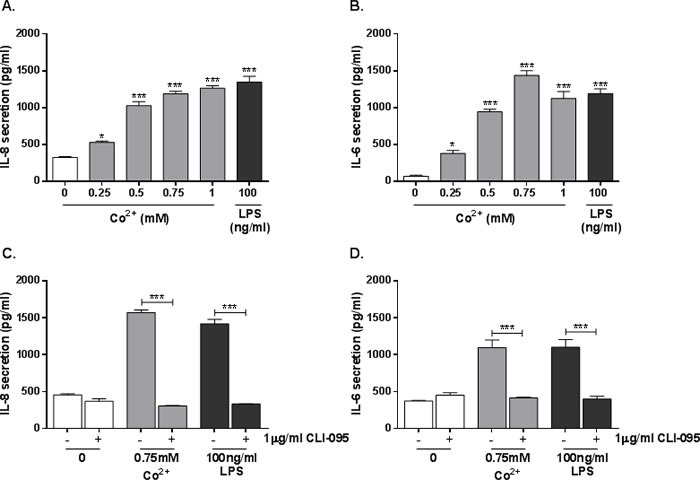
A. & B. HMEC-1 were stimulated with 0.25-1mM Co2+ or 100ng/ml LPS. A. IL-8 and B. IL-6 secretion were assessed by ELISA. C. & D. HMEC-1 were pre-treated with 1μg/ml CLI-095 followed by 24h stimulation with 0.75mM Co2+ or 100ng/ml LPS. C. IL-8 and D. IL-6 secretion was quantified by ELISA. All data is representative of three independent experiments.
To assess the role of TLR4 in the observed cytokine secretion, HMEC-1 were pre-incubated with 1μg/ml CLI-095 (a small molecule TLR4 antagonist) for 6h followed by stimulation with 0.75mM Co2+ or 100ng/ml LPS for 24h. IL-8 and IL-6 secretion were measured by ELISA. Pre-treatment with CLI-095 significantly decreased secretion of both cytokines in response to Co2+ (p < 0.001), showing that their release is TLR4-dependent. The cytokine release was not a result of Co2+-mediated cytotoxicity as trypan blue staining revealed no change in HMEC-1 viability following cobalt stimulation (Supplementary Material, Figure 6).
Co2+-mediated TLR4 activation increases ICAM1 expression in endothelial cells and macrophages
Endothelial cells are known to express adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1), which are essential in leukocyte extravasation in inflammation. We assessed the effect of Co2+ activation of TLR4 on ICAM1 expression in HMEC-1 and MonoMac 6 macrophages. Macrophages also express CAMs for cell-cell communication.
Co2+ induced a small but significant 3-fold upregulation in ICAM1 expression by HMEC-1 (p = 0.013) (Figure 2A) and a larger 35-fold upregulation in MonoMac 6 cells (p < 0.001) (Figure 2B). In both cell lines the response was found to be TLR4-dependent because it was inhibited by the TLR4 antagonist CLI-095 (both p < 0.001).
Figure 2. Effect of Co2+ and TLR4 activation on ICAM1 expression.
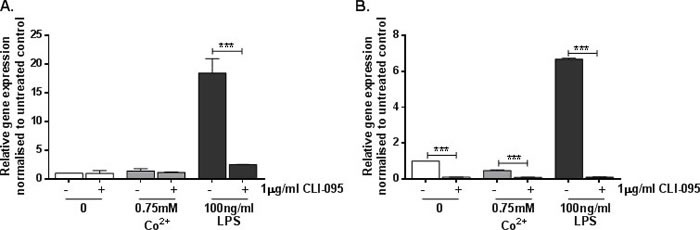
A. HMEC-1 and B. MonoMac 6 cells were stimulated with 1μg/ml CLI-095 for 6h prior to 24h stimulation with 0.75mM Co2+ or 100ng/ml LPS. RNA was isolated and cDNA synthesised by reverse transcription. ICAM1 expression was quantified by qRT-PCR. Data is representative of three independent experiments.
