

Abstract
Subarachnoid hemorrhage is a stroke subtype with particularly bad outcome. Recent findings suggest that constrictions of pial arterioles occurring early after hemorrhage may be responsible for cerebral ischemia and – subsequently – unfavorable outcome after subarachnoid hemorrhage. Since we recently hypothesized that the lack of nitric oxide may cause post-hemorrhagic microvasospasms, our aim was to investigate whether inhaled nitric oxide, a treatment paradigm selectively delivering nitric oxide to ischemic microvessels, is able to dilate post-hemorrhagic microvasospasms; thereby improving outcome after experimental subarachnoid hemorrhage. C57BL/6 mice were subjected to experimental SAH. Three hours after subarachnoid hemorrhage pial artery spasms were quantified by intravital microscopy, then mice received inhaled nitric oxide or vehicle. For induction of large artery spasms mice received an intracisternal injection of autologous blood. Inhaled nitric oxide significantly reduced number and severity of subarachnoid hemorrhage-induced post-hemorrhage microvasospasms while only having limited effect on large artery spasms. This resulted in less brain-edema-formation, less hippocampal neuronal loss, lack of mortality, and significantly improved neurological outcome after subarachnoid hemorrhage. This suggests that spasms of pial arterioles play a major role for the outcome after subarachnoid hemorrhage and that lack of nitric oxide is an important mechanism of post-hemorrhagic microvascular dysfunction. Reversing microvascular dysfunction by inhaled nitric oxide might be a promising treatment strategy for subarachnoid hemorrhage.
Keywords: Cerebral perfusion, early brain injury, microvasospasm, nitric oxide, subarachnoid hemorrhage
Introduction
Subarachnoid hemorrhage (SAH) is a subtype of stroke mainly caused by the rupture of a cerebral aneurysm. Even with treatment, the outcome following SAH is extremely poor for most patients. In the past delayed vasospasms, which develop later than five days following SAH, have been the focus of most experimental and clinical studies.1 Since, however, more than half of the hospitalized patients die within the first 48 h after SAH2 and reversing large artery spasms did not result in an improved outcome of SAH patients,3 other mechanisms potentially responsible for post-hemorrhage early brain injury (EBI) had to be taken into consideration.4 One key factor underlying experimental and clinical EBI is early cerebral ischemia.5,6 A putative mechanism responsible for cerebral ischemia following SAH are constrictions of pial arterioles as demonstrated in SAH patients7,8 and in experimental animals.9 So far, however, it is unknown which mechanisms cause microvascular dysfunction after SAH and whether restoration of the cerebral microcirculation affects outcome after SAH.
Several groups have suggested a possible role for nitric oxide (NO) for post-hemorrhage microvascular dysfunction.10–12 Due to the lack of techniques to specifically deliver NO to the microcirculation this hypothesis has not been investigated so far. Recently, we and others reported that inhaled nitric oxide (iNO) reaches the cerebral vasculature and is preferentially released in hypoxic/ischemic microvessels, where it induces arteriolar vasodilation.13 Due to this selectivity, iNO – unlike systemically applied NO donors – does not affect systemic blood pressure and has therefore no deleterious effect on cerebral perfusion pressure even under conditions of impaired cerebral autoregulation. Accordingly, the aim of the current study was to use iNO and investigate whether NO is involved in post-hemorrhage microvasospasms and whether microvascular dysfunction is responsible for the unfavorable outcome after SAH.
Materials and methods
All procedures performed on animals, group size calculation, and all statistical methods used to analyze in vivo data were reviewed and approved by the Government of Upper Bavaria (protocol numbers Az. 06-04 and Az. 66-08). Sample size was calculated using a standard statistical software package (Sigma Stat 3.0.; Jandel Scientific, Erkrath, Germany). The results of the study are reported in accordance with the ARRIVE guidelines.
Randomization and blinding
Following the induction of experimental SAH, each animal was randomly assigned to either the treatment group or the control group by drawing lots; NO inhalation was added by a person not involved in the performance of the experiment, data collection, or analysis. All surgical preparations were performed in a blinded fashion. Neurological testing, data collection, and data analysis were performed by a researcher who was blinded with respect to the treatment of the mice.
Husbandry
Mice were housed in groups of four in standard cages (207 × 140 × 265 mm, Macrolon II, Ehret Life Science Solutions, Emmendingen, Germany) under a 12-h day/12-h night cycle with free access to food and water. Cages contained cellulose tissue and a MouseHouse as an enrichment. Health screens and hygiene management checks were performed in accordance with FELASA (Federation of Laboratory Animal Science Associations) guidelines and recommendations.
