

Abstract
Cerebral edema represents a major threat following traumatic brain injury. However, therapeutic measures for control of intracranial pressure alone have failed to restore cerebral metabolism and improve neurological outcome. Since mitochondrial damage results in ATP depletion and deactivation of membrane ionic pumps, we hypothesized that modulation of ATP bioavailability may directly affect cytotoxic edema. Intracranial pressure measurements were performed in Sprague-Dawley rats treated by intraperitoneal injection of dimethylsulfoxide (vehicle), cyclosporine A (CsA), or Oligomycin B (OligB) following cortical contusion and further correlated with water content, mitochondrial damage, and electron microscopic assessment of neuronal and axonal edema. As hypothesized, ultra-structural figures of edema closely correlated with intracranial pressure elevation, increased water content and mitochondrial membrane permeabilization expressed by loss of transmembrane mitochondrial potential. Further, mitochondrial damage evidenced ultra-structurally by figures of swollen mitochondria with severely distorted cristae correlated with both cytotoxic edema and mitochondrial dysfunction. Importantly, cerebral edema and mitochondrial impairment were significantly worsened by treatment with OligB, whereas a noticeable improvement could be observed in animals that received injections of CsA. Since OligB and CsA are responsible for symmetrical and opposite effects on oxidative metabolism, these findings support the hypothesis of a causative relationship between edema and mitochondrial function.
Keywords: Cerebral metabolism, cytotoxic edema, mitochondria, mitochondrial permeability transition, traumatic brain injury
Introduction
Traumatic brain injury (TBI) represents the leading cause of death and disability caused by all injuries in western countries, resulting in estimated annual direct and indirect costs of $76.5 billion. Although traumatic and non-recursive in nature, TBI truly represents an evolving disease responsible for a wide array of secondary cellular and molecular events that may considerably worsen the initial condition of the patient as pointed by Reilly et al.1 in their famous observation: the patient who talk and die. Among the deleterious events triggered by the injury, cerebral edema is considered to be one of the most prominent threats during the clinical course.2,3 Responsible for swelling of the brain encased within the skull, edema results in elevation of the intracranial pressure (ICP) with subsequent impairment of cerebral perfusion and metabolism.4–6 Accordingly, ICP control has remained over the years the mainstay of management of neurotrauma, either based on the use of hyperventilation, hypertonic solutions, or decompressive craniectomy. Accumulating evidence, however, has shown that failure of oxidative metabolism is not of ischemic origin in most instances7–9 or may even precede ICP elevation.10 This may explain, in turn, why the current therapeutic approach to TBI, mechanistic in nature, has failed in recent studies to show a definite impact on cerebral metabolism7,11,12 or neurological outcome.13
Cerebral edema represents the consequence of two distinct biological events: disruption of the blood–brain barrier known as vasogenic edema and cellular or cytotoxic edema. Both events were recently shown to be equally common during the course of post-traumatic illness,14 although cytotoxic edema is considered as prominent during the early post-traumatic period.15 Cytotoxic edema represents the consequence of a massive cellular influx of Na+, Ca+, and water triggered by glutamate release and subsequent disruption of phospholipid membranes by calcium-activated phospholipase. As a protective physiological mechanism, calcium is stored within the mitochondrial matrix, where its accumulation eventually results in induction of the mitochondrial permeability transition pore (mPTP) with loss of mitochondrial membrane integrity and transmembrane potential (ΔΨm) reflecting the proton gradient essential for activation of ATP synthase.16 ATP depletion, in turn, will be responsible for failure of energy-dependent ionic pumps, leaving the influx of water and solutes uncontrolled.17,18 Cytotoxic edema and failure of oxidative metabolism may thus represent two different but interconnected aspects of mitochondrial damage. Accordingly, it may be hypothesized that therapeutic mito-protective measures may both contribute to restoration of ATP cellular levels and subsequent control of cytotoxic edema through re-activation of cation pumps.
The purpose of the present study was to further investigate the relationship between cerebral metabolism, ATP production, and cellular edema.
Materials and methods
Brain injury model
All animal procedures were approved by the Bar-Ilan University Animal Care Committee and were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals as well as Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. During the study, the animals were housed in groups of 2–3 rats in a sterilized solid bottom cage with contact bedding under controlled temperature and 12:12 h light/dark cycle and maintained on standard pellet diet and water supplied ad libitum. All efforts were made to maintain animals suffering to minimum and lower the number of animals used as much as possible.
The model of brain injury used was based on a modification of the impact acceleration model described by Marmarou et al.19 Sprague-Dawley male rats (250–300 g) were anesthetized by intraperitoneal injection of Equithesin (4 ml/kg of body weight) after diamond burr in a circular fashion in order to create a 4 mm diameter hole without injuring the dura. A stainless steel blunt rivet of 1 cm diameter and 2 mm depth was then attached to the skull opening, while the head was placed on a foam bed with the disc centered immediately under the lower end of a plexiglass tube (Figure 1). Through the tube, a 900 g weight was dropped from a 60 cm height down to the rivet. The rivet was then removed and the scalp was then sutured. The rat was allowed to recover from anesthesia in an individual cage, under 12 h light/dark cycles with free access to food and water. No mortality was related to the model used.