Co2+ increases secretion of sICAM-1 in a TLR4-dependent manner
In addition to its membrane-bound form, ICAM-1 can also be secreted as soluble ICAM-1 (sICAM-1). Given the TLR4-dependent increase in ICAM1 expression described in Figure 2, we hypothesised that sICAM-1 release would also be affected by Co2+. Secretion of sICAM-1 by stimulated HMEC-1 and MonoMac 6 cells was investigated by ELISA. Cells were stimulated with 0.25-1mM Co2+ or 100ng/ml LPS for 24h. In HMEC-1, sICAM-1 secretion was increased by Co2+ concentrations of 0.5mM and above (all p < 0.001), peaking at 900pg/ml with 1mM Co2+ stimulation (Figure 3A). LPS increased sICAM-1 release to more than 1000pg/ml (p < 0.001). In MonoMac 6 cells sICAM-1 release was elevated across all Co2+ concentrations, peaking at 1000pg/ml with 0.5mM treatment (p < 0.001) (Figure 3B). As in HMEC-1, LPS elicited more sICAM-1 release than Co2+.
Figure 3. Effect of Co2+ and TLR4 activation on sICAM-1 secretion.
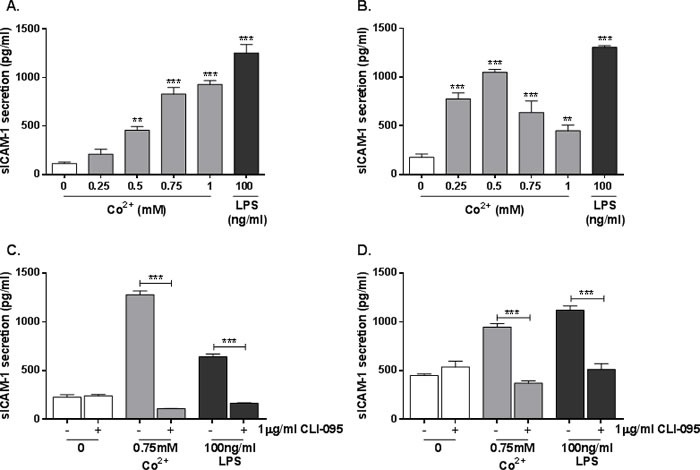
A. HMEC-1 or B. MonoMac 6 cells were stimulated with 0.25-1mM Co2+ for 24h and sICAM-1 secretion was measured by ELISA. C. HMEC-1 or D. MonoMac 6 cells were pre-incubated with 1μg/ml CLI-095 for 6h before stimulation with 0.75mM Co2+ or 100ng/ml LPS. All data is representative of three independent experiments.
HMEC-1 and MonoMac 6 cells were then pre-incubated with 1μg/ml CLI-095 for 6h followed by 24h stimulation with either 0.75mM Co2+ or 100ng/ml LPS. There was a significant decrease in Co2+ and LPS-mediated sICAM-1 secretion in both HMEC-1 (Figure 3C) and MonoMac 6 cells (Figure 3D) (p < 0.001 in all cases). This shows that sICAM-1 release in response to Co2+ and LPS is TLR4-dependent.
Co2+-mediated sICAM-1 secretion is not a result of MMP-9 activity
Previous studies have shown that sICAM-1 can be produced when mICAM-1 is cleaved by the gelatinase matrix metalloprotease-9 (MMP-9) [7]. LPS stimulates MMP-9 activity through activation of TLR4. We therefore investigated whether or not Co2+ activation of TLR4 also increases MMP-9 expression. HMEC-1 and MonoMac6 cells were pre-treated with 1μg/ml CLI-095 for 6h followed by 24h stimulation with 0.75mM Co2+ or 100ng/ml LPS. MMP9 expression was assessed using qRT-PCR.
HMEC-1 exhibited a significant 16-fold increase in MMP9 expression following stimulation with 100ng/ml LPS (p < 0.001) (Figure 4A). This was inhibited by CLI-095, showing that it is a TLR4-dependent effect (p < 0.001). In contrast there was no change in MMP9 expression in response to Co2+ (p = 0.999) (Figure 4A). A similar pattern was observed in MonoMac 6 cells; following LPS stimulation there was a 7-fold increase in MMP9 expression by (p < 0.001) (Figure 4B). CLI-095 inhibited this upregulated expression, showing that it is TLR4-dependent. However there was no increase in MMP9 expression in response to Co2+, although CLI-095 decreased its expression further.