Anesthesia and surgical preparation
Male C57BL/6 mice (6–8 weeks, weight 24–26 g; Charles River, Kisslegg, Germany) were anesthetized in a chamber containing 4% isoflurane (Halocarbon Laboratories, River Edge, NJ). Anesthesia was then continued which an intraperitoneal injection of medetomidine (0.5 mg/kg), midazolam (5 mg/kg), and fentanyl (0.05 mg/kg). The mice were oro-tracheally intubated and mechanically ventilated with 30% O2 and 70% room air (Minivent 845, Hugo Sachs Elektronik, March Hungstetten, Germany) as described previously.14 Ventilation was continuously monitored with microcapnometry (Micro Capnograph CI240, Columbus Instruments, Columbus, OH), with end-tidal CO2 held at 35–45 mmHg by actively adjusting respiration parameters. Systemic blood pressure and arterial blood gases were monitored via a catheter placed in the femoral artery. Body temperature was held at 37℃ using a feedback-controlled heating pad. After induction of SAH and a 15-min post-induction observation period, anesthesia was reversed via an intraperitoneal injection of the antagonists atipamezole (2.5 mg/kg), naloxone (1.2 mg/kg), and flumazenil (0.5 mg/kg), after which the animals were placed in an incubator at 33℃ for 24 h.
Measurement of regional cerebral blood flow
Regional cerebral blood flow (rCBF) was measured as described previously.15 Briefly, a laser Doppler probe (Perimed, Järfälla, Sweden) was placed perpendicular to the skull over the ipsilateral middle cerebral artery territory and fixed with cyanoacrylate glue. LD flux was measured and recorded continuously. CBF was recorded in all SAH experiments 10 min prior to and for 30 min after induction of SAH. In intravital epifluorescence microscopy (IVM) experiments (3 h after SAH), CBF measurement was re-applied after anesthesia induction and recorded continuously up to the end of the observation period.
Measurement of intracranial pressure (ICP)
ICP was measured prior and up to 15 min following SAH using a microchip-based probe (Codman ICP monitor, Codman, Ascot, U.K.) placed in the epidural space over the contralateral hemisphere as described previously.13 ICP was recorded in all SAH experiments 10 min prior to and for 30 min after induction of SAH.
Blood gas analysis
Arterial blood samples were collected prior to SAH and at the end of the observation period in all IVM animals; the samples were analyzed using a blood gas analyzer (Nr. 860, Chiron Diagnostics Corporation, East Walpole, MA).
Induction of subarachnoid hemorrhage
SAH was induced using the endovascular perforation technique, as described previously.9,15 In brief, a 5-0 filament was inserted into the left external carotid artery and advanced towards the Circle of Willis. Hemorrhage was confirmed by a rapid increase in intracranial pressure with a corresponding decrease in cerebral blood flow. The filament was then removed, and the external carotid artery was ligated. Sham-operated animals were treated identically, with the exception that the filament was not advanced far enough to induce hemorrhage.
The effects of iNO on macrovasospasm were measured using the cisterna magna blood injection model. First, 60 µl of autologous blood was collected by tail vein puncture; this blood was then injected into the cisterna magna under continuous ICP and cerebral blood flow control in anaesthetized, ventilated mice (see above); control animals received an injection of 60 µl physiological saline.
Intravital microscopy
IVM was performed as described previously.9,13 In brief, after induction of anesthesia and surgical preparation as described above a cranial window was prepared over the left middle cerebral artery territory (supplementary Figure 4A). Pial microvessels (10–80 µm in diameter, 15–30 vessel segments per animal, at least four measurement for each vessel category, n = 8 per group) were visualized by an intravenous injection of 150 µl FITC-dextran (MW 150 kDa, Sigma Aldrich, Deisenhofen, Germany, see supplementary Figure 4B for exemplary screenshot). For the iNO experimental series animals (were subjected to IVM 3 h after SAH; after recording baseline values for 30 min, the mice received either NO by inhalation (in 30% O2 and 70% room air; see below) or 30% O2 in room air for 60 min. After NO or control inhalation, the mice in both groups received 30% O2 in room air for an additional 30 min. The diameter of each vessel segment was recorded every 10 min and analyzed off-line using an imaging analysis software program (Leica Application Suite V3.8.0, Leica Microsystems, Solms, Germany) as described previously.9,12
Nitric oxide inhalation
Three hours following SAH induction, NO gas (264 mg/m3 NO in N2; Linde AG, Unterschleissheim, Germany) was delivered to anesthetized, intubated mice for 60 min at 50 ppm in 30% O2 and 70% room air as described previously.13,16 Control animals received 30% O2 and 70% room air (control gas).