Figure 1.
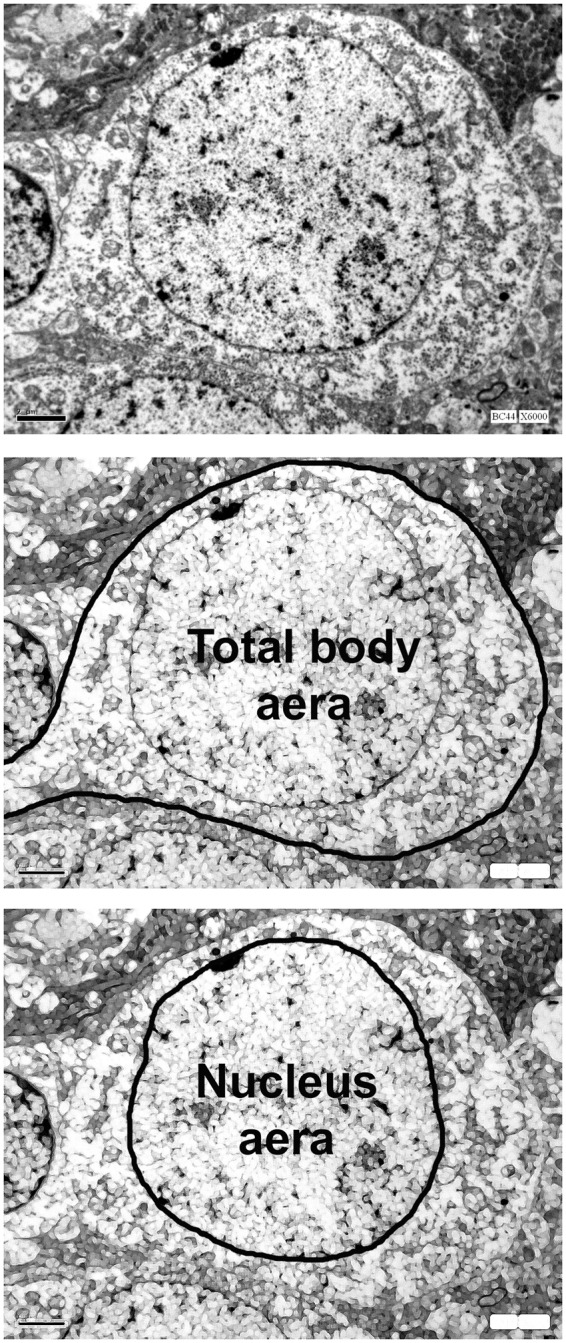
Quantitative assessment of neuronal cytotoxic edema was based on measurements of the respective surface areas of the neuron body (middle) and the nucleus (low) performed on microphotographs obtained at ×6000 magnification. Cytoplasm area was defined as the difference between them. Finally, cellular edema was expressed as the ratio between cytoplasm area over that of the nucleus.
Study design
Following the injury, animals were randomly allocated to three groups of eight rats each as follows: group 1 — dimethylsulfoxide 1% in saline (DMSO–vehicle); group 2 — Oligomycin B (OligB) in saline. group 3 — Cyclosporine A (Sandimmune®, Novartis, Switzerland) dissolved in saline. In addition, a fourth sham group of eight rats was added, in which animals were anesthetized prior to surgery that consisted the same craniectomy but without the opening of the dura and without cortical injury. Animals received two intraperitoneal injections at 30 min and 24 h post-injury. Dosage for CsA was 20 mg/kg based on numerous previous studies.20–22 The dosage for OligB was based on the results of the toxicity study of oligomycin in intact rats as reported by Kramar et al.,23 showing that intraperitoneal injection of 0.5 mg/kg reduced oxygen consumption by 50% with one-third mortality. Accordingly and since the purpose of the study was to observe the consequence of enhanced ATP depletion on brain injury without adding drug-related mortality, the dosage selected for OligB was half the LD33 dosage defined by Kramar et al.23
Electron microscopy
At 48 h post-injury, animals were re-anesthetized. Brains were harvested and dissected on ice. Tissue blocks (2 × 2 × 2 mm) were dissected from the cortex next to the injury core and the white matter immediately below. Samples were fixed in glutaraldehyde solution (3%) overnight and post-fixed in osmium tetroxide for 1 h thereafter. Samples were then rinsed in cacodylate buffer (0.1 mol/L, pH = 7.4), incubated in uranyl acetate, dehydrated with ascending ethanol concentrations, and finally embedded in 4.8 mL of Epon, 1.7 mL of DDSA, 3.5 mL of Epon hardener MNA, and 0.2 mL BDMA. Semi-thin sections were obtained from the prepared blocks and stained with toluidine blue for selection of the regions of interest under optic microscope. Ultra-thin sections (75 nm) were then obtained from prepared blocks with an ultra-microtome (Leica EM UC7), mounted on mesh copper grids and evaluated under electron microscopy (JEM-1011, Jeol, Japan).