Figure 4. Effect of Co2+ and LPS on MMP9 expression.
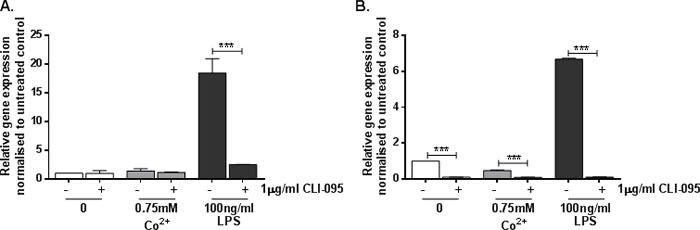
A. HMEC-1 or B. MonoMac6 cells were pre-treated with 1μg/ml CLI-095 for 6h prior to stimulation with 0.75mM Co2+ or 100ng/ml LPS for 24h. MMP9 expression was assessed by qRT-PCR. All data is representative of at least two independent experiments.
DISCUSSION
In the present study we describe a TLR4-dependent inflammatory response to Co2+ in human endothelial cells and macrophages. HMEC-1 exhibited significant increases in secretion of inflammatory cytokines IL-8 and IL-6 when stimulated with Co2+. This was inhibited by the TLR4 antagonist CLI-095, showing that the receptor is central to the responses. Previous studies have shown that Co2+ upregulates adhesion molecule expression [8-10], but have not demonstrated the exact signalling pathways involved. The data obtained in this study supports the findings of these studies and also indicates a previously unidentified role for TLR4 in Co2+-mediated ICAM1 expression in both endothelial cells and macrophages. Furthermore, for the first time a soluble adhesion molecule, sICAM-1, was detected in conditioned media from Co2+ and LPS-stimulated HMEC-1 and MonoMac 6 cells. CLI-095 inhibited sICAM-1 changes and consequently they are TLR4-dependent.
We investigated the effect of Co2+ on MMP9 expression because MMP-9 can cleave membrane-bound ICAM-1 resulting in the release of its soluble form, sICAM-1. In addition, MMP-9 can be regulated by LPS activation of TLR4 [11] and therefore it is possible that Co2+-mediated TLR4 activation results in MMP-9 activity and sICAM-1 generation. However, although LPS increased MMP9 expression in a TLR4-dependent manner, there was no change in expression in response to Co2+. The absence of any effect was consistent between HMEC-1 and MonoMac 6 cells. The lack of change in MMP9 expression following Co2+ stimulation suggests that the enzyme is not responsible for the changes in sICAM-1 secretion observed in response to Co2+. Other proteolytic enzymes potentially involved in sICAM-1 cleavage include serine proteases [12], neutrophil elastase [13], and cathepsin G [14]. However the effect of Co2+ on these factors remains to be elucidated.
sICAM-1 has previously been proposed as a marker of inflammation [15] that is cleaved to regulate inflammatory responses but studies are now reporting a broader role for sICAM-1, including promotion of angiogenesis and neovascularisation [16]. This is of particular interest to the present study because blood vessel formation is required for pseudotumour development, which is a major factor in ARMD. Soft tissue necrosis is also a common feature of ARMD and can result from vascular inflammation restricting oxygen supply to the tissues. The ability of Co2+ to cause an inflammatory response, including pro-inflammatory cytokine release, in endothelial cells may indicate that similar effects occur in vitro, which could result in ischaemia and subsequent tissue death.
A limitation of the present study is the high Co2+ concentrations that we have used to stimulate the cells. Even the concentrations at the lower end of the range are considerably higher than those detected in the serum and synovial fluid of patients with failed MoM implants [17-19]. However, the Co2+ concentrations used in our study are in line with those of similar in vitro studies of the inflammatory effects of metal ions [3, 10, 20, 21]. Hence, they are appropriate and relevant for this study.
A working model of the possible mechanisms indicated by our results is shown in Figure 5. In summary, we have shown that Co2+ has the potential to induce an inflammatory response in the endothelium through activation of TLR4. It also shows for the first time that Co2+ increases sICAM-1 secretion in a TLR4-dependent manner. Although the exact mechanism of its release remains unclear, sICAM-1 is an interesting target for further investigation in ARMD because of its previously described roles in angiogenesis, neovascularisation and tumour formation [16].
