

Abstract
Subarachnoid hemorrhage causes acute and long-lasting constrictions of pial arterioles. Whether these vessels dilate normally to neuronal activity is of great interest since a mismatch between delivery and consumption of glucose and oxygen may cause additional neuronal damage. Therefore, we investigated neurovascular reactivity of pial and parenchymal arterioles after experimental subarachnoid hemorrhage. C57BL/6 mice were subjected to subarachnoid hemorrhage by filament perforation or sham surgery. Neurovascular reactivity was assessed 3 h later by forepaw stimulation or inhalation of 10% CO2. Diameters of cerebral arterioles were assessed using two-photon microscopy. Neurovascular coupling and astrocytic endfoot Ca2+ were measured in brain slices using two-photon and infrared-differential interference contrast microscopy. Vessels of sham-operated mice dilated normally to CO2 and forepaw stimulation. Three hours after subarachnoid hemorrhage, CO2 reactivity was completely lost in both pial and parenchymal arterioles, while neurovascular coupling was not affected. Brain slices studies also showed normal neurovascular coupling and a normal increase in astrocytic endfoot Ca2+ acutely after subarachnoid hemorrhage. These findings suggest that communication between neurons, astrocytes, and parenchymal arterioles is not affected in the first few hours after subarachnoid hemorrhage, while CO2 reactivity, which is dependent on NO signaling, is completely lost.
Keywords: Subarachnoid hemorrhage, neurovascular coupling, CO2, parenchymal vessels, in vivo
Introduction
Activation of specific regions of the brain induces a focal increase in cerebral blood flow (CBF) in order to match the delivery of oxygen and glucose to the increased metabolic demand of depolarized neurons.1 This process is called NVC and is mediated through a not yet completely understood interplay between neurons, astrocytes, and cerebral vessels.2,3 Disturbances of NVC, as observed after cerebral ischemia or brain trauma, may compromise the metabolic status of neurons and further aggravate brain damage. Accordingly, it is of great scientific and clinical interest to understand the mechanisms of NVC in the healthy and diseased brain.
Subarachnoid hemorrhage (SAH) is a subtype of stroke which accounts for 5% of all first-ever strokes.4 Despite its relatively low incidence, SAH is responsible for 20% of all cerebrovascular-related deaths,5 and only 10% of all SAH patients recover completely. In most cases, SAH is caused by the rupture of a cerebral aneurysm located at the base of the skull. Experimental data show that immediately after vessel rupture the subarachnoid space fills with blood and intracranial pressure (ICP) increases to levels that blunt cerebral perfusion pressure (CPP) and causes global cerebral ischemia. If patients survive this acute spike in ICP, lasting approximately 5 min, ICP decreases and CPP normalizes. Despite normal CPP, however, many SAH patients still suffer from severe and widespread reductions in CBF.6 Since large brain surface arteries become spastic only after several days of exposure to subarachnoid blood, these early reductions in CBF are most likely caused by disturbances at the level of the cerebral microcirculation. Direct visualization of cerebral microvessels within the first few hours after SAH in patients and experimental animals indeed demonstrated microvasospasms of pial arterioles that reduced cerebral perfusion by 60–80%.7–9 These reductions in CBF trigger a cascade of events resulting in brain edema formation, neuronal cell death, and unfavorable neurological outcome referred to as “early brain injury”.10–12
Despite the importance of cerebral microvessels in the pathophysiology of SAH, relatively little is known about the consequences of SAH on neurovascular function in vivo. Recently, we demonstrated that NVC is impaired 96 h after SAH in rat brain slice preparations.13 This suggests that neuron activation may not dilate cerebral vessels after SAH to elicit an adequate increase in CBF, thereby significantly increasing the risk for further neuronal damage and loss of function. However, it is not known if NVC is impaired immediately after SAH in vivo. Therefore, the aim of the current study was to subject mice to experimental SAH for 3 h and use two-photon microscopy to investigate the reactivity of pial and parenchymal arterioles to sensory stimulation and CO2, two well established in vivo paradigms of neurovascular communication.
