

Abstract
Matrix metalloproteinases (MMPs), particularly gelatinases (MMP-2/-9), are involved in neurovascular impairment after stroke. Detection of gelatinase activity in vivo can provide insight into blood–brain barrier disruption, hemorrhage, and nerve cell injury or death. We applied gelatinase-activatable cell-penetrating peptides (ACPP) with a cleavable l-amino acid linker to examine gelatinase activity in primary neurons in culture and ischemic mouse brain in vivo. We found uptake of Cy5-conjugated ACPP (ACPP-Cy5) due to gelatinase activation both in cultured neurons exposed to n-methyl-d-aspartate and in mice after cerebral ischemia. Fluorescence intensity was significantly reduced when cells or mice were treated with MMP inhibitors or when a cleavage-resistant ACPP-Cy5 was substituted. We also applied an ACPP dendrimer (ACPPD) conjugated with multiple Cy5 and/or gadolinium moieties for fluorescence and magnetic resonance imaging (MRI) in intact animals. Fluorescence analysis showed that ACPPD was detected in sub-femtomole range in ischemic tissues. Moreover, MRI and inductively coupled plasma mass spectrometry revealed that ACPPD produced quantitative measures of gelatinase activity in the ischemic region. The resulting spatial pattern of gelatinase activity and neurodegeneration were very similar. We conclude that ACPPs are capable of tracing spatiotemporal gelatinase activity in vivo, and will therefore be useful in elucidating mechanisms of gelatinase-mediated neurodegeneration after stroke.
Keywords: Focal ischemia, matrix proteins, molecular imaging, MRI, neurodegeneration
Introduction
Ischemic infarction is the most common form of stroke, occurring due to thromboembolic blockade of a cerebral artery, and results in oxygen deprivation and neurovascular impairment with parenchymal destruction and frequent hemorrhagic transformation (HT).1 Although the molecular mechanisms involved in these outcomes remain controversial, evidence supports the role of matrix metalloproteinases (MMPs), particularly gelatinases, in mediating excitotoxicity-induced disruption of cell-matrix homeostasis and ischemia-related impairment of the neurovascular unit.2,3 Several reports4–7 have demonstrated significantly elevated MMP-9 levels after stroke in humans.
MMPs, a family of 26 zinc-dependent endopeptidases, regulate tissue homeostasis and remodeling.1,3 MMPs contain a conserved N-terminal prodomain with a critical cysteine that chelates a catalytic zinc ion, keeping the proMMP zymogens inactive. We and subsequently others reported that nitric oxide (NO), which can be generated in neurons by stimulation of N-methyl-d-aspartate (NMDA)-type glutamate receptors, induces gelatinase (MMP-2 and -9) activation via S-nitrosylation; excessive activation of MMPs results in apoptosis,8–10 disruption of the blood–brain barrier (BBB), hemorrhage, neuroinflammation, and extracellular matrix (ECM) turnover.11,12
In vivo detection of proteolysis by gelatinases after cerebral ischemia may provide mechanistic insight into MMP signaling during aberrant ECM remodeling that contributes to neurovascular impairment. Such detection might offer critical assessment and validation of new therapeutic interventions. A prior report demonstrated in vivo imaging of protease activity using fluorescence probes in a xenograft tumor model.13 However, although gelatinases could be detected, cathepsins gave more robust signals and became the preferred imaging target. This approach, however, with low sensitivity for MMPs, was not applied to neurovascular disease. Another technique for in vivo imaging of MMP activity uses multiplex FRET-quenched probes, but its use is limited to transparent species and small embryos.14 Brain MRI is widely used for stroke diagnosis because of its high-resolution tomographic signal. However, small molecule gadolinium-based contrast agents (GBCAs) manifest limited sensitivity for detection in the range ≤10−5 M, making it difficult to obtain sufficient contrast enhancement for imaging.15
In the present study, we implemented a new strategy using gelatinase-activatable cell-penetrating peptides (ACPPs) tagged with multiple fluorescence and/or GBCAs to investigate gelatinase proteolysis in cultured neurons as well as in intact mice after focal cerebral ischemia. Detection of in vivo gelatinolytic activity by ACPPs could provide molecular insight into the underlying mechanism(s) of MMP proteolysis that mediates ischemia-related neurovascular impairment, and could serve as a surrogate indicator of brain damage after stroke.
Materials and methods
ACPP syntheses
ACPPs for fluorescence imaging, ACPPs attached to dendrimers (ACPPDs) for MRI, and their negative controls, containing relatively uncleavable d-amino acids (ucACPP and ucACPPD), were synthesized as previously described.16–18 ACPPs and ucACPPs consist of polycationic CPPs (arginine9) labeled with Cy5 and fused using cleavable (-PLGC(Me)AG-) or resistant (-plgc(Me)ag-) linkers, respectively, to matching polyanions (succinyl-glutamate8). This strategy reduces the net charge towards zero and inhibits adhesion and uptake into cells (Scheme 1), allowing improved tissue distribution. If the linkers are cleaved by MMP-2/-9, the polyanions are released, locally unmasking the polycations with their inherent adhesiveness, whereas corresponding d-amino acids in the linkers of ucACPPs resist cleavage. The ACPPDs and ucACPPDs consist of generation five poly(amido)amine (Gen 5 PAMAM) dendrimers labeled with an average of six ACPPs or ucACPPs, 30 Gd-DOTA moieties, and three Cy5. ACPPDs work by the same principle as non-dendrimeric ACPPs, but the 5:1 Gd-DOTA:ACPP ratio increases Gd uptake and the higher molecular weight slows pharmacokinetics.16–18
Scheme 1.
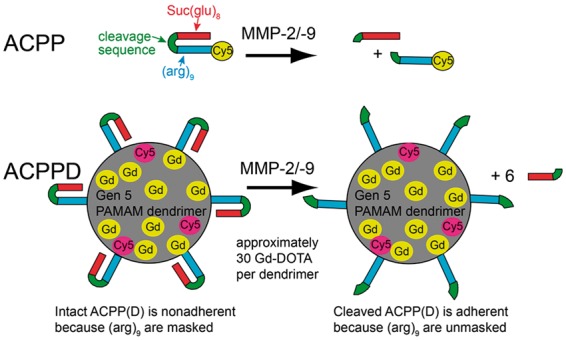
Structure of ACPPs and ACPPDs. While the cleavage sequences (green) between the polyanion (Suc(glu)8, red) and polycation ((arg)9, blue) of ACPP (top row) or ACPPD (bottom row) remain intact, the ACPP(D) remains nonadherent due to the masking of (arg)9. Once MMP-2/-9 cleaves the linkers, the polyanions are released, allowing the polycations and their payloads (Cy5 and Gd) to adhere to cells and become endocytosed. Only nine out of approximately 30 Gd-DOTA are depicted in ACPPDs.