For experiments requiring long-term iNO application (>3 h), awake animals were placed in custom-made air-tight glass containers containing 50 ppm NO in 30% oxygen and 70% room air. NO and NO2 concentrations were continuously monitored using an electrochemical sensor (ITX, Industrial Scientific Corporation, Oakdale, PA).
Assessment of brain water content
Brains were removed 24 h after induction of SAH and immediately cooled to 4℃ (n = 6/iNO group, n = 7/control group). The olfactory bulb and the cerebellum were removed, the cerebral hemispheres were separated, and the wet weight was measured. Dry weight was measured after drying the tissues at 100℃ for 24 h. Brain water content was then calculated as percent of total weight.
Quantification of neuronal damage
As previously described,17 the filament perforation model used in the current study leads to significant neuronal damage in the hippocampus which is thought to be responsible for long-term neurocognitive and behavioral deficits in SAH survivors. Three days after SAH, the animals (n = 8 mice per group) were transcardially perfused with 4% paraformaldehyde under deep chloralhydrate anesthesia. After the brains were removed and embedded in paraffin, 14 serial coronal sections (4 µm thickness) were cut and Nissl-stained. Pyknotic and viable neurons were then imaged in the CA1, CA2, and CA3 regions of the hippocampus, at 200 × magnification and quantified using a standard analysis software program (Olympus DP-Soft, analySIS, Olympus, Hamburg, Germany) as described previously.17
Bleeding time
Tail bleeding time was measured in anesthetized mice 30 min after inhalation of either NO (50 ppm) or control gas (n = 8 mice per group). A 0.5-cm segment of the tail was amputated, and the tail was placed horizontally in saline that was pre-warmed to 37℃. Bleeding was visually monitored, and the time until the bleeding stopped was measured and recorded.
Measurement of basilar artery diameter
Basilar artery spasm was evaluated 1 and 12 h following SAH induction (n = 8 mice per time point). Animals were exposed to NO (50 ppm) or control gas for 1 or 12 h. The animals were then transcardially perfused with 4% paraformaldehyde under deep chloralhydrate anesthesia. The brains were removed carefully and photographed using a dissecting microscope. The diameter of the basilar artery was measured using a calibrated imaging software program (Olympus DP-Soft, analySIS, Olympus, 4-6 separate measurements over 100 µm vessel per animal).
Neurological function
Neurological outcome (n = 9/control group, n = 8 iNO group) was assessed by testing motor function (e.g., the grasping reflex and the righting reflex), parameters reflecting general well-being (e.g., body weight and fur grooming), and vigilance as described previously.15 Animals that died before the conclusion of the assessment period were included in the analysis by receiving the highest score obtained on the respective day.
Statistical analysis
Except where indicated otherwise, all summary data are presented as the mean ± the standard error of the mean (SEM). For comparisons between two groups, we used the Mann Whitney rank sum test. Multiple groups were analyzed using the Kruskal-Wallis ANOVA on ranks, and repeated measurements were analyzed using the Friedman test. In cases in which a statistically significant difference was detected between groups, a post-hoc Dunnet test analysis was performed. All calculations were performed using a standard statistical software package (SigmaStat 3.0, Jandel Scientific, Erkrath, Germany). Differences between groups were considered to be significant at p < 0.05. Sample size calculations were performed with the following parameters: alpha error = 0.05, beta error = 0.2, calculated standard deviation ranged from 15 to 20% (depending on the parameter investigated), and biologically relevant difference = 30%.
Results
ICP, rCBF, and vessel diameter before initiation of treatment
Induction of SAH caused an immediate, large increase in ICP (supplementary Figure 1A) and a concomitant reduction in rCBF (supplementary Figure 1B) due to reduced cerebral perfusion pressure. ICP and rCBF did not differ between the groups.
Three hours after SAH and before randomization to treatment (i.e. in the baseline measurement) both experimental groups had the same degree of microvasospasm (supplementary Figure 1C). All arteriolar segments investigated displayed constriction of 20–30% compared with baseline; pre-capillary (i.e. diameter 10–20 µm) arterioles tended to be more constricted than larger arterioles. These findings are consistent with our previously published findings.9
NO inhalation reduces SAH-induced vasospasm in cerebral microvessels
We next examined the effect of iNO treatment on spastic cerebral arterioles induced by SAH. Treating these spastic vessels with iNO for 30 min (Figure 1(a)) significantly dilated cerebral arterioles which were spastic before the treatment. A time course shows that almost immediately after induction of iNO treatment, a large number of previously spastic vessels sections began to dilate and this was reflected by a decrease in the number of microvasospasms compared with baseline (Figure 1(b); open circles). Following NO inhalation, the number of microvasospasms was reduced by approximately 80% (i.e., no vasospasms were detectable in more than 80% of the vessels measured. Interestingly, the microvessels remained dilated even after cessation of iNO treatment, suggesting that dilatation of spastic microvessels by exogenous NO restored normal vessel function, at least temporarily. In contrast, the number of vessels with microvasospasms did not change significantly in the control group (Figure 1(b); closed circles).