Materials and methods
Animal breeding, housing, and all experimental procedures were conducted according to institutional guidelines of the University of Munich and University of Vermont and were approved by the Ethical Review Board of the Government of Upper Bavaria (protocol number 220-13) and the Institutional Animal Care and Use Committee at the University of Vermont. Male C57BL/6 mice (20 to 23 g body weight Charles River Laboratory, Sulzfeld, Germany and Charles River Laboratory, Saint Constant, Quebec, Canada) were used for this study. Experiments were planned, carried out, and reported according to the ARRIVE guidelines.14
Animal preparation and monitoring
Experimental animals had free access to food and water prior and after surgery. For induction of SAH, anesthesia was induced by intraperitoneal injection of midazolam (5 mg/kg; Braun, Melsungen, Germany), fentanyl (0.05 mg/kg; Jansen-Cilag, Neuss, Germany), and medetomidine (0.5 mg/kg; Pfizer, Karlsruhe, Germany) as previously described.15,16 Mice were orotrachealy intubated and mechanically ventilated (Minivent, Hugo Sachs, Hugstetten, Germany). End-tidal pCO2 was measured with a microcapnometer (Capnograph, Hugo Sachs, Hugstetten, Germany) and kept constant between 30 and 40 mmHg by respective adjustments to the ventilation.16 A thermostatically regulated, feedback-controlled heating pad (FHC, Bowdoin, ME, USA) was used to maintain body temperature at 37℃. ICP was measured in each animal for 15 min after SAH using a microsensor-based ICP probe (Codman & Shurteff Inc, Raynham, MA) to prove successful induction of SAH as described before.17 For continuous monitoring of regional cerebral blood flow (rCBF), a flexible laser-Doppler probe (Periflux 4001 Master, Perimed, Stockholm, Sweden) was glued onto the skull above the territory of the left middle cerebral artery (MCA). Blood gases and electrolytes were determined at the end of each experiment.
For experiments on neurovascular reactivity mice were initially anesthetized with 2% isoflurane in 70% N2O and 30% O2. Later on, isoflurane was gradually reduced over the course of 10 min to a range of 0.5 to 0.9% in 70% room air and 30% O2. At the same time, a continuous intra-arterial infusion of ketamine (30 mg/kg/h, Inresa, Freiburg, Germany) was initiated.18
Induction of SAH
SAH was induced using the filament perforation model as previously described.17,19,20 Briefly, a 5-0 monofilament was introduced via the left external carotid artery into the internal carotid artery and advanced intracranially. SAH induction was indicated by a sudden increase of ICP. Immediately thereafter the filament was withdrawn, and the external carotid artery was ligated. Sham-operated mice were treated in the same way with the only exception that the filament was not advanced far enough to induce hemorrhage. Anesthesia was terminated by intraperitoneal injection of atipamezole (2.5 mg/kg; Pfizer) naloxone (1.2 mg/kg; Inresa, Freiburg, Germany), and flumazenil (0.5 mg/kg; Hoffmann-La-Roche, Grenzach-Wyhlen, Germany). Thereafter, mice were kept in an incubator at 33℃ for 2.5 h.
Forepaw-evoked NVC
Mice were re-anesthetized 2.5 h after SAH, and the CBF response after NVC was evaluated as previously described.18 Briefly, the left forepaw was stimulated with two subdermally inserted needle electrodes with a diameter of 0.2 mm (Hwato, Suzhou, China) at an intensity of 2 mA for 0.3 ms (Digitimer Ltd, Hertfordshire, England). One stimulation cycle contained 96 stimulations and lasted for 16 s (6 Hz). The interval between two stimulation cycles was 40 s. CBF was assessed at five different locations covering the whole somatosensory cortex. The region with the strongest CBF response was also continuously stimulated (2 mA) for 1 min in order to evaluate the response to a tonic stimulus. This region was then considered for analysis and further for assessment of the microvascular response by two-photon microscopy. A graphical representation of the experimental protocol is shown in Figure 1(a).
Figure 1.

SAH induction and experimental design. (a) Schematic representation of the experimental design for SAH (or sham) surgery and forepaw stimulation followed by 10% CO2 inhalation. (b) Intracranial pressure and (c) cerebral blood flow over time, starting 5 min before SAH induction until 20 min thereafter. A sudden increase in ICP and drop in CBF confirm vessel perforation. Mean±SD, n = 10 each.
Two-photon microscopy
After assessing the CBF response to forepaw stimulation, i.e. 4 h after SAH, a cranial window (2 × 1 mm) was drilled under constant cooling above the area of the somatosensory cortex associated to the forepaw leaving the dura mater intact as previously described.21 Mice were placed under a two-photon microscope (Zeiss LSM-7 MP, Oberkochen, Germany) equipped with a Li:Ti laser (Chameleon, Coherent, USA) as described previously,22 and the exposed dura mater was kept wet with isotonic saline. The fluorescent plasma dye, fluorescein isothiocyanate (FITC-dextran; molecular weight 150 kDa) was given systematically via femoral artery injection (0.05 ml of a 0.5% solution; Sigma, Deisenhofen, Germany), and all parenchymal (diameter: 5–20 µm; depth: 100 µm) and pial arterioles (diameter: 20 to 40 µm) in the region previously selected (see above) were visualized using two-photon fluorescence microscopy and a 10× Zeiss EC Plan-NeoFluar objective. Pial arterioles were followed into the parenchyma along an axis normal to the brain surface. Arterioles were distinguished from venules on the basis of velocity and direction of the blood flow.