Cell cultures
Primary cultures of the enriched neurons were prepared from embryonic (day 16 to 18 in gestation) Sprague–Dawley rats as described previously.8,19 Following the dissection of cerebral cortex and hippocampus in Hank’s buffer, cells were dispersed into a single-cell suspension. Cerebral cortical or hippocampal neurons were plated at density of 6.7 × 105 per 35-mm dish containing poly-l-lysine-coated 12-mm glass cover-slips. The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) with Ham’s F12 nutrient medium and heat-inactivated bovine calf serum (BCS) at a ratio of 8:1:1. After 10 days, the neurons were enriched by treatment with cytosine arabinoside for 72 h. The cultures were incubated at 37℃ in a 5% CO2, 95% air humidified atmosphere and used for experiments approximately two weeks after plating. Activation of gelatinases in neurons was achieved by S-nitrosylation of MMP via exposure to a neurotoxic concentration of NMDA.8 As negative controls, we applied cleavable-resistant ACPP-Cy5 (ucACPP) with d-amino acid linker plglag, and used a non-selective metal chelator and broad-spectrum MMP inhibitor, calcium diethylenetriaminepenta-acetate (Ca-DTPA, 5 mM), to inhibit MMP activity. After washing three times with EBSS containing Ca2+ and equilibrating with 5 µM glycine for 15 min, 300 µM NMDA was added for a 10-min incubation. The cells were incubated for 3 h at 37℃ in either the absence or presence of Ca-DTPA. Cells were then fixed with 4% paraformaldehyde (PFA), permeabilized with 0.3% Triton X-100 for 5 min. Neurons could be reliably recognized by morphological criteria under phase-contrast microscopy, as later confirmed by immunocytochemistry of neuron specific markers.
Experimental models of focal cerebral ischemia
A total of 71 mice were used in this study; 18 mice were excluded due to unexpected death or because of pre-specified exclusion criteria, including cerebral hemorrhage. Two mouse models of focal cerebral ischemia were used in this study: a filament-occlusion model and an embolic ischemia model, representing mechanical blockage and clot-induced occlusion, respectively, as we have previously described.20,21 All animal experiments were approved by the University of Missouri-Columbia Animal Care and Usage Committee and performed in accordance with the ARRIVE guidelines. Adult male C57BL/6J mice (8–10 weeks old from The Jackson Laboratory, Bar Harbor, ME), weighing 20–25 g, were housed in a 12-h light/dark cycle and permitted food and water intake ad libitum. Anesthesia was induced with 5.0% gaseous isoflurane in a nitrogen–oxygen mixture, and maintained with 1.5% gaseous isoflurane in a nitrogen/oxygen mixture. The body temperature was maintained at 37℃ during surgery. For the filament-induced transient middle cerebral artery (MCA) occlusion, a silicon-coated 6-0 monofilament was introduced from external carotid artery into the circle of Willis under isoflurane anesthesia to block the origin of the MCA. After a 2-h occlusion, the filament was then removed for 24 h of reperfusion. For embolic cerebral ischemia model, we first prepared emboli by obtaining blood from a donor mouse in a 3-foot long sterile PE-50 polyethylene tube, with subsequent incubation at 37℃ for 2 h and cooling at 4℃ overnight to form fibrin-rich blood clots. Blood clots were washed with sterile phosphate buffered saline (PBS) in 2-foot long PE-10 tubing for 6–8 rounds. After ejection from the PE-10 tubing, clots were cut into 10-mm long (0.02 µL) pieces. A single piece of autologous clot was inserted via a PE-10 catheter with a modified tip of ∼0.20 mm outer diameter from the external carotid artery into the circle of Willis to the origin of the MCA.
A laser Doppler flowmeter (Moor Laboratory, London, UK) with a probe fixed on the skull surface (3 mm lateral to midline and 2 mm posterior to the bregma), was positioned at the distal arterial supply of the MCA to measure regional CBF (rCBF). The initial reading of rCBF was assigned a value of 100%, and subsequent readings were expressed relative to this value. Monitoring of rCBF showed that mice sustained cerebral ischemia to less than 25% of pre-ischemic baselines. The gelatinase selective inhibitor SB-3CT (12.5 mg/mL) was prepared as a suspension formulated in vehicle [25% DMSO/65% polyethylene glycol 200 (PEG-200)/10% H2O], as described previously.20 Mice were intraperitoneally injected with 2 µL/gram body weight of this solution (equivalent to 25 mg/kg) 2 h after embolic ischemia, followed by an additional dose at 4 h. Animals were randomly assigned to different experimental groups.
Assessment of gelatinase activity by ACPPs compared to in situ zymography
The uptake of ACPP-Cy5 was compared to the benchmark assay—in situ zymography—using gelatinase-specific fluorogenic substrate DQ-gelatin-FITC (Thermo Fisher Scientific, Waltham, MA, USA) on both primary cultured neurons and brain sections, as described previously.8,22 Active gelatinases, in cultured neurons exposed to NMDA or in ischemic regions, attack the intramolecularly quenched DQ-gelatin-FITC and yield fluorescent gelatin-FITC peptides, which can be viewed under epifluorescence microscopy. For in situ zymography on brain sections, 8 µm thick cryosections were cut from OCT-embedded fresh mouse brain and incubated with 10 µg/ml DQ-gelatin-FITC in TBS overnight at 37℃ with broad-spectrum MMP inhibitor GM6001 (50 µM) or 1 × non-MMP cocktail of protease inhibitors (PIC). Cells and cryosections were then post fixed with 4% PFA.
Quantification of ACPP-Cy5 uptake in ischemic brains
A highly sensitive fluorescence assay monitoring the uptake of ACPPs was developed based on a previously described method23 with the following modifications. After euthanasia, brains were harvested, and tissues were separated into left and right cortex, hippocampus and striatum. The tissue was then cut into 2 × 2 mm pieces and homogenized using an Eppendorf grinder in three volumes of 50 mM Tris lysis buffer (e.g. 300 µL of lysis buffer/100 mg of tissue) with 1% SDS at pH 7.6. Striata or hippocampi of two animals were pooled together to achieve a final tissue weight of ≥30 mg. Tissue homogenates were sonicated three times for 10 s each at power level 2. The samples were heated at 80℃ for 10 min, mixed well, and transferred to black 96-well plates at 100 µL/well in triplicate for each sample. Fluorescent ACPPs were detected with a Synergy-4 multi-mode microplate reader (BioTek, Winooski, VT, USA) at excitation/emission wavelengths of 650/680 nm with sensitivity set at 150. To generate a standard curve, ACPP-Cy5 at concentrations of 0, 0.63, 1.25, 2.5, 5, 10, 20, 50, 100, and 200 nM was mixed with brain tissue lysates, and their fluorescence intensities were determined. The uptake of ACPP-Cy5 in brain tissue was converted to femtomole (fmol) concentrations of ACPPs per mg tissue.