Figure 1.

NO inhalation improves posthemorrhagic microcirculation. NO inhalation caused a near immediate and long-lasting vasodilation of spastic vessels. (a) Left: a baseline scan after SAH and prior to iNO treatment; right: the same vessel segment at t = 60 min, i. e. after 30 min of iNO treatment. (b) The number of spastic vessel segments (expressed as percent relative to baseline) was significantly decreased by iNO treatment. (c) 60 min after the start of IVM, i.e. 4 h after onset of SAH, spasm severity (i.e., the grade of constriction) was significantly lower in the iNO-treated vessels compared to control-treated vessels; this difference in constriction was largest in the smallest-diameter vessels but was significant in all vessel segments examined.
In the remaining vessels in which microvasospasms were still detectable, the degree of vasoconstriction was also significantly reduced in the iNO-treated vessels compared with the control vessels (Figure 1(c)). Whereas the severity of vasoconstriction in the control vessels was unchanged over time (20–30% constriction; Supplementary Figures 1 and 1C; closed bars), the degree of microvasospasm in the mice that received iNO was reduced to only 10–15%.
In contrast to the arterial vessels, the diameter of the venous vessels was unchanged throughout the observation period (supplementary Figure 2). In addition, physiological parameters measured at the end of the experiments did not differ between the iNO-treated and control groups (supplementary Table 1). Importantly, iNO had no effect on systemic blood pressure (Figure 2(a)) or hemostasis (Figure 2(b)), suggesting that delivering NO by inhalation may be safer than systemically applied NO donors, which can cause systemic hypotension.
Figure 2.
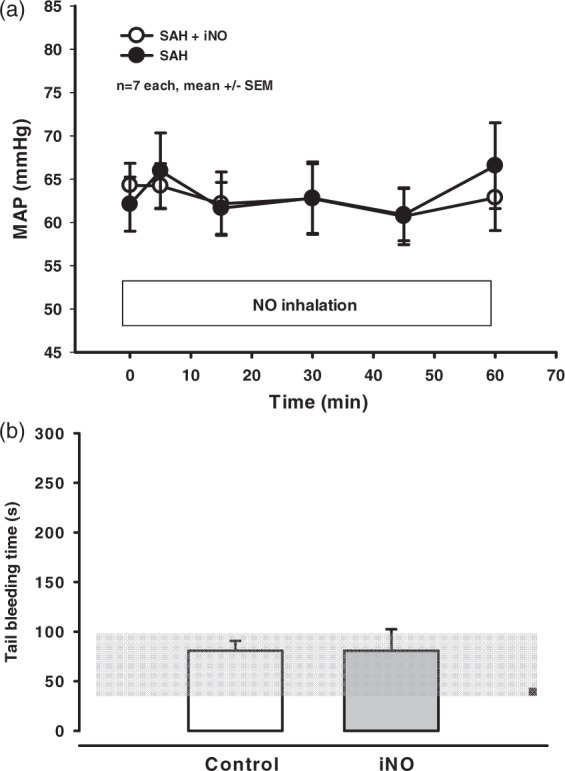
iNO treatment does not affect MAP or systemic coagulation. (a) Mean arterial blood pressure (MAP) was measured over time in iNO-treated and control-treated mice. (b) Tail bleeding time was measured in mice treated with iNO for 60 min and control mice (n = 5−7 each, mean +/− SEM).
NO inhalation reduces vasospasm in large cerebral arteries
Since next to spasms of the cerebral microcirculation SAH causes also acute macrovasospasms, we also investigated the effect of NO inhalation on spasms in larger cerebral vessels. Because the intraluminal filament model does not reliably cause spasms in large vessels, we induced SAH using the cisterna magna injection model instead, which produces a consistent spastic constriction of the basilar artery. The diameter of the basilar artery was significantly decreased within 1 h of blood injection and decreased further by 12 h post-injection (Figure 3). Treating mice with iNO continuously starting 10 min after SAH induction significantly decreased the vasoconstriction at 12 h post-injection (Figure 3). However, the effect is lost 24 h after SAH (Figure 3, bars on the right).