Neurovascular reactivity to CO2
Diameters of both parenchymal and pial arterioles were examined under physiological conditions in order to obtain baseline values. Thereafter, arteriolar diameter was observed during inhalation of 10% CO2 for 10 min. The amount of inhaled CO2 was measured by microcapnometry (Hugo-Sachs Elektronik, March-Hugstetten, Germany). Arteriolar diameters were quantified with a calibrated image analysis software (Zen, Zeiss, Oberkochen, Germany) and expressed in percentage of baseline as previously described.18
Vessel diameter during forepaw-evoked NVC
For evaluation of changes in vessel diameter after forepaw stimulation, the same protocol as for assessment of CBF was used. Briefly, the region yielding the most pronounced CBF response was stimulated with one stimulation cycle, contained 96 stimulations and lasted for 16 s (6 Hz). The interval between two stimulation cycles was 40 s. Thereafter, the same region was also continuously stimulated (2 mA) for 1 min in order to evaluate the response to a tonic stimulus.
Ex vivo NVC in freshly prepared brain slices
To examine NVC ex vivo, parenchymal arteriolar diameter and astrocytic endfoot Ca2+ were simultaneously measured in freshly prepared cortical brain slices using a combination of two-photon and infrared-differential interference contrast (IR-DIC) microscopy as described previously.13 Four hours after SAH induction mice were euthanized by decapitation under deep pentobarbital anesthesia (60 mg/kg). Coronal sections of somatosensory cortex (160 µm thick), prepared from the MCA region using a vibratome (Leica VT1000S), were loaded with the Ca2+ indicator dye, fluo-4 (Invitrogen), for 1.5 h at 29℃. Throughout the experiment, brain slices were superfused with artificial cerebral spinal fluid (aCSF; 125 mM NaCl, 3 mM KCl, 18 mM NaHCO3, 1.25 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, and 5 mM glucose, aerated with 5% CO2/20% O2/75% N2, pH ∼7.35, 35–37℃) containing 125 Nm U46619 (a thromboxane A2 analog, Calbiochem) and 0.4 mM ascorbic acid. U46619 was added to establish a physiological level of arteriolar tone, and ascorbic acid was used as a supplement to prevent swelling of brain slices13,23 The following criteria was used to a elect the recording region within brain slices for NVC: (1) brain parenchymal arterioles encased by astrocyte endfeet (i.e. arterioles past the end of Virchow-Robin space, or at least 50 µm from the brain surface), (2) Arterioles that are no greater than 300 µm from brain surface, and (3) Arterioles with adjacent astrocyte endfeet with detectable fluorescent Ca2+ indicator. NVC was initiated using electrical field stimulation (EFS, 50 Hz, 0.3-ms alternating square pulse, 3 s duration). IR-DIC and fluorescent (Ex 820 nm, Em 525/50 nm) images were simultaneously acquired at ∼1 Hz using a Zeiss LSM-7 multiphoton imaging system. Custom software SparkAn (written by Dr. Bonev, University of Vermont) was used to measure arteriolar diameter at 3 points along the segment (∼10 µm) of the arteriole exhibiting the largest response to EFS, and are expressed as percent diameter change compared to the first image of the recording (prior to EFS). Estimated Ca2+ concentration in the astrocyte endfoot adjacent to the arteriolar segment of interest was obtained using the maximal fluorescent method.23
Statistical analysis
For the in vivo study, statistical analysis was performed with a standard statistical software package (Sigma Plot 12.5; Systat Software, Erkrath, Germany). Results for CO2 reactivity experiments are presented as mean ± standard error of the mean (SEM). Results for somatosensory stimulation are presented as median ± 75/25 percentile. Differences across groups were evaluated using the Mann-Whitney Rank Sum test with the Bonferroni correction.
For the ex vivo study, Origin 9.1 software (Origin Lab, Northampton, MA, USA) was used for statistical analysis. EFS-induced dilations (control vs. SAH) were compared by unpaired t-test. Ca2+ concentrations in astrocyte endfoot (control vs. SAH, before and after EFS) were analyzed by one-way ANOVA followed by post hoc comparison of means using the Tukey test. Data, in this case, are presented as mean ± SEM.