Quantitative assessment of neuronal cytotoxic edema was based on measurements of the respective surface areas of the neuron body and the nucleus performed on microphotographs obtained at ×6000 magnification using Image J software (NIH Image J, National Institutes of Health, USA) and expressed as the ratio between the cytoplasm area (total neuron body area minus the nucleus area) over that of the nucleus (Figure 1). For each animal, microphotographs from at least 10 grids containing 1–3 neurons each were analyzed by two different operators blinded from grouping.
ICP measurements and assessment of water content
At 48 h, animals were re-anesthetized as described above and the wound opened. An ICP microsensor catheter introduced intracranially and connected to a dedicated monitor (ICP express®, Codman, Depuy Synthes, West Chester, PA). After stabilization, ICP levels were recorded every 2 min for 1 h and averaged. Brains were than harvested for assessment of water content. For that purpose, large tissue samples from the injured hemisphere centered at the lesion core were harvested at 48 h post-injury and then weighted before and after drying at 75℃ for 48 h. Water content was then calculated according to the following formula: wet-dry/dry × 100.
Brain tissue partial oxygen tension measurements
Since ischemia triggered by ICP increase may represent a possible confounding factor of mitochondrial damage, brain perfusion was estimated by measurement of brain tissue partial oxygen tension (PtbO2) measurements. PtbO2 assessment was chosen as a simple and convenient tool that emerged as accurate surrogate of cerebral perfusion in several recent studies performed in head-injured patients.23,24 PtbO2 was measured in rats randomly allocated to vehicle and CsA groups of eight animals each, using NEUROVENT-PTO neuro-monitoring catheters (RAUMEDIC AG, Helmbrechts, Germany). At 48 h post-injury, the animals were re-anesthetized, the wound opened and the catheter introduced in the brain parenchyma through the craniectomy. In order to disclose absolute as well as relative cerebral hypoperfusion, measurements were made in both hemispheres. For this purpose, a second small burr hole was created in a symmetric fashion over the midline and the PtbO2 sensor introduced. After stabilization, PtbO2 levels were recorded every 2 min for 1 h and averaged. PtbO2 readings were performed by an operator blinded to animal grouping.
Measurements of Δψm
For each animal, a tissue sample ranging in weight from 100 to 150 mg was harvested from the parietal region. Tissue samples were washed in phosphate buffer solution for 5 min at 4℃ and then handled using a specially designed mitochondria isolation kit (MITO-ISO1, Sigma-Aldrich, Saint Louis, MO) according to the manufacturer instructions. Briefly, the collected tissue was homogenized on ice with 10 volumes of ice-cold extraction buffer (10 mM HEPES, pH 7.5, containing 200 mM mannitol, 2 mg/ml bovine serum albumin, 70 mM sucrose, and 1 mM EGTA) and centrifuged at 1000 × g for 10 min at 4℃. The pellets were discarded and the supernatant re-centrifuged at 3500 × g for 10 min at 4℃. Pellets obtained from this centrifugation were re-suspended with 10 volumes of a second ice-cold extraction buffer (20 mM MOPS, pH 7.5, containing 110 mM KCl and 1 mM EGTA) and treated by sequential centrifugation as above. Protein concentrations were measured at this point and pellets were then re-suspended in storage buffer accordingly in order to obtain closely similar amounts of mitochondria in all wells (10 mM HEPES, pH 7.4, containing 250 mM sucrose, 1 mM ATP, 0.08 mM ADP, 5 mM sodium succinate, 2 mM K2HPO4, and 1 mM DTT), ranging between 25 and 30 mg/ml.
Mitochondrial transmembrane potentials were assessed in freshly prepared mitochondrial pellets immediately after their re-suspension as described. For this purpose, we measured the uptake of the cationic carbocyanine dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenz-imidazolcarbocyanine iodide) into the mitochondrial matrix. The lipophylic fluorophore cation JC-1 crosses the mitochondrial membrane in direct relation with the Δψm.25 Its accumulation into the matrix results in formation of aggregates that can be easily characterized by a red-orange fluorescence at 590 nm when excited at 490 nm. In return, dissipation of Δψm will result in reduced J-aggregates within the mitochondrial matrix so that the intensity of fluorescence at 590 nm can be used to quantitatively assess the loss of mitochondrial transmembrane potential. Fluorescence intensity characteristically increases fast for a short while and then progressively reaches a plateau. A quantitative assessment was therefore initiated immediately after adding JC-1 to the storage buffer and maintained for 15 min. As a validation tool, a separate measurement of mitochondrial sample was performed in presence of carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (Fccp). The values from the last 3 min of stable fluorescence measurements were averaged and used for statistical analysis. For each rat, two samples of mitochondrial pellets were simultaneously tested.