Figure 3.

NO inhalation reduces basilar arterial spasm. (a) SAH was induced by injecting blood into the cisterna magna; saline was injected as a sham treatment. Representative photomicrographs of a saline-injected brain (left), an SAH-induced brain (middle), and the brain of an animal that received iNO treatment for 12 h following SAH induction (right). (b) Quantification of the diameter of the basilar artery in saline-injected (Sham) mice and SAH-induced mice after 1 and 12 h with or without iNO treatment. The positive effect of iNO observed 12 h after SAH is abated at 24 and 48 h (data not shown).
NO inhalation reduces the development of brain edema and neuronal cell death
We next examined whether the positive effect with respect to cerebral microvasospasms translated into improved outcome. We induced SAH, treated the mice for 24 h with either iNO or control gas (starting 1 h after induction of hemorrhage) and then measured brain edema formation and neuronal viability three and seven days after SAH, respectively. As above, SAH induction and severity were similar between the iNO and control groups (supplementary Figure 3). SAH-induced brain edema (measured as brain water content) was significantly lower in the iNO-treated animals compared with control-treated mice (Figure 4(a)). In addition, neuronal survival was significantly higher in the CA3 hippocampal region of the iNO-treated group (Figure 4(b)).
Figure 4.

iNO treatment reduces SAH-induced brain damage. (a) 72 h after SAH, brain edema (measured as brain water content) was significantly higher in control animals compared with sham (non-SAH) animals (dotted line). iNO treatment significantly reduced brain water content in both hemispheres. (b) 7 days after SAH, neuronal survival (measured as the number of viable cells per ROI) was significantly increased in the CA3 hippocampal region.
NO inhalation improves neurological function and increases survival following SAH
In control mice, SAH caused long-lasting neurological deficits (Figure 5(a); closed circles) and 30% mortality within two days of hemorrhage (Figure 5(b); dashed line). In contrast, treating mice with NO inhalation significant reduced these neurological deficits (Figure 5(a); open circles), after seven days the iNO-treated mice exhibited virtually no neurological impairment. This improved neurological function was reflected by a lack of mortality (i.e. 100% survival) in the iNO-treated group (Figure 5(b); solid line).
Figure 5.

iNO treatment improves outcome following experimental SAH. (a). After SAH (day 0), control animals developed severe neurological deficits measured using a multivariate neurological score (higher scores indicate poorer performance). This impaired neurological performance remained significant through the observation period. In contrast, neurological outcome was significantly better in iNO-treated animals from the third day after SAH, with the iNO-treated mice reaching baseline performance by day 6. (b) Survival plots of mice that were subjected to SAH, then iNO-treated or control-treated for 23 h. The 72-h survival rate in the control-treated mice was 70%, whereas the survival rate in the iNO-treated mice was 100%.
Discussion
Our results show that treating mice subjected to SAH with iNO reduces the number of spastic microvessels by more than 80%, decreases the formation of brain edema, prevents neuronal cell death and mortality, and improves outcome. The reduction in the severity and number of microvasospasms persisted even after iNO treatment was stopped, suggesting that inhaling NO may not only transiently dilate spastic microvessels but also has a long-term effect by replenishing depleted endothelial NO stores in the early post-hemorrhagic phase. The present data corroborate the previously shown protective effect of iNO after SAH, cerebral ischemia, and traumatic brain injury13,16,18 and demonstrate the underlying mechanisms of this protection, i.e. the restoration of the cerebral microcirculation. Thereby the current results demonstrate the utmost importance of an impaired cerebral microcirculation for the development of post-hemorrhage brain damage. To our knowledge, this is the first report demonstrating that reversing spasms of pial arterioles within the first few hours after hemorrhage blunts mortality and significantly improves outcome after SAH.