Randomization and blinding
All animals were randomly assigned to the procedures; the surgical preparation and data analysis were performed by a researcher blinded towards the treatment of the animals.
Results
In vivo physiological parameters and mortality
Body temperature, systemic blood pressure, blood pH, pCO2, and pO2 – factors shown to have strong effects on CBF24 – were carefully monitored in all investigated mice. Parameters did not differ between groups with the exception of the blood pressure which, as expected, was significantly increased in the SAH group (P = 0.03) as a consequence of the Cushing’s reflex due to high ICP that results from the induction of SAH25 (Figure 1(b) and Supplementary Table S1). Despite this increase in systemic blood pressure, the high ICP observed immediately after SAH caused a reduction in CPP that resulted in reduction of CBF by almost 80%. After ICP decreased to values around 30 mmHg, CBF normalized and remained at this level for the remaining observation period (Figure 1(c)). After SAH, animals showed reduced motor activity. Two mice from each group (4/24) died before imaging could be performed, i.e. within 3 h after SAH.
In vivo CO2 reactivity in cerebral pial arteries and parenchymal arterioles is abolished after SAH
In sham-operated mice, pial arterioles dilated by 17–20% upon inhalation of 10% CO2 (Figure 2(a) and (b) white symbols). A similar, though stronger, response was observed in parenchymal arterioles (Figure 2(a) and (c) white symbols). Following SAH, pial vessels of a diameter of 20 to 40 µm were non-reactive to the increase in pCO2 compared to the sham group (Figure 2(a) and (b), gray symbols). Given this lack of pial microvascular CO2 reactivity 3 h after SAH, we also investigated the response of parenchymal arterioles. Following SAH, parenchymal arterioles were also non-reactive to CO2 (Figure 2(a) and (c), gray symbols). These results demonstrate that exposure to CO2, which results in vasodilation in healthy mice is abolished in pial and parenchymal arterioles after SAH.
Figure 2.

Pial and parenchymal arteries do not dilate in response to hypercapnia after SAH. (a) Representative two-photon microscopy images of pial (top) and parenchymal arteries (bottom) of SAH (left) and sham-operated (right) mice before and during 10% CO2 inhalation. (b) Surface and (c) parenchymal artery diameter during hypercapnia of mice subjected to sham surgery (white symbols) or SAH (gray symbols). Pial and parenchymal vessel dilated normally in response to inhalation of 10% CO2 in sham-operated mice (white symbols), while no response was observed after SAH (gray symbols). Mean ± SEM; Mann-Whitney Rank Sum test; 16 to 37 arteries (b) and 25 to 34 arteries (c) in n = 6–7 mice per group. ** P < 0.01, *** P < 0.001.
In vivo NVC is maintained in acute SAH animals
Three hours after hemorrhage sensory stimulation of the forepaw resulted in a comparable increase of CBF and vessel diameter in both sham-operated and SAH mice (Figure 3(a) to (c)). In response to the stimulation, the increase in artery diameter of parenchymal vessels reached values of 20–25% in the sham-operated group (Figure 3(c), white symbols) while after SAH arterioles dilated by 30–40% (Figure 3(c), gray symbols). Continuous sensory stimulation of the forepaw for 1 min increased CBF by 15% (Figure 3(d), white and gray symbols) and the diameter of parenchymal arteries by approximately 40% in sham and SAH mice (Figure 3(e)). These results point to a preserved NVC early after SAH in vivo.
Figure 3.

Neurovascular coupling is not altered 3 h after SAH. (a,b) Box plots showing CBF increase in response to the first four (a) and last six (b) discrete electrical stimuli to the forepaw, in sham-operated mice (white symbols) and after SAH (gray symbols). No significant effect was found between the experimental groups. Median ± 75/25, n = 10 each. (c) Artery dilation in response to a discrete electrical stimulation shows no difference between the experimental groups. Mean ± SEM; 22 to 30 arteries from n = 7–8 mice. (d) CBF increase in response to continuous electrical stimulation shows no difference between sham-operated mice and mice subjected to SAH. Mean ± SEM; Mann-Whitney Rank Sum test n = 3 mice each. (e) Artery dilation in response to a continuous electrical stimulation shows no difference between SAH or sham-operated mice. Mean ± SEM; Mann-Whitney Rank Sum test; 9 to 13 arteries from n = 3 mice each.