Histology and immunohistochemistry
After transcardiac perfusion fixation with 4% paraformaldehyde, mouse brains were dissected and cut into serial coronal sections of 40 -µm thickness. Ischemic brain damage was identified by detecting neuronal cell death with cresyl violet staining. Cells or tissue sections were immunostained with the following antibodies: neuronal markers microtubule associated protein-2 (MAP-2, 1:200, M4403, Clone HM-2; Sigma-Aldrich, St. Louis, MO) and NeuN (1:200, MAB377, Clone A60; Millipore, Temecula, CA); ECM basement membrane component laminin (1:200, L9393; Sigma-Aldrich, St. Louis, MO, USA); microglia marker (Iba-1, 1:500, 019-19741;Wako, Richmond, VA, USA) and astrocyte marker (GFAP, 1:400, G3893; Sigma-Aldrich, St. Louis, MO). Cells or sections were visualized with fluorophore-conjugated secondary antibodies (1:200, goat anti-mouse IgG-Alexa488, A11001; goat anti-mouse IgG-Alexa594, A11005; or goat anti-rabbit IgG-Alexa488, A110034; Invitrogen, San Diego, CA, USA). Cells and sections were counterstained with nuclear DNA dye Hoechst 33342. Brain sections were examined by an Olympus IX81 spinning-disc confocal microscopy (Olympus America Inc., Center Valley, PA, USA) using Slidebook 5.1 (Intelligent Imaging Innovations, Denver, CO, USA) “Montage” image re-construction software.
In vivo MRI analysis of brain ischemia
In vivo MRI was performed using a 7-T Varian Unity Inova MRI equipped with a Millipede quadrature RF coil (40 mm ID), as described in detail previously.24 Mice were anesthetized with 1.5% isoflurane in oxygen. A T1-weighted multislice spin echo sequence was performed to record images pre- and post-infusion of contrast agents. The imaging parameters were TR = 500 ms, TE = 15 ms for T1WI (T1-weighted imaging); TR = 2500 ms, TE = 15 ms for T2WI (T2-weighted imaging); slice thickness = 1 mm, 12 slices, average = 4, matrix = 256 × 128, and FOV = 25 × 25 mm (axial) or 40 × 25 (coronal). Initially, we examined ischemic mouse brains at various time points including 4 and 24 h, and 2, 4 and 7 days to determine the intensity profiles of MRI T1WI and T2WI as well as diffusion-weighted images (DWI) without any contrast agent. We then applied ACPPD consisting of (on average) six cleavable ACPPs, three Cy5 and 30 Gd-DOTA per generation 5 dendrimer (ACPPD-Cy5/Gd). As a baseline for T1WI intensity, pre-infusion imaging had been acquired two days after a 60-min transient MCA occlusion. ACPPD-Cy5/Gd (400 nmol/kg body weight) or ucACPPD was slowly infused into the femoral vein in 200 µL saline for 30 min; ucACPPD with an equimolar amount of Gd served as a negative control. Mice were imaged before infusion and two days post-infusion (corresponding to two and four days post-ischemia). Vital signs were continually monitored, and mice were returned to their cage after each imaging session to fully recover. Two mice with high T1WI intensity in the ischemic region were excluded because of spontaneous cerebral hemorrhages that were confirmed after sacrifice.
Image analysis and processing were performed with ParaVision 5.1 (Billerica, MA, USA), ImageJ (National Institute of Health), or VnmrJ2.1D software (Palo Alto, CA, USA) by an individual blinded treatment group. Regions of interest (ROIs) were manually drawn on the ischemic brain region of T1-weighted imaging (T1WI) scans for each time point. Control values were determined from ROIs drawn on the corresponding anatomical structure in the contralateral side. Signal intensity (SI) was measured and signal enhancement ratio (SER) was calculated according to the following equation, as described previously.25,26
Quantitative analysis of Gd uptake in ischemic brains
Inductively coupled plasma mass spectrometry (ICP-MS) was performed for quantification of Gd uptake in ischemic brains as previously described.16 Mice were sacrificed with an overdose of isoflurane two days post-infusion of contrast agents (four days post-ischemia). The brain tissues were collected and wet weight determined. The samples were then freeze-dried and digested with 300 µL HNO3 (Fisher brand Optima grade, concentrated) at 190℃ in an ETHOS Plus microwave unit using vessel inserts in a SK-12 medium pressure rotor with temperature control (Milestone, Shelton, CT, USA). Ultrapure water was added to the digested brain tissues to bring the volume to 10 mL. Internal standards consisting of indium (In) and thallium (Tl) were added to all samples. Samples were analyzed using a NexION ICP-MS (PerkinElmer, Waltham, MA, USA) calibrated with five Gd linearity standards in 3% HNO3. The sample limits of detection were calculated based on the instrument limit of detection as three times the standard deviation of the Gd concentration in multiple analyses of zero point standards (blank 3% HNO3 with internal standards). The dilution of samples due to the digestion process was taken into account when calculating the limit of sample detection. The content of Gd in the tissue was measured based on sample wet weight. Gd contents for individual mice were calculated as follows: ng Gd/g wet tissue in ischemic hemisphere − ng Gd/g wet tissue in contralateral hemisphere. As controls, there was no detectable Gd in the digestion blanks, and multiple isotopes of Gd were monitored to confirm the results. Measurements were performed by an individual blinded to the identity of the samples.
Statistical analysis
Ex vivo ACPP-Cy5 fluorescence intensities in mouse brain lysates were plotted against mean fluorescence intensities, followed by a linear regression analysis to calculate R2 and to determine the regression equation. Differences in the mean fluorescence intensities were measured over the ischemic and contralateral hemispheres. Data are presented as mean ± SEM. Based on predictions formulated with prior data, comparisons between two samples were made using a one-tailed Student’s t-test. A value of p < 0.05 was considered statistically significant.