Despite significant therapeutic advances, e.g. endovascular techniques for the prevention of re-bleedings, SAH-related mortality has not decreased significantly in recent years.19 One of the main reasons for this situation is that the mechanisms responsible for post-hemorrhage cerebral ischemia, a crucial event in the pathophysiology of SAH, are still unknown.20 For many years, it was believed that the large artery spasms occurring later than five days after the bleeding are the main reason for cerebral ischemia in SAH patients.21 Recent clinical trials using the endothelin receptor antagonist clazosentan, however, showed that while the drug effectively treated angiographic vasospasm patient outcome was not significantly improved.3 Since delayed ischemia does not seem to be responsible for the majority of SAH-induced mortality, the interest shifted towards earlier ischemic changes, which occur within the first few hours and days after SAH. Early post-hemorrhage ischemia is independent of cerebral perfusion pressure and in the majority of reported cases the available diagnostic tools did not reveal any obvious pathology of large cerebral vessels such as vasospasm or thrombosis.22 Thus, the cerebral microcirculation was hypothesized to be a possible vascular bed responsible for early ischemia following subarachnoid hemorrhage.21,23,24
Evidence that the cerebral microcirculation is indeed impaired already within the first hours after SAH came from studies performed by Uhl and colleagues7 and later Pennings et al.,8 who described “pearl string-like spasms” of pial arteries in patients undergoing surgery after SAH. We recently studied this phenomenon experimentally and found persistent constrictions of pial arterioles in mice subjected to SAH.9 This effect was mainly observed in pial arterioles which came into contact with subarachnoid blood. It appeared as early as 3 h and lasted for up to 72 h after hemorrhage.9 Attempts to dilate these vessels with inhalation of CO2, a selective and potent dilator of cerebral vessels, revealed that pial vessels becoming spastic after SAH were unresponsive to CO2.12 Since CO2 mediates vasodilatation though the activation of nitric oxide synthases, we hypothesized that lack of NO might be the underlying mechanism of post-hemorrhage pial artery spasms.
Actually, several other reports also suggested that local NO depletion may be involved in the pathogenesis of cerebral ischemia after SAH. Studies of ischemic stroke25,26 and traumatic brain injury27 have shown that cerebral ischemia causes a rapid decrease in cerebral NO concentrations by inhibiting the expression and activity of endogenous nitric oxide synthase (NOS). Following subarachnoid hemorrhage, reduced NO levels and impaired function of endothelial NOS (eNOS) and neuronal NOS (nNOS) – both of which are expressed constitutively in the brain – play a key role in the pathogenesis of delayed vasospasm, which, however, occurs only later than five days after hemorrhage.28,29 Furthermore, genetic polymorphisms of eNOS that lead to enzyme dysfunction increase the susceptibility to delayed post-hemorrhagic vasospasm, resulting in poorer neurological outcome following SAH.30 The lack of NO seems to start very early after SAH; Sehba and coworkers showed that NO concentrations in brain tissue drop almost immediately after the induction of experimental SAH.31,32
Based on these considerations, several groups investigated whether replenishing NO stores after SAH may be neuroprotective. Administering the NO precursor L-arginine,32,33 the NO donor S-nitrosoglutathione (GSNO),34 or giving NO by inhalation18 significantly improved histopathological or behavioral outcome following experimental SAH. However, GSNO caused a sharp – albeit transient – decrease in systemic blood pressure,34 a phenomenon that also occurs following the application of L-arginine.35 Furthermore, none of the studies using NO administration performed so far investigated by which mechanism NO exerted its neuroprotective effect. Hence, it is still unclear why administration of NO improved the outcome after SAH.
The current study addressed this issue by administering NO by inhalation while observing the pial microcirculation. Thereby we were able to show that iNO reduced the number of pial artery spasms by more than 80% and dilated the remaining 20% of spastic vessels by approximately 50%. This demonstrates that restoration of pial artery diameter 3 h after the bleeding results in less brain edema formation, lack of mortality, reduced hippocampal damage, and improved neurological outcome seven days after SAH. Accordingly, this is the first study suggesting that early pial artery spasms are responsible for late brain damage and unfavorable outcome following SAH.
In addition to showing the importance of pial arteriolar perfusion for the outcome after SAH, the results of the current study suggest that the mechanism responsible for SAH-induced microvasospasms is indeed a lack of NO at the neuro-vascular unit. This conclusion is based on the fact that inhalation of NO delivers NO to vascular beds which are hypoperfused thereby dilating these vessels and increasing perfusion.13 Accordingly, successful dilatation of spastic vessels with iNO is a strong indication that these vessels lack NO. Admittedly, this is just indirect evidence and further experiments directly measuring NO in vivo or in isolated vessels are necessary to ultimately prove this assumption.
In practical terms iNO has the great advantage that it is an FDA and EMA approved drug. It was originally developed to treat pulmonary vasoconstriction and proved for 15 years to be safe for the treatment of pulmonary hypertension and acute respiratory distress syndrome in newborn and adult patients.36 As mentioned before and as shown in the current study, another practical advantage of iNO over all other systemic NO delivery strategies is its lack of adverse effects on systemic blood pressure as demonstrated experimentally13,16 and clinically.37 Increased bleeding time was initially reported as a possible side-effect of iNO;38 however, later on it was shown that iNO has no negative effects on coagulation39 and that it can even be used safely during major surgery,40 findings corroborated by our current results.