Preserved ex vivo NVC in brain slices from 4 h SAH animals
To examine the acute effects of SAH on ex vivo NVC, two-photon imaging was performed on cortical brain slices prepared from mice 4 h after induction of SAH via endovascular perforation. In brain slices from un-operated control animals, local neuronal activation by EFS caused the anticipated increase in astrocytic endfoot Ca2+ followed by vasodilation of the adjoining parenchymal arteriole (18 ± 1% increase in diameter, n = 5; Figure 4(a) to (c)). EFS also induced vasodilation in brain slices obtained 4 h after SAH (22 ± 2% increase in diameter, n = 5; Figure 4(b) and (c)), similar to that observed in brain slices from control mice. Further, basal endfoot Ca2+ concentration (control: 118 ± 11 nM, SAH 121 ± 16 nM, n = 5 each; Figure 4(d)) and endfoot Ca2+ concentration after EFS (control: 394 ± 32 nM, SAH 360 ± 29 nM n = 5 each) were not significantly different between groups. These results demonstrate preserved NVC in cortical brain slices 4 h after SAH.
Figure 4.
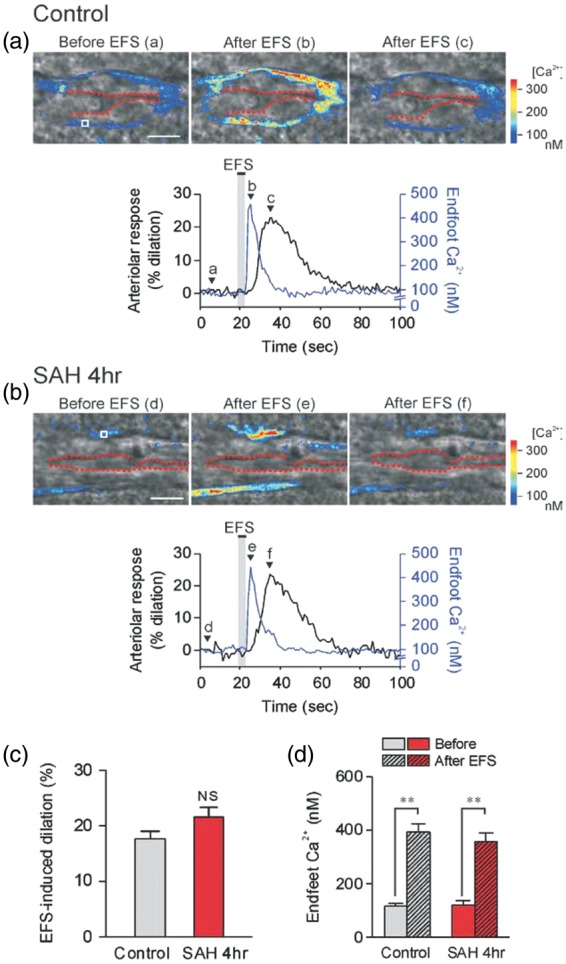
EFS-induced vasodilation is unaltered in brain slices 4 h after SAH. (a,b upper panels) Infrared-differential interference contrast (IR-DIC) images of cortical brain slices from control and 4 h SAH mice. The time points corresponding to IR-DIC images (a–f) are indicated by alphabetically labeled arrows in the lower panels. Red dashes outline the intraluminal diameter of parenchymal arterioles. Pseudocolor regions overlapping IR-DIC images depict estimated Ca2+ levels in astrocyte endfeet, which were imaged simultaneously with arteriolar diameter using the fluorescent Ca2+ indicator Fluo-4 and two-photon imaging. Scale bars, 10 µm. (a,b lower panels) EFS-induced changes in arteriolar diameter (black lines) and endfoot Ca2+ concentration (blue lines) obtained from brain slices depicted in upper images. Regions of interest (ROI) used for endfoot Ca2+ calculation are shown in images (a) and (d) labeled “Before EFS”. (c,d) Summary of EFS-evoked changes in arteriolar diameter (c) and astrocytic endfoot Ca2+ (d) in control and 4 h SAH animals (n = 5 each). Average diameters before EFS were 4.08 ± 0.44 µm (control) and 3.84 ± 0.50 µm (SAH 4 h), and were not significantly different between groups.
NS: not significant.