Functional neurological assessment
Functional performance status was assessed at days 1, 2, 3, 4, and 6 post-injury, according to the modified neurological severity score (mNSS).22 Scores were quoted by an operator blinded to animal grouping. In order to rule out a possible behavioral bias in the evaluation, animals were trained on a daily basis to all the mNSS tasks prior surgery and all reached a zero score.
Statistical analysis
Differences between the different measured parameters were analyzed by means of either one-way analysis of variance (ANOVA), two-way repeated measures ANOVA, or Kruskal–Wallis one-way ANOVA on ranks in presence of non-normal distribution of samples. Post hoc pair wise comparison between groups was performed by either Dunn, Holm-Sidak, or Tukey-Kramer test when appropriate. A P value of less than 0.05 was considered as significant.
Results
ICP
As anticipated, brain injury was associated with a significant ICP elevation in non-treated rats in comparison with sham-operated animals (Figure 2(a), Kruskal–Wallis one-way ANOVA on ranks P < 0.001; sham vs. vehicle P < 0.05 Tukey-Kramer). In contrast, the magnitude of ICP increase was significantly lowered in CsA-treated rats, whereas ICP levels in animals treated with OligB were 2.5 fold higher as that of CsA-treated rats (CsA vs. OligB, P < 0.05 Tukey-Kramer).
Figure 2.

(a) In comparison with sham-operated animals, injured rats suffered from significantly higher levels of ICP. Treatment with CsA an OligB resulted in opposite effects with a significant ICP reduction and further but non-significant ICP elevation, respectively. *: P < 0.05 Tukey-Kramer pair wise multiple groups comparison. Bars represent mean ICP values with standard errors. (b) Expectedly, analysis of water content in the different groups showed results closely similar to that found with ICP with comparable trends for cerebral edema observed with treatment with CsA and OligB. *: P < 0.05 Tukey-Kramer pair wise multiple groups comparison. Bars represent mean values of water content with standard errors. (c) Although injury resulted in brain edema and ICP increase, there was no evidence of reduced value of brain oxygenation that could be indicative of cerebral ischemia induced by elevation of cerebral peripheral vascular resistance. Accordingly, mitochondrial functional alteration could not be attributed to compromised oxygen delivery. As shown, normal PtbO2 values (>20 mm Hg) could be found in both injured (black bars) and non-injured (gray bars) cerebral hemispheres as well as in vehicle and CsA-treated animals with no difference.
Bars represent mean values of water content with standard errors.
Water content
Brain water content proved to be moderately increased after the injury in non-treated animals in comparison with sham-operated rats (Figure 2(b), Kruskal–Wallis one-way ANOVA on ranks P < 0.001; sham vs. vehicle P < 0.05 Tukey-Kramer). No difference in water content, however, could be found between the CsA-treated and the sham-operated groups, both with significantly lower than that of the vehicle group. In contrast, treatment with OligB was associated with a marked and significant increase in brain water content in comparison with CsA-treated rats (CsA vs. OligB P < 0.05 Tukey-Kramer).
PtbO2
Despite significant elevation in ICP in the vehicle group, no difference at all could be noticed between brain oxygen levels in animals of the vehicle-treated and that of CsA-treated animals. Further, there was no difference between PtbO2 levels in the injured and non-injured hemispheres in the two studied groups (Figure 2(c)).
Mitochondrial damage
Following the injury, mitochondrial damage could be evidenced both morphologically and physiologically. Structural damage was characterized by numerous figures of severely swollen and/or ruptured mitochondria either shown within mitochondrial pellets (Figure 3(a)) or within neurons (Figure 3(b)) and axons (Figure 4). Physiologically, mitochondrial membrane permeabilization resulted in a significant loss of Δψm as the consequence of the injury (Figure 5(a), one-way ANOVA P < 0.0001, Sham vs. Vehicle Tukey-Kramer P < 0.001), the amplitude of which could be significantly attenuated by CsA treatment (Figure 5(b), CsA vs. Vehicle, Tukey-Kramer P < 0.001). In contrast, OligB caused an additional and significant loss of Δψm in comparison with animals of the vehicle group (OligB vs. Vehicle Tukey-Kramer P < 0.01).
Figure 3.

(a) Within mitochondrial pellets, structural damage was characterized by severely swollen with distorted cristae and/or blasted mitochondria (B) profoundly different from that of sham-operated animals with normal tubular cristae and small compact mitochondria with no signs of matrix change (A). In contrast, treatment with CsA (C) resulted in some degree of preservation of normal mitochondrial morphology, whereas mitochondria from OligB-treated animals (D) showed extreme structural alterations. (b) High magnification EM micrographs of neurons (×20,000) disclosed the same morphological alterations within mitochondria as shown within mitochondrial pellets. A: sham-operated group; B: vehicle group; C: CsA group; D: OligB group.