In summary, our results demonstrate that administration of NO by inhalation (iNO) resolves microvasospasms of pial arterioles and dramatically improves outcome following experimental SAH. From these findings, we conclude that early spasms of the cerebral microcirculation play a causal role for the outcome after SAH and that these spasms are caused by a lack of vascular NO. Based on its excellent safety profile, iNO may therefore represent a novel, safe, and effective therapeutic concept for the treatment of brain damage following subarachnoid hemorrhage with high translational potential.
Supplementary Material
Acknowledgements
Data contained in this paper are part of the doctorial theses of S. Feiler, A. Dienel, and F. Müller.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the GEMI Fund and the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
NAT, SF, AD, FM, NH, BF performed all experiments, NAT, SF, AD, FM, KS, ST are responsible for data analysis, the manuscript was written by NAT and NP and edited by JS; NAT, NP, KS, ST planned experiments and did sample size calculation. NP developed the initial idea of inhaled NO as a cerebrovascular agent and ideated all subsequent experiments.
Supplementary information
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data.
References
- 1.Mayberg MR, Batjer HH, Dacey R, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage. A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Circulation 1994; 90: 2592–2605. [DOI] [PubMed] [Google Scholar]
- 2.Hop JW, Rinkel GJ, Algra A, et al. Case-fatality rates and functional outcome after subarachnoid hemorrhage: a systematic review. Stroke 1997; 28: 660–664. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Li YM, Li W, et al. Effect of clazosentan in patients with aneurysmal subarachnoid hemorrhage: a meta-analysis of randomized controlled trials. PLoS One 2012; 7: e47778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sehba FA, Hou J, Pluta RM, et al. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol 2012; 97: 14–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke 1995; 26: 1086–1091. [DOI] [PubMed] [Google Scholar]
- 6.Schubert GA, Seiz M, Hegewald AA, et al. Acute hypoperfusion immediately after subarachnoid hemorrhage: a xenon contrast-enhanced CT study. J Neurotrauma 2009; 26: 2225–2231. [DOI] [PubMed] [Google Scholar]
- 7.Uhl E, Lehmberg J, Steiger HJ, et al. Intraoperative detection of early microvasospasm in patients with subarachnoid hemorrhage by using orthogonal polarization spectral imaging. Neurosurgery 2003; 52: 1307–1315. [DOI] [PubMed] [Google Scholar]
- 8.Pennings FA, Bouma GJ, Ince C. Direct observation of the human cerebral microcirculation during aneurysm surgery reveals increased arteriolar contractility. Stroke 2004; 35: 1284–1288. [DOI] [PubMed] [Google Scholar]
- 9.Friedrich B, Muller F, Feiler S, et al. Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: an in-vivo microscopy study. J Cereb Blood Flow Metab 2012; 32: 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sehba FA, Bederson JB. Nitric oxide in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl 2011; 110: 99–103. [DOI] [PubMed] [Google Scholar]
- 11.Hanggi D, Steiger HJ. Nitric oxide in subarachnoid haemorrhage and its therapeutics implications. Acta Neurochir (Wien) 2006; 148: 605–613. [DOI] [PubMed] [Google Scholar]
- 12.Friedrich B, Michalik R, Oniszczuk A, et al. CO2 has no therapeutic effect on early microvasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab 2014; 34: e1–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terpolilli NA, Kim SW, Thal SC, et al. Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circ Res 2012; 110: 727–738. [DOI] [PubMed] [Google Scholar]
- 14.Thal SC, Sporer S, Schmid-Elsaesser R, et al. Inhibition of bradykinin B2 receptors before, not after onset of experimental subarachnoid hemorrhage prevents brain edema formation and improves functional outcome. Crit Care Med 2009; 37: 2228–2234. [DOI] [PubMed] [Google Scholar]
- 15.Feiler S, Friedrich B, Scholler K, et al. Standardized induction of subarachnoid hemorrhage in mice by intracranial pressure monitoring. J Neurosci Methods 2010; 190: 164–170. [DOI] [PubMed] [Google Scholar]
- 16.Terpolilli NA, Kim SW, Thal SC, et al. Inhaled nitric oxide reduces secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab 2013; 33: 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thal SE, Zhu C, Thal SC, et al. Role of apoptosis inducing factor (AIF) for hippocampal neuronal cell death following global cerebral ischemia in mice. Neurosci Lett 2011; 499: 1–3. [DOI] [PubMed] [Google Scholar]