** P < 0.01 vs. endfoot Ca2+ before EFS in each group.
Discussion
In this study, we have examined the acute effects, within the first 3–4 h, of SAH on in vivo CO2 reactivity and in vivo and ex vivo NVC using a mouse SAH model. Our data show that within 3 h after SAH pial as well as intraparenchymal arterioles show a complete loss of reactivity to CO2, a specific cerebral vasodilator, while NVC, that is the dilatation of cerebral vessels upon neuronal activation, was completely preserved. Thus, our data demonstrate that (1) after SAH not only pial vessels but also intraparenchymal vessels lost in vivo CO2 reactivity and (2) in vivo and ex vivo NVC remains unaffected in the acute phase of SAH. Since CO2 reactivity depends, among others, on proper function of constitutive nitric oxide synthases (NOS),26,27 our data suggest that SAH may cause a rapid dysfunction of NO signaling throughout brain pial and parenchymal arterioles.
Disturbances of the cerebral microcirculation after SAH were described for the first time by Herz et al., when they observed acute vasoconstriction of pial vessels after vascular micropuncture in guinea pigs.28 Since then, little research has been done to understand the role of the cerebral microcirculation after SAH. The majority of studies investigating pial and parenchymal arterioles used in vitro systems,29–35 which require experimental conditions not necessarily reflecting microvascular function in vivo. The few published in vivo studies investigating the cerebral microcirculation after SAH used conventional epifluorescence microscopy and were due to the limited penetration depth of this technology limited to pial vessels.36–38 These vessels were of specific interest in the context of SAH since these are the only cerebral microvessels coming in direct contact with extravasated blood after subarachnoid bleeding. Indeed, pial vessels were shown to constrict after SAH in experimental animals models and in SAH patients thereby suggesting that spasms of cerebral microvessels are one of the main reasons for the cerebral ischemia observed after SAH.7–9 Pial microvessels, however, are morphologically distinct from parenchymal arterioles in that pial vessels have perivascular innervation and lack of astrocytic endfeet coverage.39 Most importantly, parenchymal, not pial, arterioles are in direct contact with the brain parenchyma, directly communicate with astrocytes and neurons, and increase their diameter upon neuronal activation. However, parenchymal arterioles within the brain were beyond the range of conventional epifluorescence microscopy. Here, we applied the novel technology, i.e. two-photon microscopy in combination with neuronal stimulation paradigms, to provide the first measurements of parenchymal arteriolar functionality acutely after SAH.
Using this approach, our results show that SAH results in loss of CO2 reactivity of inparenchymal arterioles in addition to pial arteries 3 h after SAH, confirming our previous results on the lack of CO2 reactivity in pial arterioles 3 and 24 h after SAH.37 Our results show that not only microvessels located in the subarachnoid space and coming in direct contact with blood but also arterioles in the parenchyma where blood is not present lose their ability to dilate in the face of increased levels of CO2. Thus, the brain as a whole seems to lose its ability to regulate blood flow in response to metabolic stress and may thus become increasingly vulnerable to additional damage. Since CO2 reactivity is at least partly mediated through the activation of constitutive NO synthases,26,27 our results point towards a possible lack of proper NO signaling in this process in the acute phase of SAH. Further experiments need to clarify if targeting NO signaling or other signaling pathways downstream of CO2 have the potential to restore microvascular function and to improve outcome after SAH.
Interestingly, NVC, the process coupling neuronal activation to vasodilatation of neighboring vessels, was not affected in the first few hours after SAH, a time point associated with pial microvasospasms, pial microthrombosis, and decrease of CBF in experimental SAH and in SAH patients.6,7 Mechanistically, this suggests that CO2 reactivity and NVC are at least partly mediated by different signaling pathways and that these pathways are differentially affected by SAH. Another important point is that SAH seems to affect in vivo CO2 reactivity much earlier than NVC since ex vivo studies report a paradoxical inversion of NVC from dilation to constriction that emerges 24 to 96 h after SAH.40 In the current study, our in vivo and ex vivo data univocally demonstrate that NVC coupling is not impaired within the first few hours after SAH. Thus, targeting SAH-induced impairment of NVC, that develops with a delayed onset (i.e. longer therapeutic window) may also have clinical potential. The mechanisms underlying SAH-induced inversion of NVC are not fully understood but are likely to involve an increase in the amplitude of spontaneous Ca2+ release events in astrocyte endfeet leading to elevation of K+ in the restricted perivascular space around parenchymal arterioles.13,37
Taken together, the current study investigated for the first time the consequences of SAH on the functional integrity of parenchymal microvessels. Pial and parenchymal microvessels show a very early loss of CO2-induced vasodilation in vivo but display normal in vivo and ex vivo NVC within the first few hours after SAH. These results suggest an early dysfunction of NO signaling in cerebral vessels after SAH. Together with our previous results on delayed dysfunction of NVC later than 24 h after SAH, we conclude that multiple molecular pathways seem to be involved in microvascular dysfunction after SAH. Further research is needed to decipher these mechanisms thereby paving the way for novel therapeutic strategies for SAH patients who suffer from early and delayed cerebral ischemia.