Results
Detection of in situ gelatinolytic activity by ACPPs in cultured neurons
We first examined the uptake of ACPPs in neurons responding to NMDA-induced gelatinase activation. Deconvolution microscopy revealed that the fluorescence intensity of ACPP-Cy5 was greater in neurons exposed to NMDA compared to vehicle-treated neurons (Figure 1(a)). The broad-spectrum MMP inhibitor, Ca-DTPA (5 mM), abolished ACPP uptake. We validated uptake of ACPP-Cy5 by immunocytochemistry. After exposure to NMDA, ACPP-Cy5 uptake was observed mainly in neurons, as confirmed with the specific neuronal markers MAP-2 and NeuN (Figure 1(b)), with Ca-DTPA significantly attenuating the ACPP-Cy5 fluorescence. Under similar conditions, there was no apparent uptake of the cleavage-resistant ucACPP. These results are consistent with prior findings that ACPPs are cleaved by active gelatinases and the released CPPs taken up into tumor cells.16–18,23
Figure 1.

Detection of gelatinase activity by ACPPs in cultured neurons exposed to NMDA. (a) and (b), deconvolution microscopy reveals greater uptake of the gelatinase-cleavable ACPP-Cy5 (red) in cultured neurons after gelatinase activation by NMDA compared to vehicle-treated neurons. The broad-spectrum MMP inhibitor Ca-DTPA (5 mM) reduced ACPP-Cy5 uptake. Neurons were identified by immunostaining with the specific neuronal markers MAP-2 and NeuN (green); DAPI (blue) labeled nuclei. As a control, cleavage-resistant ucACPP was not detected in neurons exposed to NMDA. (c) Gelatinase activity by in situ zymography at 0, 3, 24 h after incubation in 10 µg/ml DQ-gelatin-FITC compared to a 3 h incubation in 1 μM ACPP-Cy5. Scale bar, 40 µm (a), 20 µm (b), and 250 µm (c).
To further validate the specificity of ACPP cleavage by active gelatinases, we examined uptake of ACPP-Cy5 after NO-mediated gelatinase activation in HT1080 cells, a cell line derived from human fibrosarcoma that is capable of secreting substantial amounts of gelatinases.27 We found a significant increase in the uptake of ACPP-Cy5 in HT1080 cells exposed to the physiological NO donor GSNO, with Ca-DTPA significantly attenuating the ACPP-Cy5 fluorescence. Under similar conditions, there was no apparent uptake of cleavage-resistant ACPP-Cy5 (Supplementary Figure S1). These results suggest that active forms of gelatinases can be detected by ACPP cleavage.
We next examined the uptake of ACPP-Cy5 for its sensitivity compared to the benchmark assay of in situ zymography using DQ-gelatin-FITC for the detection of MMP activity. After incubation of cultured neurons with DQ-gelatin-FITC, gelatinase fluorescent activity was barely detectable at 3 h, but clearly observable by ∼24 h (Figure 1(c)). In contrast, uptake of ACPP-Cy5 was detected within 3 h at a level equivalent to that of the DQ-gelatin-FITC value at 24 h.
Detection of in vivo gelatinolytic activity by ACPPs in ischemic mouse brain
We first evaluated ACPP-Cy5 uptake to detect in vivo gelatinolytic activity in ischemic mouse brain. ACPP-Cy5 uptake was observed throughout the microvasculature of the brain within 10 min of injection and dissipated shortly thereafter. Within 4 h, ACPP-Cy5 was evident in the ischemic penumbra, but not the contralateral cerebral cortex (Figure 2). In mice with embolic ischemia, uptake of ACPP-Cy5 was detected in cells in the ischemic region, including the cortex, striatum, and hippocampus seven days after ischemia (Figure 3). Neuronal cell death in the ischemic region was confirmed by cresyl violet staining. Mice treated with the highly specific thiirane gelatinase inhibitor SB-3CT (25 mg/kg) showed attenuation in ACPP-Cy5 uptake. Mice injected with the cleavage-resistant ucACPP showed no preferential fluorescence uptake in ischemic regions.
Figure 2.
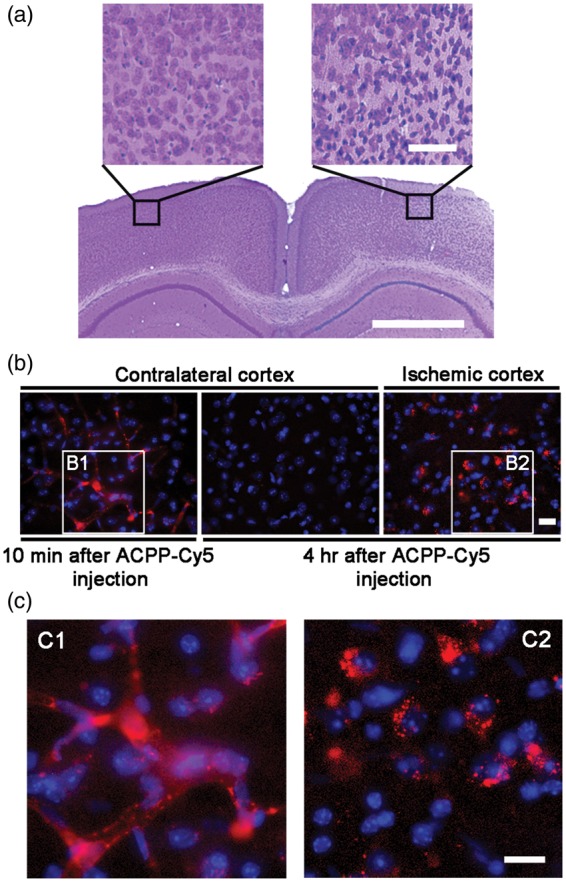
Detection of ACPP-Cy5 distribution in ischemic mouse brains. (a) Representative microphotographs of the cresyl violet staining show the contralateral and ischemic penumbra areas in cortex. (b) Deconvolution microscopy reveals that ACPP-Cy5 remained in brain microvasculature 10 min after intravenous (i.v.) injection of 2 nmol ACPP-Cy5, but was not detected in contralateral cortex 4 h later. ACPP-Cy5 was taken up into the ischemic penumbra area in cortex 4 h after i.v. infusion. (c) Enlarged microphotographs (C1 and C2) of the outlined areas in (b) panels (B1 and B2), respectively. Scale bar, 50 µm (a) and 20 µm (b, c).
Figure 3.

Association of ACPP-Cy5 uptake with neuronal cell death seven days after embolic MCA occlusion in mice. ACPP-Cy5 or uncleavable ACPP-Cy5 (2 nmol) was injected i.v. seven days after embolus-induced MCA occlusion and 4 h prior to sacrifice of the mice. (a) Deconvolution microscopy reveals ACCP-Cy5 (red) uptake in ischemic region. Gelatinase selective inhibitor SB-3CT (25 mg/kg) attenuated the uptake of ACPP-Cy5 in ischemic regions. The cleavage-resistant ACPP (ucACPP) control was not detectable in the ischemic regions. (b) Neuronal cell death in the ischemic region was confirmed by cresyl violet staining, which revealed irregular, condensed cell bodies compared to round, faintly-stained healthy neurons in the contralateral region. ACPP-Cy5 uptake into the ischemic hemisphere correlated with neuronal cell death identified by cresyl violet staining in cortex, striatum, and hippocampus. (c) Corresponding regions of the contralateral hemisphere are shown for comparison. Scale bar, 1 mm (a) and 100 µm (b and c).