N: nucleus; m: mitochondria.
Figure 4.

Axonal damage as evidenced by EM was characterized by axolemmal increased permeability and swelling, myelin decompaction, and numerous figures of swollen or blown-up mitochondria (B), profoundly different from the even aspect of axons harvested from injured white matter of sham-operated rats (A). As for other parameters, treatment with CsA (C) and OligB (D) showed symmetrical and opposite effects on axonal damage.
Left column: low magnification (×6000); Right column: high magnification (×12000).
m: mitochondria
Figure 5.
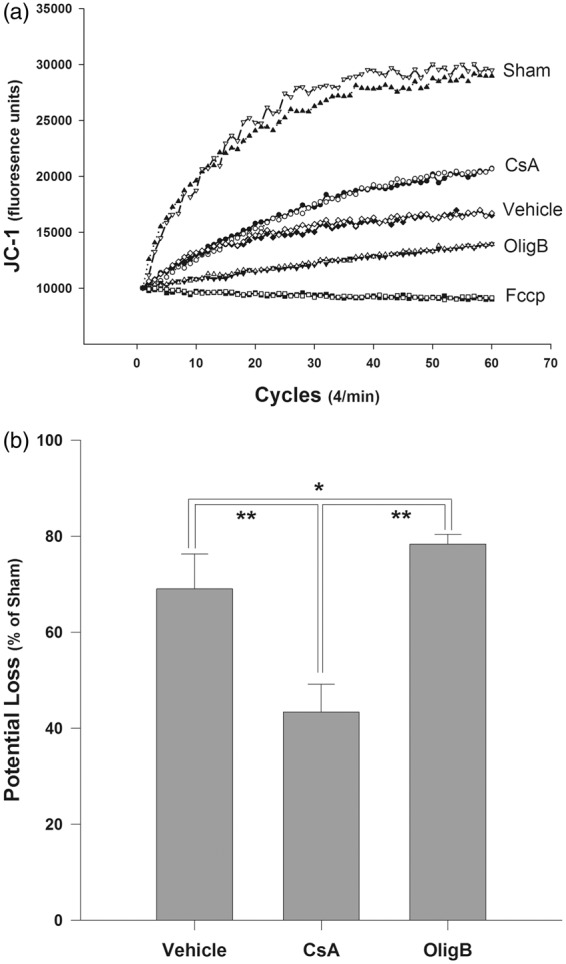
(a) Trends of JC-1 aggregation within the mitochondrial matrix in the different groups. The lipophilic fluorophore cation JC-1 crosses the mitochondrial membrane in direct relation with the Δψm and then aggregates within the mitochondrial matrix emitting a characteristic by a red-orange fluorescence in response with excitation. Each graph represents a grand average of all tests that were performed twice for each sample of each group. (b) Physiologically, mitochondrial membrane permeabilization resulted in a significant loss of Δψm as the consequence of the injury. Loss of Δψm could be significantly attenuated by CsA treatment. In contrast, OligB caused an additional and significant loss of Δψm in comparison with animals of the vehicle group. Although, OligB is usually added in small doses to freshly prepared normal mitochondria in order to create an hyperpolarization of the membrane with high Δψm values, in the present situation damaged mitochondria were exposed in vivo to high doses of OligB for 48 h, responsible for further exhaustion of mitochondrial respiration.
*: P < 0.05 and **: P < 0.001 Tukey-Kramer pair wise multiple groups comparison. Loss of Δψm is expressed as the percentage of the sham value as mean values with standard errors.
Electron microscopy
Electron microscopic micrographs revealed a significant and opposite effect of CsA and OligB with severely swollen neurons in the OligB group contrasting with near to normal appearance of neurons from the CsA group (Figure 6(a)). Analysis of the neuronal cytoplasm area in respect with that of the nucleus (cytoplasm/nucleus ratio) showed differences between the different groups similar to that found for ICP levels and water content (Figure 6(b), Kruskal–Wallis one-way ANOVA on ranks P < 0.001).
Figure 6.

(a) Electron microscopic micrographs revealed prominent alterations of cellular membrane permeability in neurons that appeared obviously swollen as the consequence of the injury (B) in comparison with sham-operated rats (A). CsA and OligB resulted in a significant and opposite effect in cellular edema, with severely swollen neurons in the OligB group (D) contrasting with near to normal appearance of neurons from the CsA group (C). (b) Analysis of the neuronal cytoplasm area in respect with that of the nucleus (cytoplasm/nucleus ratio) showed similar differences between the different groups to that found for ICP levels and water content.
*: P < 0.05 Tukey-Kramer pair wise multiple groups comparison. Bars represent cytoplasm/nucleus area ratio mean values with standard errors.