- 18.Li YS, Shemmer B, Stone E, et al. Neuroprotection by inhaled nitric oxide in a murine stroke model is concentration and duration dependent. Brain Res 2013; 1507: 134–145. [DOI] [PubMed] [Google Scholar]
- 19.Sandvei MS, Mathiesen EB, Vatten LJ, et al. Romundstad PR Incidence and mortality of aneurysmal subarachnoid hemorrhage in two Norwegian cohorts, 1984-2007. Neurology 2011; 77: 1833–1839. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa M, Kusaka G, Yamaguchi N, et al. Platelet and leukocyte adhesion in the microvasculature at the cerebral surface immediately after subarachnoid hemorrhage. Neurosurgery 2009; 64: 546–553. [DOI] [PubMed] [Google Scholar]
- 21.Pluta RM, Hansen-Schwartz J, Dreier J, et al. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res 2009; 31: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schubert GA, Seiz M, Hegewald AA, et al. Hypoperfusion in the acute phase of subarachnoid hemorrhage. Acta Neurochir Suppl 2011; 110: 35–38. [DOI] [PubMed] [Google Scholar]
- 23.Plesnila N. Pathophysiological role of global cerebral ischemia following subarachnoid hemorrhage: the current experimental evidence. Stroke Res Treat 2013; 2013: 651958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction? Stroke 2009; 40: S86–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalkara T, Moskowitz MA. The complex role of nitric oxide in the pathophysiology of focal cerebral ischemia. Brain Pathol 1994; 4: 49–57. [DOI] [PubMed] [Google Scholar]
- 26.Huang Z, Huang PL, Ma J, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab 1996; 16: 981–987. [DOI] [PubMed] [Google Scholar]
- 27.Cherian L, Hlatky R, Robertson CS. Nitric oxide in traumatic brain injury. Brain Pathol 2004; 14: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pluta RM. Dysfunction of nitric oxide synthases as a cause and therapeutic target in delayed cerebral vasospasm after SAH. Acta Neurochir Suppl 2008; 104: 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vellimana AK, Milner E, Azad TD, et al. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage-induced cerebral vasospasm. Stroke 2011; 42: 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khurana VG, Fox DJ, Meissner I, et al. Update on evidence for a genetic predisposition to cerebral vasospasm. Neurosurg Focus 2006; 21: E3. [DOI] [PubMed] [Google Scholar]
- 31.Sehba FA, Schwartz AY, Chereshnev I, et al. Acute decrease in cerebral nitric oxide levels after subarachnoid hemorrhage. J Cereb Blood Flow Metab 2000; 20: 604–611. [DOI] [PubMed] [Google Scholar]
- 32.Sun BL, Zhang SM, Xia ZL, et al. L-arginine improves cerebral blood perfusion and vasomotion of microvessels following subarachnoid hemorrhage in rats. Clin Hemorheol Microcirc 2003; 29: 391–400. [PubMed] [Google Scholar]
- 33.Yang MF, Sun BL, Xia ZL, et al. Alleviation of brain edema by L-arginine following experimental subarachnoid hemorrhage in a rat model. Clin Hemorheol Microcirc 2003; 29: 437–443. [PubMed] [Google Scholar]
- 34.Sehba FA, Ding WH, Chereshnev I, et al. Effects of S-nitrosoglutathione on acute vasoconstriction and glutamate release after subarachnoid hemorrhage. Stroke 1999; 30: 1955–1961. [DOI] [PubMed] [Google Scholar]
- 35.Bath PM, Willmot M, Leonardi-Bee J, et al. Nitric oxide donors (nitrates), L-arginine, or nitric oxide synthase inhibitors for acute stroke. Cochrane Database Syst Rev 2002CD000398. [DOI] [PubMed]
- 36.Bloch KD, Ichinose F, Roberts JD, Jr., et al. Inhaled NO as a therapeutic agent. Cardiovasc Res 2007; 75: 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frostell C, Fratacci MD, Wain JC, et al. Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation 1991; 83: 2038–2047. [DOI] [PubMed] [Google Scholar]
- 38.Samama CM, Diaby M, Fellahi JL, et al. Inhibition of platelet aggregation by inhaled nitric oxide in patients with acute respiratory distress syndrome. Anesthesiology 1995; 83: 56–65. [DOI] [PubMed] [Google Scholar]
- 39.Albert J, Norman M, Wallen NH, et al. Inhaled nitric oxide does not influence bleeding time or platelet function in healthy volunteers. Eur J Clin Invest 1999; 29: 953–959. [DOI] [PubMed] [Google Scholar]
- 40.Della RG, Coccia C. Nitric oxide in thoracic surgery. Minerva Anestesiol 2005; 71: 313–318. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.