ACPP-Cy5 uptake was then compared with the conventional method in detecting gelatinase activity, in situ zymography, on the same brain section after MCA occlusion in mice. Increased fluorescence intensity of both DQ-gelatin-FITC and ACPP-Cy5 was observed in the ischemic zone, including the hippocampal CA3 region (Figure 4(a)), compared to the contralateral side. Cell death in the ischemic region was confirmed by Hoechst dye staining, which revealed condensed cell nuclei when compared to the contralateral side. We found that ACPP-Cy5 fluorescence localized to the para-nuclear area (Figure 4(a), inset). Furthermore, the broad-spectrum MMP inhibitor GM6001, when applied at 50 µM to sections of the ischemic brain, resulted in attenuation of proteolytic activity, as detected by in situ zymography performed ex vivo. GM6001, applied ex vivo, did not affect ACPP-Cy5 uptake as expected. Additionally, gelatinase activity was not inhibited by a non-MMP cocktail of protease inhibitors (PIC). ACPP-Cy5 accumulated in subcellular compartments as punctate fluorescent aggregates in the same distribution as the ischemic region. In contrast, the more diffuse fluorescent pattern, detected by ex vivo proteolysis of DQ-gelatin-FITC in unfixed tissue, can occur due to diffusion of the label.
Figure 4.

Detection of gelatinase activity by ACPPs in mouse brain after MCA occlusion. (a) Representative micrographs of ACPP-Cy5 uptake and in situ zymography (ISZ) in the hippocampal CA3 subregion. ACPP-Cy5 (2 nmol) was injected i.v. after 2-h filament-induced MCA occlusion in mice, which were sacrificed 24 h later. ACPP-Cy5 uptake by cells in the brain parenchyma displayed a similar pattern of distribution to increased DQ-gelatin-FITC labeling in the ischemic CA3 region, but not in the contralateral hippocampus. MMP inhibitor GM6001applied ex vivo to brain frozen sections attenuated the ISZ fluorescence of DQ-gelatin-FITC in the ischemic hippocampus, but did not affect uptake of ACPP-Cy5. As a negative control, the non-MMP protease inhibitor cocktail (PIC) did not reduce ISZ fluorescence intensity. Brain sections were counterstained with Hoechst to visualize nuclei (blue). Scale bars, 20 µm, and 10 µm (inset). (b) ACPP-Cy5 uptake was quantified ex vivo in different brain regions 24 h post-ischemia. ACPP-Cy5 uptake was detected at fmol/mg levels and significantly increased in the ischemic cortex (**p < 0.005, n = 8) and striatum (*p < 0.05, n = 10) compared to contralateral control. (c) Differences in ACPP-Cy5 uptake between ischemic and contralateral regions were compared after SB-3CT treatment or vehicle-treated control. ACPP-Cy5 uptake was significantly attenuated by the gelatinase selective inhibitor SB-3CT in the cerebral cortex (*p < 0.05, n = 8 vehicle-treated mice, n = 6 SB-3CT treated mice). Data are expressed as mean ± SEM. Statistics determined by one-tailed Student’s t-test.
To quantify ACPP uptake, fluorescence intensity from various regions of ischemic and non-ischemic brains were analyzed ex vivo. A standard curve indicated a linear range over ACPP-Cy5 concentrations from 0 to 50 nM (data not shown). We were able to detect ACPP update at fmol levels in brain tissue, representing a 2.1- to 7.6-fold increase in the ischemic cortex and striatum compared to the contralateral region (Figure 4(b)). The gelatinase inhibitor SB-3CT attenuated ACPP-Cy5 uptake into the ischemic cortex by 78% (Figure 4(c)). These results suggest that the peptide is not only selective but also specific for gelatinases under these conditions.
Correlation of the increased gelatinase activity indicated by ACPP-Cy5 fluorescence to subsequent neurodegeneration in ischemic mouse brain
In the present study, 4-channel immunofluorescent images were obtained in order to demonstrate the link between gelatinase activity and neurovascular impairment after ischemia. In the ischemic cortex (Figure 5(a)) and hippocampus (Figure 5(b)), we observed an increase in ACPP-Cy5 fluorescence intensity correlated with loss in neuronal laminin and neurodegeneration—indicated by loss of neuronal cell bodies and dendrites. In contrast, the contralateral cortex and hippocampus showed little if any ACPP-Cy5 uptake, intact neurons, and typical ring-shaped laminin structures. Additionally, we observed that ACPP-Cy5 uptake correlated with microglial activation, but not astrocytic proliferation after focal ischemia (Figure 6).
Figure 5.

Uptake of ACPP-Cy5 is associated with loss of neuronal laminin and neurodegeneration in ischemic brain. ACPP-Cy5 (2 nmol) was injected i.v. after 2-h filament-induced MCA occlusion in mice, which were sacrificed 24 h later. Sections were immunostained with the neuronal markers NeuN and MAP-2 (red) to visualize cell bodies and processes; ECM laminin was labeled with anti-laminin antibody (green) and cell nuclei with Hoechst dye (blue). (a) Neuronal loss (bottom panel) and adjacent laminin staining in the vasculature (middle panel) correlate with uptake of ACPP-Cy5 (cyan, top panel) in the ischemic cortex (separated by a white dashed line). (b) In ischemic hippocampus, white arrows indicate areas of increased ACPP-Cy5 uptake (top panel), correlating with loss of laminin immunoreactivity (middle panel) and neuronal degeneration (loss of neuronal markers in bottom panel). In contrast, green arrows indicate areas of decreased ACPP-Cy5 uptake, correlating with relatively intact laminin and less impaired neuronal processes. The contralateral hippocampus manifested minimal ACPP-Cy5 uptake. Scale bar, 30 µm (top, inset) and 75 µm (bottom).
Figure 6.
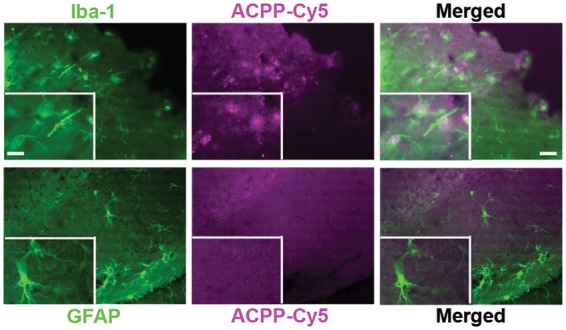
Detection of ACPP-Cy5 uptake with activated microglia after MCA occlusion. ACPP-Cy5 in 2 nmol contents was i.v. injected 10 min after ischemia and the mice were sacrificed 24 h later. Representative photomicrographs show that immunoreactivity of microglial marker Iba-1, but not astrocytic marker GFAP, correlates with uptake of ACPP-Cy5 in ischemic cortex. Scale bar, 30 µm (right) and 15 µm (left, inset).
