

Abstract
Risk factors and cognitive sequelae of brain arteriolosclerosis pathology are not fully understood. To address this, we used multimodal data from the National Alzheimer's Coordinating Center and Alzheimer's Disease Neuroimaging Initiative data sets. Previous studies showed evidence of distinct neurodegenerative disease outcomes and clinical-pathological correlations in the “oldest-old” compared to younger cohorts. Therefore, using the National Alzheimer's Coordinating Center data set, we analyzed clinical and neuropathological data from two groups according to ages at death: < 80 years (n = 1008) and ≥80 years (n = 1382). In both age groups, severe brain arteriolosclerosis was associated with worse performances on global cognition tests. Hypertension (but not diabetes) was a brain arteriolosclerosis risk factor in the younger group. In the ≥ 80 years age at death group, an ABCC9 gene variant (rs704180), previously associated with aging-related hippocampal sclerosis, was also associated with brain arteriolosclerosis. A post-hoc arterial spin labeling neuroimaging experiment indicated that ABCC9 genotype is associated with cerebral blood flow impairment; in a convenience sample from Alzheimer's Disease Neuroimaging Initiative (n = 15, homozygous individuals), non-risk genotype carriers showed higher global cerebral blood flow compared to risk genotype carriers. We conclude that brain arteriolosclerosis is associated with altered cognitive status and a novel vascular genetic risk factor.
Keywords: Arteriosclerosis, CVD, HS-Aging, TDP-43, VCID
Introduction
Cerebrovascular pathologies affecting small arteries and arterioles are common findings in the aged brain1–3 and are often seen in brains of persons with dementia.4–7 Here, we focused on brain arteriolosclerosis (B-ASC), a subtype of cerebral small vessel pathology characterized by degenerative wall thickening of arterioles.8–10 The true prevalence of B-ASC is unknown but has been observed in 39–80% of autopsied elderly individuals.11,12 The pathologic changes of B-ASC include hypertrophy and/or death of vascular smooth muscle cells and extracellular deposition of elastin and collagen.1,2,13,14 In the present study, we sought to better understand the risk factors and global cognitive status associated with B-ASC pathology among aged individuals.
Hypertension and diabetes are presumed risk factors for arteriolosclerosis (ASC) in the brain and other organs. Chronic hypertension is associated with ASC pathology in the brain, kidney, and other organs.15–17 Hypertensive animal models have thicker cerebral arteriolar walls, larger vessel cross-sectional areas, and smaller inner arteriolar diameters compared to control animals.18,19 Diabetic patients have thicker subcutaneous gluteal arteriolar walls and larger cross-sectional areas compared to controls.20,21 Diabetic animal models have thicker retinal capillary walls22 and renal arteriolar glomerular basement membranes23 compared to controls.
In addition to clinical risk factors, recent studies suggest that a single nucleotide polymorphism (SNP) located in ABCC9, ATP-binding cassette sub-family C member 9, may be a genetic risk factor for B-ASC pathology in older elderly individuals. Evidence in support of the link between an ABCC9 SNP and B-ASC pathology are as follows: (1) The rs704180 SNP located in ABCC9 is associated with hippocampal sclerosis of aging (HS-Aging),24,25 a neurodegenerative disease affecting individuals >80 years at death.8,26 (2) Individuals with HS-Aging have worse B-ASC pathology compared to individuals without HS-Aging pathology.8 (3) The gene product of ABCC9 is a subunit of ATP-sensitive potassium channels found in vascular smooth muscle cells, including arterioles.27 Thus, by extension, ABCC9 gene variants may constitute a risk factor for B-ASC pathology in elderly individuals. However, this credible hypothesis has not been tested previously.
As the clinical and genetic risk factors for B-ASC are imperfectly understood, so is the cognitive impairment associated with this pathology. Studies on the global cognitive status of patients with B-ASC have included the analyses of Mini Mental State Examination (MMSE) scores,28 Clinical Dementia Rating Scale (CDR) scores,29 and CDR Sum of Boxes (CDRSUM) scores.30 The MMSE is an assessment tool used in measuring global cognitive function,31 while the CDR is a measure of a person's ability to accomplish activities of daily living.32,33 The CDRSUM score is derived by summing scores from all CDR domains.34 Prior analyses of data from 334 elderly individuals did not reveal an association between B-ASC pathology and MMSE scores.28 However, in an autopsy study with 52 cases, widespread B-ASC pathology in cases with Alzheimer's disease (AD) was associated with worse global CDR scores.29 Similarly, in an autopsy study using 715 AD cases with CDRSUM information, researchers found that high B-ASC severity was associated with worse CDRSUM scores.30 Conflicting results from these studies may be due to a number of experimental factors including small sample size (statistical power), particular cognitive domains affected by small blood vessel pathologies, frequent presence of comorbid pathologies, and parameters that vary in different parts of the human aging spectrum.