Within the white matter, injury resulted in a profound distortion of the normal axonal architecture mostly characterized by myelin decompaction and axonal swelling with numerous figures of swollen mitochondria (Figure 4). As for neuron bodies, treatment with CsA resulted in marked reduction of edema and mitochondrial damage, although figures of myelin decompaction were left unaffected. Oppositely, OligB profoundly worsened the extent and magnitude of mitochondrial damage with numerous blown-up mitochondria centered in severely swollen axons.
Neurological outcome
Analysis of the course of mNSS in vehicle and CsA-treated animals showed that reduction of cytotoxic edema was associated with improved recovery expressed by significantly lower mNSS in comparison with the vehicle group (P < 0.05, Two-Way ANOVA Repeated Measures, Figure 7) at day 6 (P < 0.05, Holm-Sidak pair wise multiple groups comparison, effect of treatment at day 6.).
Figure 7.
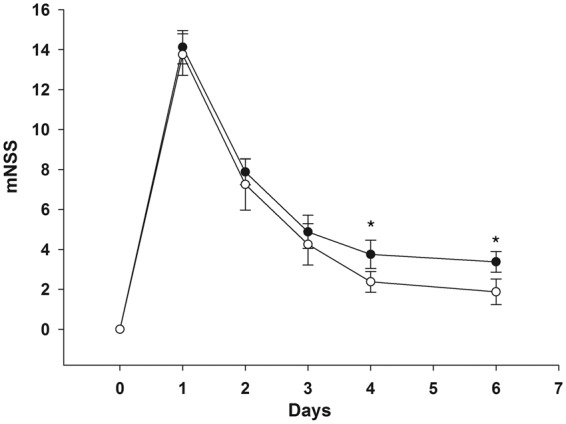
CsA-mediated reduction of cytotoxic edema proved to correlate with functional improvement expressed by significantly lower mNSS at day 6 (white dots) in comparison with animals of the vehicle group (black dots). Scores are expressed as mean with standard deviation. *: P < 0.05, Holm-Sidak pair wise multiple groups comparison, effect of treatment at day 6.
Discussion
Cerebral edema represents one of the most serious threats following brain injury.3 When the encased brain swells within the rigid boundaries of the skull, ICP increases, leading to elevated transmural pressure over brain parenchyma and cerebral vessels with subsequent impaired perfusion and cerebral damage.26 As pressure further increases, cerebral herniation may result in brainstem compression and eventually death.27,28 Accordingly, control of ICP is currently considered as one of the most prominent guidelines in the management of TBI.29 Yet, several recent studies have failed to provide evidence of any significant benefit of ICP monitoring.30,31 Moreover, radical surgical measures such as decompressive craniectomy, although highly effect effective in ICP control, have failed to improve neurological outcome.12,13 Importantly, several studies have shown that failure of oxidative metabolism was not necessarily related to ICP elevation, neither was alleviated by ICP relief.7,8,12 In fact, this could have been anticipated, since numerous studies have shown that post-traumatic failure of metabolism was of non-ischemic origin in most instances so that improvement of cerebral perfusion by ICP relief is unlikely to be crucial from this point of view.7,8,20,32 Our findings of normal PtbO2 values in both vehicle and CsA groups and both hemispheres are in accordance with these previous observations as well as with the results of a recently published study based on microdialysis and PtbO2 measurements showing that low PtbO2 values were uncommon.24 As hypothesized by Okonkwo and Povlishock33, both cerebral edema and metabolic crisis may actually share a common and pivotal event represented by mitochondrial damage. Following glutamate release, sequestration of calcium by mitochondria acts as a protective mechanism preventing its accumulation within the cytosol where it triggers osmotic water cellular influx. Whenever the buffering capacity is reached, damage to mitochondria may eventually lead to mPTP induction with subsequent loss of the proton gradient essential to activation of ATP synthase. In this scenario, cytotoxic edema may thus reflect the bioavailability of high-energy phosphates needed for activation of energy-dependent membrane ionic pumps responsible for maintenance of cellular homeostasis.33
The results of the present study establish a clear relationship between cytotoxic edema and mitochondrial damage and ATP production. As hypothesized, ultra-structural figures of edema closely correlated with ICP elevation, increased water content, and mitochondrial membrane permeabilization expressed by loss of Δψm. Further, mitochondrial damage evidenced ultra-structurally by figures of swollen mitochondria with severely distorted cristae proved to correlate with both cytotoxic edema and mitochondrial dysfunction. Importantly, cerebral edema and mitochondrial impairment were significantly worsened by treatment with OligB, whereas a noticeable improvement could be observed in animals that received injections of CsA. Since OligB and CsA are responsible for symmetrical and opposite effects on oxidative metabolism, these findings support the hypothesis of a causative relationship between edema and mitochondrial function. Accordingly, targeting mitochondrial damage may thus represent a more effective and etiological approach for management of cerebral edema. This assumption has been supported by both clinical34,35 and experimental evidence36 showing that decrease in lactate/pyruvate ratio correlated with ICP decrease.