In vivo monitoring of gelatinase activity in ischemic brain with ACPPD as an MRI contrast agent
Cerebral ischemia analyzed by MRI shows increased BBB permeability in contrast-enhanced T1WI. Aberrant activation of MMP-9 is known to contribute to this BBB leakage after ischemia.1–3 Therefore, we analyzed MRI scans using the contrast agents ACPPD or ucACPPD with dual Cy5 and Gd-DOTA to study the gelatinase activity in ischemic mouse brain. We observed contrast enhancement in ischemic regions two days after administration of ACPPD compared to ucACPPD (Figure 7(a)). For quantitative analysis, we initially determined the Signal Enhancement Ratios (SER) of T1WI intensity in ROIs for ACPPD and ucACPPD, which were almost identical before infusion (p = 0.48). The SER for ucACPPD was not significantly higher two days after infusion compared to pre-infusion (p = 0.24). In contrast, the SER for ACPPD was increased by 68.3% by two days after infusion (p = 0.0046). Moreover, the SER for ACPPD was 46.7% greater than for ucACPPD by two days after infusion (p = 0.0169; Figure 7(b)). The accumulation of ACPPD in the penumbra of ischemic cortex was further confirmed by fluorescence microscopy (Figure 7(c)).
Figure 7.

In vivo detection of gelatinase proteolysis with ACPPD, and quantification of Gd uptake by ICP-MS in mouse brain after MCA occlusion. (a) In vivo T1-weighted MRI before and two days after administration of ACPPD or ucACPPD (each containing 30 Gd-DOTA per dendrimer). Infusions were performed i.v. two days after ischemia with equivalent doses of approximately 300 nmol Gd (calculated as 400 nmol/kg × ∼30 Gd/dendrimer per 25 g mouse). Two days later, there was an increase in signal intensity contrast in the ischemic region (red arrow) of ACPPD-treated mice (mouse red#4 shown in panel (b)) compared to ucACPPD (mouse blue#1 shown in panel (b)). (b) Comparison of the signal enhancement ratio (SER) from the signal intensity (SI) measured by T1 MRIs among the various groups. Before infusion, the SER was not significantly different between the ACPPD and ucACPPD groups (p = 0.48). Within two days of infusion, the SER was significantly greater for ACPPD (*p = 0.0046), but not for ucACPPD (p = 0.24), compared to that of pre-infusion. Moreover, the SER was significantly greater for ACPPD than ucACPPD two days after infusion (#p = 0.0169 by one-tailed Student’s t-test, n = 5 in the ACPPD group; n = 3 in the ucACPPD; scattergram plots represent each mouse). (c) Ex vivo fluorescence imaging showing uptake of ACPPD in ischemic cortex compared to the contralateral side. There was virtually no uptake of ucACPPD in the ischemic cortex. Scale bars, 30 µm. (d) Quantification of ACPPD uptake in mouse brain after MCA occlusion. After MRI, mice were sacrificed and brain tissue separated into right and left hemispheres. Gd content of each sample was measured using ICP-MS and presented as the difference in ng Gd/g wet tissue between ischemic and contralateral hemispheres, which was significantly greater for ACPPD than ucACPPD (*p = 0.0134 by one-tailed Student’s t-test, n = 5 for ACPPD; n = 3 for ucACPPD). Data are expressed as mean ± SEM.
Next, in order to quantify the uptake of ACPPD in ischemic mouse brain two days after infusion, Gd content was measured by inductively coupled plasma mass spectrometry (ICP-MS) and presented as the differences of ng Gd/g wet tissue between ischemic hemispheres and contralateral hemispheres. There was a significant (p = 0.0134) increase by 1.67 fold in Gd from ACPPD (74.8 ± 6.1 ng/g) vs. ucACPPD infusions (44.7 ± 8.7 ng/g; Figure 7(d)). As controls, there was no detectable Gd in the digestion blanks, and multiple isotopes of Gd were monitored to confirm the results. Note that the Gd probably accumulated preferentially in the zone of ischemia, so that local Gd concentrations would be much higher than these averages obtained over the entire hemisphere.
Discussion
ACPPs consist of a polycationic cell-penetrating peptide (typically nine d-arginine residues or r9) connected via a cleavable l-amino acid linker (here PLGLAG) to a matching polyanionic string of d-glutamate residues (e9), which reduces the net charge to nearly zero and inhibits adhesion and uptake into cells. These peptides allowed us to analyze gelatinase activity with high spatial resolution in cell-based models and in mouse brain after cerebral ischemia in vivo. As in previous studies16–18,23 on tumor cells and other tissues, avid uptake of ACPPs is largely blocked by their polyanionic sequences, which neutralize polycations by forming intramolecular hairpins. Cleavage of the l-amino acid linkers by gelatinases dissociates the inhibitory polyanions and releases the fluorescent and/or Gd-tagged polycationic peptides, which attach to the cell surface and are taken up by cells.16,18 Analogous thrombin-specific ACPPs have been generated with DPRSFL or PPRSFL as the cleavage sequence, showing the role of this enzyme in atherosclerosis and ischemic stroke.28,29
MMP-9 has been considered as a potential biomarker to diagnose and quantify stroke in clinical research and hospital laboratories,30 especially to predict HT in acute ischemic stroke.4 However, there has not been a corresponding in vivo analysis of the role of gelatinases in ischemic stroke. In this study, we examined uptake of ACPP or ACPPD in two paradigms―NMDA-induced excitotoxicity in primary neuronal cultures and in vivo after focal cerebral ischemia in mice. In our previous studies, we demonstrated neuronal injury and death involves NMDA-mediated excessive Ca2+ influx, which in turn triggers formation of nitric oxide (NO) via neuronal NO synthase (nNOS). With gelatin zymography, we found that NMDA activates MMP-9, but not MMP-2, in neural cells.9 Additionally, Peixoto et al. tested the roles of different MMPs in NMDA stimulated neurons with broad and specific MMPs inhibitors and knockout mice. They found that only MMP-9, neither MMP-2 nor MMP-3, was involved in Ca2+/calmodulin-dependent protein kinase signaling pathway.31 For in vivo studies, using the same ischemic mouse models, we previously observed during the course of experiments that MMP-9 levels were dramatically unregulated and activated while MMP-2 levels remained similar in the ischemic cortex and the non-lesioned cortex, suggesting that MMP-9 is the predominant gelatinase involved, at least under our experimental conditions.8,20,21,32 Our observations are consistent with the studies of Lo and others,33–35 suggesting that there is only low level induction of MMP-2 after cerebral ischemia in rodent models; moreover, these levels of MMP-2 are much lower than those of MMP-9. Similar results were found in brain tissues and blood from human stroke patients showing increased MMP-9 levels.4,5,36 However, studies from the group of del Zoppo37,38 show that MMP-2 is detectably increased in the basal ganglia of baboons early after cerebral ischemia. In fact, the levels of MMPs could depend on the different animal models, brain region, and time course studied after ischemia. Nevertheless, we observed that MMP-9 was the predominant gelatinase under our experimental conditions.8,20,21,32 It should be noted, however, that our present ACPPs do not discriminate between MMP-2 and MMP-9, although we are actively developing more selective ACPPs.