Funding
This work was supported by the Solorz-Żak foundation, by the American Heart Association (14SDG20150027), National Institutes of Health (P01 HL095488, P30 RR032135, P30 GM103498 and S10 OD10583), Totman Medical Research Trust Fund, the Peter Martin Brain Aneurysm Endowment, and the Deutsche Forschungsgemeinschaft through the Munich Cluster of Systems Neurology (Synergy).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contribution
Matilde Balbi: performed all in vivo experiments and wrote the manuscript.
Masayo Koide: performed all ex vivo experiments and wrote the manuscript.
Susanne M. Schwarzmaier: provided technical input.
George C. Wellman: designed the study and edited the manuscript.
Nikolaus Plesnila: designed the study, wrote and edited the manuscript.
Supplementary Material
Disclosure/Conflict of Interest
No competing financial interests exist.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Moore CI, Cao R. The hemo-neural hypothesis: on the role of blood flow in information processing. J Neurophysiol 2008; 99: 2035–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci 2007; 10: 1369–1376. [DOI] [PubMed] [Google Scholar]
- 3.Attwell D, Buchan AM, Charpak S, et al. Glial and neuronal control of brain blood flow. Nature 2010; 468: 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction? Stroke 2009; 40(3 Suppl): S86–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver JP, Fisher M. Subarachnoid hemorrhage: an update of pathogenesis, diagnosis and management. J Neurol Sci 1994; 125: 119–131. [DOI] [PubMed] [Google Scholar]
- 6.Schubert GA, Seiz M, Hegewald AA, et al. Acute hypoperfusion immediately after subarachnoid hemorrhage: a xenon contrast-enhanced CT study. J Neurotrauma 2009; 26: 2225–2231. [DOI] [PubMed] [Google Scholar]
- 7.Friedrich B, Muller F, Feiler S, et al. Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: an in-vivo microscopy study. J Cereb Blood Flow Metab 2012; 32(3): 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pennings FA, Bouma GJ, Ince C. Direct observation of the human cerebral microcirculation during aneurysm surgery reveals increased arteriolar contractility. Stroke 2004; 35: 1284–1288. [DOI] [PubMed] [Google Scholar]
- 9.Uhl E, Lehmberg J, Steiger HJ, et al. Intraoperative detection of early microvasospasm in patients with subarachnoid hemorrhage by using orthogonal polarization spectral imaging. Neurosurgery 2003; 52: 1307–1315. discussion 1315–1317. [DOI] [PubMed] [Google Scholar]
- 10.Zhang JH. Molecular neurosciences. Neurol Res 2009; 31: 113. [DOI] [PubMed] [Google Scholar]
- 11.Sehba FA, Hou J, Pluta RM, et al. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol 2012; 97: 14–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hockel K, Scholler K, Trabold R, et al. Vasopressin V(1a) receptors mediate posthemorrhagic systemic hypertension thereby determining rebleeding rate and outcome after experimental subarachnoid hemorrhage. Stroke 2012; 43: 227–232. [DOI] [PubMed] [Google Scholar]
- 13.Koide M, Bonev AD, Nelson MT, et al. Inversion of neurovascular coupling by subarachnoid blood depends on large-conductance Ca2+-activated K+ (BK) channels. Proc Natl Acad Sci USA 2012; 109: E1387–E1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilkenny C, Browne W, Cuthill IC, et al. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacolgy 2010; 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thal SC, Plesnila N. Non-invasive intraoperative monitoring of blood pressure and arterial pCO2 during surgical anesthesia in mice. J Neurosci Methods 2007; 159: 261–267. [DOI] [PubMed] [Google Scholar]
- 16.Schwarzmaier SM, Plesnila N. Contributions of the immune system to the pathophysiology of traumatic brain injury – evidence by intravital microscopy. Front Cell Neurosci 2014; 8: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feiler S, Friedrich B, Scholler K, et al. Standardized induction of subarachnoid hemorrhage in mice by intracranial pressure monitoring. J Neurosci Methods 2010; 190: 164–170. [DOI] [PubMed] [Google Scholar]
- 18.Balbi M, Ghosh M, Longden TA, et al. Dysfunction of mouse cerebral arteries during early aging. J Cereb Blood Flow Metab 2015; 35: 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholler K, Feiler S, Anetsberger S, et al. Contribution of bradykinin receptors to the development of secondary brain damage after experimental subarachnoid hemorrhage. Neurosurgery 2011; 68: 1118–1123. [DOI] [PubMed] [Google Scholar]