Although a comparative assessment of astrocytic and neuronal edema was beyond the scope of the present study, our findings emphasize the involvement of neurons by the usually considered as more prone to swelling than neurons because of their participation in regulation of extracellular levels of K+ through activation of Na+-K+-Cl−- cotransporters (NKCC1) responsible for Na+ cellular influx.37,38 Since astrocytic but not neuronal NKCC1 are triggered by extracellular levels of K+, significant neuronal swelling was less anticipated.39 This finding may of clinical importance when interpreting the neurological status of TBI patients in presence of edema. Furthermore and as previously shown by Okonkwo and Povlishock,33 edema was not confined to neuronal bodies but extensively involved axons as well. Axonal damage was characterized by myelin decompaction and edema associated with the presence of swollen mitochondria. Both findings proved to be symmetrically and oppositely affected by OligB and CsA. These observations further reinforce the potential therapeutic benefit of targeting mitochondrial damage either for combined or isolated axonal injury.
In conclusion, the results of the present study demonstrate the existence of an intimate correlation between mitochondrial damage and cytotoxic edema within the neuron population and may therefore help to understand the recently reported failure of therapeutic measures aiming at ICP control for improvement of neurological outcome in TBI patients.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Eugene Vlodavsky: Study design, EM studies concept and analysis and manuscript preparation.
Eilam Palzur: Animal studies, data analysis.
Mona Shehadeh: Animal studies, data analysis.
Jean F Soustiel: Study design, data analysis and manuscript preparation.
References
- 1.Reilly PL, Graham DI, Adams JH, et al. Patients with head injury who talk and die. Lancet 1975; 2: 375–377. [DOI] [PubMed] [Google Scholar]
- 2.Miller JD, Becker DP, Ward JD, et al. Significance of intracranial hypertension in severe head injury. J Neurosurg 1977; 47: 503–516. [DOI] [PubMed] [Google Scholar]
- 3.Saul TG, Ducker TB. Effect of intracranial pressure monitoring and aggressive treatment on mortality in severe head injury. J Neurosurg 1982; 56: 498–503. [DOI] [PubMed] [Google Scholar]
- 4.Bouma GJ, Muizelaar JP, Bandoh K, et al. Blood pressure and intracranial pressure-volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg 1992; 77: 15–19. [DOI] [PubMed] [Google Scholar]
- 5.Jaggi JL, Obrist WD, Gennarelli TA, et al. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J Neurosurg 1990; 72: 176–182. [DOI] [PubMed] [Google Scholar]
- 6.Obrist WD, Langfitt TW, Jaggi JL, et al. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg 1984; 61: 241–253. [DOI] [PubMed] [Google Scholar]
- 7.Chieregato A, Tanfani A, Compagnone C, et al. Global cerebral blood flow and CPP after severe head injury: a xenon-CT study. Intensive Care Med 2007; 33: 856–862. [DOI] [PubMed] [Google Scholar]
- 8.Soustiel JF, Sviri GE. Monitoring of cerebral metabolism: non-ischemic impairment of oxidative metabolism following severe traumatic brain injury. Neurol Res 2007; 29: 654–660. [DOI] [PubMed] [Google Scholar]
- 9.Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab 2005; 25: 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belli A, Sen J, Petzold A, et al. Metabolic failure precedes intracranial pressure rises in traumatic brain injury: a microdialysis study. Acta Neurochir (Wien) 2008; 150: 461–469. discussion 470. [DOI] [PubMed] [Google Scholar]
- 11.Cottenceau V, Masson F, Mahamid E, et al. Comparison of effects of equiosmolar doses of mannitol and hypertonic saline on cerebral blood flow and metabolism in traumatic brain injury. J Neurotrauma 2011; 28: 2003–2012. [DOI] [PubMed] [Google Scholar]
- 12.Soustiel JF, Sviri GE, Mahamid E, et al. Cerebral blood flow and metabolism following decompressive craniectomy for control of increased intracranial pressure. Neurosurgery 2010; 67: 65–72. discussion 72. [DOI] [PubMed] [Google Scholar]
- 13.Cooper DJ, Rosenfeld JV, Murray L, et al. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med 2011; 364: 1493–502. [DOI] [PubMed] [Google Scholar]
- 14.Hudak AM, Peng L, Marquez de la Plata C, et al. Cytotoxic and vasogenic cerebral oedema in traumatic brain injury: assessment with FLAIR and DWI imaging. Brain Inj 2014; 28: 1602–1609. [DOI] [PubMed] [Google Scholar]
- 15.Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol 2010; 23: 293–299. [DOI] [PubMed] [Google Scholar]