Initially, we observed uptake of ACPP-Cy5 as early as 3 h after exposure to NMDA in neuronal cultures, indicating increased gelatinase activity. Uptake of ACPP-Cy5 into cells occurred much more rapidly than the detection of gelatinase activity by in situ zymography, which required 24 h. After the linker peptide is cleaved by gelatinases, ACPPs deliver their payload not only to the cell surface, which occurs because of the polycationic charges, but also to the cell interior via endocytosis.18
Our next objective was to develop in vivo molecular imaging to facilitate the study of neurodegeneration in ischemic mice. We found that gelatinase activity can be detected by ACPP-Cy5 as early as 4 h post-infusion. Moreover, uptake of ACPPs was detected at fmol levels in ischemic mouse brain. The distribution of ACPP-Cy5 uptake was similar to the pattern of activity observed with in situ zymography. During in situ zymography, detection of net enzymatic activity is valid only if the zymogens are not artificially activated; hence, valuable information can be lost during sample preparation.14 In contrast, ACPP is a more rapid and reliable method to detect in vivo proteolytic activity.
Since laminin is a substrate of MMPs, increased activity of MMPs (especially MMP-9) during cerebral ischemia is associated with loss of neuronal laminin.21 Thus, areas of greatest ACPP-Cy5 uptake closely correspond to regions of neuronal laminin degradation, neurovascular impairment, and subsequent cell death in ischemic regions, as we previously reported.21 Additionally, we found the ACPP colocalized with activated microglial cells in ischemic brain regions, consistent with the notion that microglia are among the sources of gelatinases under these conditions.39
MRI provides noninvasive imaging for the clinical diagnosis of acute stroke. Most clinically available contrast agents used in MRI displays to date manifest limited signal enhancement and are not target specific. For example, non-targeted GBCAs, perhaps due to their short half-life, reportedly fail to predict some cases of HT.40 Targeted MRI contrast agents are designed to localize to specific cell types or tissues through active binding mechanisms, which may exploit the conjugation of the signal enhancer with suitable markers.41 With target-specific MR contrast agents, MRI can elucidate molecular mechanisms responsible for disease progression.42
To increase the sensitivity of MRI for detecting low abundance molecules, one approach is to utilize molecular amplification – in our case using dendrimers incorporating many molecules of Gd, as well as a few fluorophores. This approach not only enhances contrast, but also slows excretion of the reagent.16 We used ACPPD to enhance the intensity of T1WI in the ischemic region, and validated the technique by fluorescence microscopy. Our interpretation of the results is that ACPPD molecules first passively cross the BBB into the ischemic region, and then undergo additional steps of gelatinase-dependent cleavage and uptake, requiring 1 to 2 days. Compared to ACPP-Cy5 (MW <3 kDa with only 1 peptide chain), ACPPDs are known to take a longer period of time to be maximally processed because of their higher MW [80 kDa with poly(ethyleneglycol) caps and six peptide chains].
We performed MRI examinations at two time points: pre-infusion and two days post-infusion of contrast agents (e.g. two and four days post-ischemia). Initially, we examined ischemic mouse brains at various time points. However, due to repeated bouts of anesthesia, the survival rate of ischemic mice was relatively low, at least under our experimental conditions (60-min MCA occlusion; 30-min anesthesia per MRI imaging). Thus, our experimental approach at present does not allow us to perform additional MRIs in order to provide finer temporal resolution of gelatinase involvement after cerebral ischemia.
In summary, we present evidence that gelatinase activity can be followed in mice after cerebral ischemia using ACPPs tagged with a fluorescence probe and/or a Gd MRI contrast agent. The combination of a sensitive fluorescent imaging agent and MRI can be used to monitor the distribution of gelatinase activity in the brain over time at the cellular level after an ischemic insult. Moreover, in vivo detection of the activity of gelatinases should provide additional molecular insight into the role of MMPs in the pathophysiology of stroke. Importantly, our findings indicate that ACPPDs can be used as gelatinase-specific, non-invasive probes to detect the pathological process of stroke in vivo, and may thus serve as a surrogate indicator for validation of potential treatments.
Supplementary Material
Acknowledgments
We thank Drs Shahriar Mobashery and Mayland Chang of the University of Notre Dame for providing the gelatinase inhibitor SB-3CT. We acknowledge the resources at the Biomolecular Imaging Center supported by the Truman VA Hospital and the University of Missouri.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from The Dana Foundation, American Heart Association National Scientist Development Award (09SDG2260983) and the University of Missouri Department Research Fund (ZG), NIH grants P01 HD29587 and P30 NS076411 (SAL), and R01 CA158448 (RYT). We acknowledge the National Science Foundation for the award BCS-0922374 to the University of Missouri, which funded the ICP-MS equipment used in this research.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: TJ and RYT own stock in (and RYT is a scientific advisor to) Avelas Biosciences, which has licensed ACPP technology from the University of California, San Diego. The other authors declare no competing financial interests.
Authors’ Contributions
ZG and RYT designed the research; SC, JC, ESO, QC, MY, WW, JMG, LM and ZG performed the research; TJ and RYT contributed reagents; SC, JC, QC, MY, JMG, JDR, LM and ZG analyzed the data; SC, JC, SAL, RYT and ZG wrote the paper with significant input from JDR and LM.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data.