Figure 5. Working model.
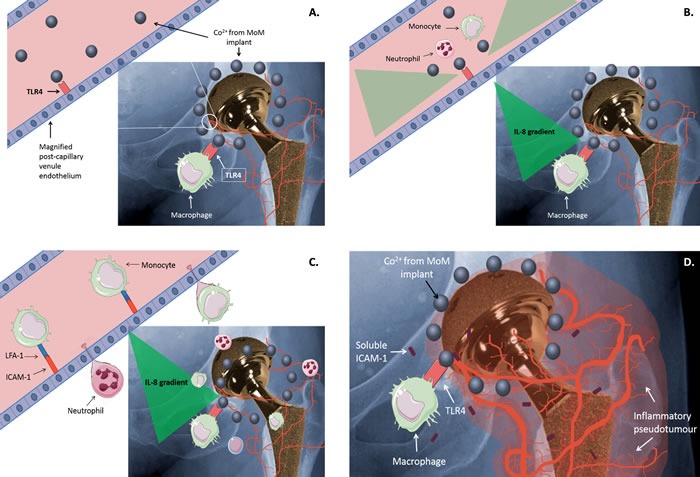
A. Co2+ released from metal-on-metal (MoM) hip implants activates TLR4 on immune cells such as macrophages. B. Co2+-mediated TLR4 activation results in the release of inflammatory cytokines and chemokines, including IL-8, by macrophages and endothelial cells. The generated cytokine/chemokine gradient attracts circulating leukocytes, such as monocytes and neutrophils. C. Co2+ activation of TLR4 on endothelial cells increases expression of ICAM-1, which promotes leukocyte binding via interaction with LFA-1. This in turn drives leukocyte extravasation. D. Migrated leukocytes, together with sICAM-1 released by endothelial cells and macrophages, promote an inflammatory response that may contribute to inflammation and pseudotumour development around a MoM implant.
MATERIALS AND METHODS
Cell culture
Human microvascular endothelial cells (HMEC-1) are derived from dermal foreskin. Cells were cultured in MCDB131 (Sigma-Aldrich, Gillingham, UK) medium supplemented with 10% foetal bovine serum (FBS), 50U/ml penicillin, 50μg/ml streptomycin, 10ng/ml epidermal growth factor (EGF) and 1μg/ml hydrocortisone (all Sigma-Aldrich).
MonoMac 6 cells are a human TLR4-expressing cell line derived from acute monocytic leukaemia. Cells were cultured as previously described [22].
Cell stimulation
Cells were stimulated with cobalt chloride hexahydrate (referred to as Co2+ in this study) in complete culture medium appropriate for each cell line. Complete culture medium was used as a negative control while 100ng/ml TLR4-specific LPS (Alexis Biochemicals, San Diego, USA) provided a positive control.
ELISA (IL-8, IL-6, sICAM-1)
Inflammatory cytokine secretion was quantified by enzyme-linked immunosorbent assay (ELISA). IL-6, IL-8 and sICAM-1 ELISA kits were purchased from Peprotech (London, UK) and assays performed as described previously [4].
qRT-PCR
Gene expression changes were assessed by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) using TaqMan primers and probes (ThermoFisher Scientific, Massachusetts, USA). RNA was isolated using a Qiagen RNeasy Mini kit (Qiagen, Venlo, Netherlands) and cDNA synthesised using Superscript III reverse transcriptase (ThermoFisher Scientific). Each qRT-PCR reaction contained 5μl TaqMan Gene Expression Mastermix (ThermoFisher Scientific), 2μl diluted cDNA template, 2.5μl nuclease-free H2O and 0.5μl TaqMan Gene Expression Assay (ThermoFisher Scientific). No template controls with nuclease-free H2O instead of cDNA were included. All reactions were performed in triplicate and target gene expression was normalised to GAPDH expression.
CLI-095
Inhibition of TLR4 was performed by pre-incubating cells for 6h with 1μg/ml CLI-095. CLI-095 (Invivogen, UK) is a small molecule TLR4 antagonist that binds to the intracellular domain of the receptor and prevents recruitment of downstream adaptor proteins.