- 16.Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 2009; 10: 481–494. [DOI] [PubMed] [Google Scholar]
- 17.Kahle KT, Simard JM, Staley KJ, et al. Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology (Bethesda) 2009; 24: 257–265. [DOI] [PubMed] [Google Scholar]
- 18.Szabo C. Mechanisms of cell necrosis. Crit Care Med 2005; 33(12 Suppl): S530–S534. [DOI] [PubMed] [Google Scholar]
- 19.Marmarou A, Foda MA, van den Brink W, et al. A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J Neurosurg 1994; 80: 291–300. [DOI] [PubMed] [Google Scholar]
- 20.Glenn TC, Kelly DF, Boscardin WJ, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab 2003; 23: 1239–1250. [DOI] [PubMed] [Google Scholar]
- 21.Fukui S, Signoretti S, Dunbar JG, et al. The effect of cyclosporin A on brain edema formation following experimental cortical contusion. Acta Neurochir Suppl 2003; 86: 301–303. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 2001; 32: 1005–1011. [DOI] [PubMed] [Google Scholar]
- 23.Kramar R, Hohenegger M, Srour AN, Khanakah G. Oligomycin toxicity in intact rats. Agents and Actions 1984; 15(5–6): 660–663. [DOI] [PubMed]
- 24.Bouzat P, Marques-Vidal P, Zerlauth JB, et al. Accuracy of brain multimodal monitoring to detect cerebral hypoperfusion after traumatic brain injury*. Crit Care Med 2015; 43: 445–452. [DOI] [PubMed] [Google Scholar]
- 25.Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 1991; 30: 4480–4486. [DOI] [PubMed] [Google Scholar]
- 26.Rosner MJ, Daughton S. Cerebral perfusion pressure management in head injury. J Trauma 1990; 30: 933–940. discussion 940–941. [DOI] [PubMed] [Google Scholar]
- 27.Alali AS, Fowler RA, Mainprize TG, et al. Intracranial pressure monitoring in severe traumatic brain injury: results from the American College of Surgeons Trauma Quality Improvement Program. J Neurotrauma 2013; 30: 1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schreiber MA, Aoki N, Scott BG, et al. Determinants of mortality in patients with severe blunt head injury. Arch Surg 2002; 137: 285–290. [DOI] [PubMed] [Google Scholar]
- 29.Bratton SL, Chestnut RM, Ghajar J, et al. Guidelines for the management of severe traumatic brain injury. VIII. Intracranial pressure thresholds. J Neurotrauma 2007; 24(Suppl 1): S55–S58. [DOI] [PubMed] [Google Scholar]
- 30.Chesnut RM, Temkin N, Carney N, et al. A trial of intracranial-pressure monitoring in traumatic brain injury. N Engl J Med 2012; 367: 2471–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan Q, Wu X, Sun Y, et al. Impact of intracranial pressure monitoring on mortality in patients with traumatic brain injury: a systematic review and meta-analysis. J Neurosurg 2015; 122: 574–5787. [DOI] [PubMed] [Google Scholar]
- 32.Soustiel JF, Glenn TC, Vespa P, et al. Assessment of cerebral blood flow by means of blood-flow-volume measurement in the internal carotid artery: comparative study with a 133xenon clearance technique. Stroke 2003; 34: 1876–1880. [DOI] [PubMed] [Google Scholar]
- 33.Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab 1999; 19: 443–451. [DOI] [PubMed] [Google Scholar]
- 34.Rockswold GL, Ford SE, Anderson DC, et al. Results of a prospective randomized trial for treatment of severely brain-injured patients with hyperbaric oxygen. J Neurosurg 1992; 76: 929–934. [DOI] [PubMed] [Google Scholar]
- 35.Rockswold SB, Rockswold GL, Zaun DA, et al. A prospective, randomized phase II clinical trial to evaluate the effect of combined hyperbaric and normobaric hyperoxia on cerebral metabolism, intracranial pressure, oxygen toxicity, and clinical outcome in severe traumatic brain injury. J Neurosurg 2013; 118: 1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soustiel JF, Vlodavsky E, Milman F, et al. Improvement of cerebral metabolism mediated by Ro5-4864 is associated with relief of intracranial pressure and mitochondrial protective effect in experimental brain injury. Pharm Res 2011; 28: 2945–2953. [DOI] [PubMed] [Google Scholar]
- 37.Su G, Kintner DB, Flagella M, et al. Astrocytes from Na(+)-K(+)-Cl(−) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol 2002; 282: C1147–C1160. [DOI] [PubMed] [Google Scholar]
- 38.Su G, Kintner DB, Sun D. Contribution of Na(+)-K(+)-Cl(−) cotransporter to high-[K(+)](o)- induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol 2002; 282: C1136–C1146. [DOI] [PubMed] [Google Scholar]
- 39.Liang D, Bhatta S, Gerzanich V, et al. Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurg Focus 2007; 22: E2. [DOI] [PMC free article] [PubMed] [Google Scholar]