References
- 1.Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol 2008; 8: 82–9. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol 2009; 8: 205–216. [DOI] [PubMed] [Google Scholar]
- 3.Yong VW. Metalloproteinases: Mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 2005; 6: 931–944. [DOI] [PubMed] [Google Scholar]
- 4.Castellanos M, Leira R, Serena J, et al. Plasma metalloproteinase-9 concentration predicts hemorrhagic transformation in acute ischemic stroke. Stroke 2003; 34: 40–46. [PubMed] [Google Scholar]
- 5.Heo JH, Kim SH, Lee KY, et al. Increase in plasma matrix metalloproteinase-9 in acute stroke patients with thrombolysis failure. Stroke 2003; 34: e48–e50. [DOI] [PubMed] [Google Scholar]
- 6.Horstmann S, Kalb P, Koziol J, et al. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: Influence of different therapies. Stroke 2003; 34: 2165–2170. [DOI] [PubMed] [Google Scholar]
- 7.Switzer JA, Hess DC, Ergul A, et al. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke 2011; 42: 2633–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu Z, Kaul M, Yan B, et al. S-Nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002; 297: 1186–1190. [DOI] [PubMed] [Google Scholar]
- 9.Manabe S, Gu Z, Lipton SA. Activation of matrix metalloproteinase-9 via neuronal nitric oxide synthase contributes to NMDA-induced retinal ganglion cell death. Invest Ophthalmol Vis Sci 2005; 46: 4747–4753. [DOI] [PubMed] [Google Scholar]
- 10.Gawlak M, Gorkiewicz T, Gorlewicz A, et al. High resolution in situ zymography reveals matrix metalloproteinase activity at glutamatergic synapses. Neuroscience 2009; 158: 167–176. [DOI] [PubMed] [Google Scholar]
- 11.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 2001; 17: 463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ Res 2003; 92: 827–839. [DOI] [PubMed] [Google Scholar]
- 13.Weissleder R, Tung CH, Mahmood U, et al. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nat Biotechnol 1999; 17: 375–378. [DOI] [PubMed] [Google Scholar]
- 14.Vandooren J, Geurts N, Martens E, et al. Zymography methods for visualizing hydrolytic enzymes. Nat Meth 2013; 10: 211–220. [DOI] [PubMed] [Google Scholar]
- 15.Khemtong C, Kessinger CW, Ren J, et al. In vivo off-resonance saturation magnetic resonance imaging of alphavbeta3-targeted superparamagnetic nanoparticles. Cancer Res 2009; 69: 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson ES, Jiang T, Aguilera TA, et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci USA 2010; 107: 4311–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguilera TA, Olson ES, Timmers MM, et al. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr Biol (Camb) 2009; 1: 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang T, Olson ES, Nguyen QT, et al. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci USA 2004; 101: 17867–17872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei SZ, Pan ZH, Aggarwal SK, et al. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 1992; 8: 1087–1099. [DOI] [PubMed] [Google Scholar]
- 20.Cui J, Chen S, Zhang C, et al. Inhibition of MMP-9 by a selective gelatinase inhibitor protects neurovasculature from embolic focal cerebral ischemia. Mol Neurodegener 2012; 7: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu Z, Cui J, Brown S, et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci 2005; 25: 6401–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh LY, Larsen PH, Krekoski CA, et al. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci 1999; 19: 8464–8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olson ES, Aguilera TA, Jiang T, et al. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr Biol (Camb) 2009; 1: 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma L, Yu P, Veerendra B, et al. In vitro and in vivo evaluation of Alexa Fluor 680-bombesin[7-14]NH2 peptide conjugate, a high-affinity fluorescent probe with high selectivity for the gastrin-releasing peptide receptor. Mol Imaging 2007; 6: 171–180. [PubMed] [Google Scholar]
- 25.Song SS, Latour LL, Ritter CH, et al. A pragmatic approach using magnetic resonance imaging to treat ischemic strokes of unknown onset time in a thrombolytic trial. Stroke 2012; 43: 2331–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goswami LN, Ma L, Cai Q, et al. cRGD peptide-conjugated icosahedral closo-B12(2-) core carrying multiple Gd3+-DOTA chelates for α(v)β3 integrin-targeted tumor imaging (MRI). Inorg Chem 2013; 52: 1701–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada Y, Gonoji Y, Naka K, et al. Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibrosarcoma cells. Purification and activation of the precursor and enzymic properties. J Biol Chem 1992; 267: 21712–21719. [PubMed] [Google Scholar]
- 28.Olson ES, Whitney MA, Friedman B, et al. In vivo fluorescence imaging of atherosclerotic plaques with activatable cell-penetrating peptides targeting thrombin activity. Integr Biol (Camb) 2012; 4: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen B, Friedman B, Whitney MA, et al. Thrombin activity associated with neuronal damage during acute focal ischemia. J Neurosci 2012; 32: 7622–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sacco RL, Kasner SE, Broderick JP, et al. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44: 2064–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peixoto RT, Kunz PA, Kwon H, et al. Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 2012; 76: 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Meng F, Chen Z, et al. Two-dimensional zymography differentiates gelatinase isoforms in stimulated microglial cells and in brain tissues of acute brain injuries. PLoS One 2015; 10: e0123852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke 2002; 33: 831–836. [DOI] [PubMed] [Google Scholar]
- 34.Asahi M, Asahi K, Jung JC, et al. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab 2000; 20: 1681–1689. [DOI] [PubMed] [Google Scholar]
- 35.Park KP, Rosell A, Foerch C, et al. Plasma and brain matrix metalloproteinase-9 after acute focal cerebral ischemia in rats. Stroke 2009; 40: 2836–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosell A, Ortega-Aznar A, Alvarez-Sabin J, et al. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke 2006; 37: 1399–1406. [DOI] [PubMed] [Google Scholar]
- 37.Heo JH, Lucero J, Abumiya T, et al. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab 1999; 19: 624–633. [DOI] [PubMed] [Google Scholar]
- 38.Chang DI, Hosomi N, Lucero J, et al. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J Cereb Blood Flow Metab 2003; 23: 1408–1419. [DOI] [PubMed] [Google Scholar]
- 39.del Zoppo GJ, Milner R, Mabuchi T, et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke 2007; 38: 646–651. [DOI] [PubMed] [Google Scholar]
- 40.Hjort N, Wu O, Ashkanian M, et al. MRI detection of early blood–brain barrier disruption: Parenchymal enhancement predicts focal hemorrhagic transformation after thrombolysis. Stroke 2008; 39: 1025–1028. [DOI] [PubMed] [Google Scholar]
- 41.Mazzucchelli S, Colombo M, De Palma C, et al. Single-domain protein A-engineered magnetic nanoparticles: toward a universal strategy to site-specific labeling of antibodies for targeted detection of tumor cells. ACS Nano 2010; 4: 5693–5702. [DOI] [PubMed] [Google Scholar]
- 42.Tan M, Lu ZR. Integrin targeted MR imaging. Theranostics 2011; 1: 83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.