Cytotoxicity assay
Cytotoxicity was assessed by trypan blue staining. Stimulated cells were resuspended in a small volume of supernatant and 10μl cell suspension was mixed with 10μl trypan blue dye. Staining was visualised on a Luna II automated cell counter (Logos Biosystems, Virginia, USA)
Statistical analysis
Statistical significance was calculated using a one-way analysis of variance (ANOVA). When samples were compared to an untreated control (Figures 1A, 1B, 3A, and 3B), Dunnett's test for multiple comparisons was performed. When comparing all samples to each other, Tukey's test for multiple comparisons was performed.
SUPPLEMENTARY MATERIALS FIGURES
Footnotes
CONFLICTS OF INTEREST
There is no conflict of interest
REFERENCES
- 1.Tyson-Capper AJ, Lawrence H, Holland JP, Deehan DJ, Kirby JA. Metal-on-metal hips: Cobalt can induce an endotoxin-like response. Annals of the Rheumatic Diseases. 2013;72(3):460–461. doi: 10.1136/annrheumdis-2012-202468. [DOI] [PubMed] [Google Scholar]
- 2.Potnis PA, Dutta DK, Wood SC. Toll-like receptor 4 signaling pathway mediates proinflammatory immune response to cobalt-alloy particles. Cellular Immunology. 2013;282(1):53–65. doi: 10.1016/j.cellimm.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Raghavan B, Martin SF, Esser PR, Goebeler M, Schmidt M. Metal allergens nickel and cobalt facilitate TLR4 homodimerization independently of MD2. EMBO Reports. 2012;13(12):1109–1115. doi: 10.1038/embor.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence H, Deehan D, Holland J, Kirby J, Tyson-Capper A. The immunobiology of cobalt: Demonstration of a potential aetiology for inflammatory pseudotumours after metal-on-metal replacement of the hip. Bone and Joint Journal. 2014;69B(9):1172–1177. doi: 10.1302/0301-620X.96B9.33476. [DOI] [PubMed] [Google Scholar]
- 5.Oblak A, Pohar J, Jerala R. MD-2 determinants of nickel and cobalt-mediated activation of human TLR4. PLoS ONE. 2015;10(3):e0120583. doi: 10.1371/journal.pone.0120583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodríguez De La Flor M, Hernández-Vaquero D, Fernández-Carreira JM. Metal presence in hair after metal-on-metal resurfacing arthroplasty. Journal of Orthopaedic Research. 2013;31(12):2025–2031. doi: 10.1002/jor.22450. [DOI] [PubMed] [Google Scholar]
- 7.Fiore E, Fusco C, Romero P, Stamenkovic I. Matrix metalloproteinase 9 (MMP-9/gelatinase B) proteolytically cleaves ICAM-1 and participates in tumor cell resistance to natural killer cell-mediated cytotoxicity. Oncogene. 2002;21(34):5213–5223. doi: 10.1038/sj.onc.1205684. [DOI] [PubMed] [Google Scholar]
- 8.Caicedo MS, Pennekamp PH, McAllister K, Jacobs JJ, Hallab NJ. Soluble ions more than particulate cobalt-alloy implant debris induce monocyte costimulatory molecule expression and release of proinflammatory cytokines critical to metal-induced lymphocyte reactivity. Journal of Biomedical Materials Research - Part A. 2010;93(4):1312–1321. doi: 10.1002/jbm.a.32627. [DOI] [PubMed] [Google Scholar]
- 9.Goebeler M, Meinardus-Hager G, Roth J, Goerdt S, Sorg C. Nickel chloride and cobalt chloride, two common contact sensitizers, directly induce expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and endothelial leukocyte adhesion molecule (ELAM-1) by endothelial cells. Journal of Investigative Dermatology. 1993;100(6):759–765. doi: 10.1111/1523-1747.ep12476328. [DOI] [PubMed] [Google Scholar]