- 20.Buhler D, Schuller K, Plesnila N. Protocol for the induction of subarachnoid hemorrhage in mice by perforation of the Circle of Willis with an endovascular filament. Transl Stroke Res 2014; 5: 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terpolilli NA, Kim SW, Thal SC, et al. Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circ Res 2012; 110: 727–738. [DOI] [PubMed] [Google Scholar]
- 22.Schwarzmaier SM, Zimmermann R, McGarry NB, et al. In vivo temporal and spatial profile of leukocyte adhesion and migration after experimental traumatic brain injury in mice. J Neuroinflammation 2013; 10: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girouard H, Bonev AD, Hannah RM, et al. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci USA 2010; 107: 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev 1998; 78: 53–97. [DOI] [PubMed] [Google Scholar]
- 25.Ayling J. Managing head injuries. Emerg Med Serv 2002; 31: 42. [PubMed] [Google Scholar]
- 26.Iadecola C. Does nitric oxide mediate the increases in cerebral blood flow elicited by hypercapnia? Proc Natl Acad Sci USA 1992; 89: 3913–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iadecola C, Zhang F. Nitric oxide-dependent and -independent components of cerebrovasodilation elicited by hypercapnia. Am J Physiol 1994; 266(2 Pt 2): R546–R552. [DOI] [PubMed] [Google Scholar]
- 28.Herz DA, Baez S, Shulman K. Pial microcirculation in subarachnoid hemorrhage. Stroke 1975; 6: 417–424. [DOI] [PubMed] [Google Scholar]
- 29.Kajita Y, Dietrich HH, Dacey RG., Jr Effects of oxyhemoglobin on local and propagated vasodilatory responses induced by adenosine, adenosine diphosphate, and adenosine triphosphate in rat cerebral arterioles. J Neurosurg 1996; 85: 908–916. [DOI] [PubMed] [Google Scholar]
- 30.Park KW, Metais C, Dai HB, et al. Microvascular endothelial dysfunction and its mechanism in a rat model of subarachnoid hemorrhage. Anesth Analg 2001; 92: 990–996. [DOI] [PubMed] [Google Scholar]
- 31.Park KW, Dai HB, Metais C, et al. Isoflurane does not further impair microvascular vasomotion in a rat model of subarachnoid hemorrhage. Can J Anaesth 2002; 49: 427–433. [DOI] [PubMed] [Google Scholar]
- 32.Katusic ZS, Milde JH, Cosentino F, et al. Subarachnoid hemorrhage and endothelial L-arginine pathway in small brain stem arteries in dogs. Stroke 1993; 24: 392–399. [DOI] [PubMed] [Google Scholar]
- 33.Nystoriak MA, O'Connor KP, Sonkusare SK, et al. Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am J Physiol Heart Circ Physiol 2011; 300: H803–H812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koide M, Wellman GC. SAH-induced suppression of voltage-gated K(+) (K (V)) channel currents in parenchymal arteriolar myocytes involves activation of the HB-EGF/EGFR pathway. Acta Neurochir Suppl 2013; 115: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishiguro M, Puryear CB, Bisson E, et al. Enhanced myogenic tone in cerebral arteries from a rabbit model of subarachnoid hemorrhage. Am J Physiol Heart Circ Physiol 2002; 283: H2217–H2225. [DOI] [PubMed] [Google Scholar]
- 36.Sun BL, Zheng CB, Yang MF, et al. Dynamic alterations of cerebral pial microcirculation during experimental subarachnoid hemorrhage. Cell Mol Neurobiol 2009; 29: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedrich B, Michalik R, Oniszczuk A, et al. CO2 has no therapeutic effect on early microvasospasm after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab 2014; 34: e1–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishikawa M, Kusaka G, Yamaguchi N, et al. Platelet and leukocyte adhesion in the microvasculature at the cerebral surface immediately after subarachnoid hemorrhage. Neurosurgery 2009; 64: 546–553; discussion 553–554. [DOI] [PubMed] [Google Scholar]
- 39.Cipolla MJ. The cerebral circulation, San Rafael, CA: Morgan & Claypool Life Sciences, 2009. [PubMed] [Google Scholar]
- 40.Koide MDK, Bulkeley EA, Nelson MT, et al. In vivo and ex vivo dysfunction of neurovascular coupling in a mouse model of subarachnoid hemorrhage. FASEB 2014; 28(1 Suppl): 676. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.