- 10.Ninomiya JT, Kuzma SA, Schnettler TJ, Krolikowski JG, Struve JA, Weihrauch D. Metal Ions Activate Vascular Endothelial Cells and Increase Lymphocyte Chemotaxis and Binding. Journal of Orthopaedic Research. 2013;31(9):1484–1491. doi: 10.1002/jor.22377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Xu H, Sun B. Lipopolysaccharide regulates MMP-9 expression through TLR4/NF-κB signaling in human arterial smooth muscle cells. Molecular Medicine Reports. 2012;6(4):774–778. doi: 10.3892/mmr.2012.1010. [DOI] [PubMed] [Google Scholar]
- 12.Mendez MP, Morris SB, Wilcoxen S, Du M, Monroy YK, Remmer H, Murphy H, Christensen PJ, Paine R., 3rd Disparate mechanisms of sICAM-1 production in the peripheral lung: contrast between alveolar epithelial cells and pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294(4):L807–814. doi: 10.1152/ajplung.00398.2007. [DOI] [PubMed] [Google Scholar]
- 13.Champagne B, Tremblay P, Cantin A, St. Pierre Y. Proteolytic cleavage of ICAM-1 by human neutrophil elastase. Journal of Immunology. 1998;161(11):6398–6405. [PubMed] [Google Scholar]
- 14.Robledo O, Papaioannou A, Ochietti B, Beauchemin C, Legault D, Cantin A, King PD, Daniel C, Alakhov VY, Potworowski EF, St-Pierre Y. ICAM-1 isoforms: Specific activity and sensitivity to cleavage by leukocyte elastase and cathepsin G. European Journal of Immunology. 2003;33(5):1351–1360. doi: 10.1002/eji.200323195. [DOI] [PubMed] [Google Scholar]
- 15.Kovacs E. The serum levels of soluble intercellular adhesion molecule-1 (sICAM-1) and soluble gp130 (sgp130) in different tumour stages. Correlation between the two parameters in progression of malignancy. Biomedicine and Pharmacotherapy. 2005;59(9):498–500. doi: 10.1016/j.biopha.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 16.Yong Song G, Kim PN, Li HC, Elkin M, Kleinman HK. Stimulation of tumor growth by human soluble intercellular adhesion molecule-1. Cancer Research. 2001;61(10):4253–4257. [PubMed] [Google Scholar]
- 17.Langton DJ, Jameson SS, Joyce TJ, Webb J, Nargol AVF. The effect of component size and orientation on the concentrations of metal ions after resurfacing arthroplasty of the hip. Journal of Bone and Joint Surgery - Series B. 2008;90(9):1143–1151. doi: 10.1302/0301-620X.90B9.20785. [DOI] [PubMed] [Google Scholar]
- 18.Andrews RE, Shah KM, Wilkinson JM, Gartland A. Effects of cobalt and chromium ions at clinically equivalent concentrations after metal-on-metal hip replacement on human osteoblasts and osteoclasts: Implications for skeletal health. Bone. 2011;49(4):717–723. doi: 10.1016/j.bone.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Kwon YM, Ostlere SJ, McLardy-Smith P, Athanasou NA, Gill HS, Murray DW. “Asymptomatic” Pseudotumors After Metal-on-Metal Hip Resurfacing Arthroplasty. Prevalence and Metal Ion Study. Journal of Arthroplasty. 2011;26(4):511–518. doi: 10.1016/j.arth.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 20.Rachmawati D, Bontkes HJ, Verstege MI, Muris J, Von Blomberg BME, Scheper RJ, Van Hoogstraten IMW. Transition metal sensing by Toll-like receptor-4: Next to nickel, cobalt and palladium are potent human dendritic cell stimulators. Contact Dermatitis. 2013;68(6):331–338. doi: 10.1111/cod.12042. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt M, Raghavan B, Muller V, Vogl T, Fejer G, Tchaptchet S, Keck S, Kalis C, Nielsen PJ, Galanos C, Roth J, Skerra A, Martin SF, Freudenberg MA, Goebeler M. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nature Immunology. 2010;11(9):814–819. doi: 10.1038/ni.1919. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence H, Mawdesley A, Holland J, Kirby J, Deehan D, Tyson-Capper A. Targeting Toll-like receptor 4 prevents cobalt-mediated inflammation. Oncotarget. 2016;7(7):7578. doi: 10.18632/oncotarget.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.