In order to gain insight into B-ASC risk factors and global cognitive status while factoring in other dementia-associated pathologies, we analyzed a subset of individuals from the National Alzheimer's Disease Coordinating Center (NACC) data set. Because there is evidence of distinct neurodegenerative outcomes and clinical-pathological correlation differences between the “younger-old” and “oldest-old” persons,35–41 we analyzed groups separately according to ages at death: < 80 years and ≥80 years. The goals of the study were to determine if autopsy-verified B-ASC is associated with global cognitive status, to assess the association between vascular risk factors and B-ASC pathology, and to determine the relationship between ABCC9 HS-Aging risk genotype and B-ASC pathology. In order to further test the association between the ABCC9 HS-Aging risk genotype and B-ASC pathology, we studied the relationship between the ABCC9 HS-Aging risk genotype and cerebral blood flow (CBF) (a possible in vivo manifestation of B-ASC pathology). Genetic and neuroimaging data on a sub-sample of individuals from the Alzheimer's Disease Neuroimaging Initiative (ADNI) data set were used to test this association.
Methods
This study used data from the NACC and ADNI data sets. Patient recruitment and data collection in the NACC data set have been previously described including details related to institutional review board approval and patient consent.42–45 Research using NACC data was approved by the University of Washington Human Subjects Division. Patient recruitment, neuroimaging acquisition, and data collection on individuals included in the ADNI data set have been previously described with approval by local ethics review boards at each participating site.46,47
Study subjects
The NACC data set contains research subject information collected from 34 past and present Alzheimer's Disease Centers (ADCs) across the United States, comprising clinical, genetic, and neuropathological (autopsy) data.43 More detailed information on data collection is available online (https://www.alz.washington.edu/WEB/dataforms_main.html). Archival data from September 2005 through December 2013 were obtained for this study.
The initial pulled archival data from the NACC data set comprised 29,483 cases. Living cases (n = 26,686) and autopsied cases with missing information which would preclude making an assessment of B-ASC status (n = 407) were excluded from the analysis. For analysis, cases were split into two age at death groups: < 80 years (n = 1008) and ≥80 years (n = 1382).
The ADNI data set is from a multicenter longitudinal study in the United States and Canada in which subjects with normal cognition, MCI and AD were followed with cognitive testing, neuroimaging techniques, and other biomarker.46 A convenience sample (n = 15) of rs704180 homozygous (A_A or G_G genotype) individuals was used for CBF analysis. Neither other scans nor SNPs were assessed from ADNI.
Cognitive and functional assessment in the NACC data set
MMSE (0–30; 30 = no global impairment) and CDRSUM (0–18; 0 = no global impairment) scores were used as measures of global cognitive status.31–33 Scores from the most recent ADC clinical visit before each individual's death were used (median interval between final visit and death: 0.83 years).
Clinical, neuropathologic, and genetic parameters in the NACC data set
Clinical data were obtained from each participant's final ADC clinical visit before death. During clinical research visits, medical histories were obtained from subjects, caregivers (particularly if the subject was cognitively impaired), and/or patient records. The following self-reported vascular risk factors and cerebrovascular diseases were used in the analyses: medical histories of hypertension, diabetes, hypercholesterolemia, sex, smoking, stroke, and atrial fibrillation. Responses were coded initially as unknown, absent, recent/active, or remote/inactive, and subsequently, “recent” and “remote” responses were combined into one category (i.e. history of a condition) for analytical purposes. Body mass index (BMI) values were derived from height and weight measurements. Pack years were derived from self-reported cigarette packs smoked per day and years of smoking.
Neuropathologic details from all cases included Braak staging,48 CERAD neuritic plaque densities,49 and other parameters as described in detail previously.44 In the NACC Neuropathology Data Set Coding Guidebook version 9.1 (https://www.alz.washington.edu/WEB/forms_np.html), B-ASC was described as “hyalinosis of the media and adventitia of small parenchymal and/or leptomeningeal vessels.” B-ASC pathology was diagnosed using a semi-quantitative four-tier categorization system with responses scored to indicate “none”, “mild”, “moderate”, or “severe” B-ASC pathology. In the NACC guidebook, neuropathologists were instructed to estimate the overall severity of B-ASC pathology. No specific brain region for B-ASC pathological diagnosis was mentioned; thus, this diagnostic methodology was left to the discretion of each individual neuropathologist and/or research center.
Genetic data were obtained and analyzed as described previously.24,25 Briefly, the Alzheimer's Disease Genetic Consortium (ADGC) accrued genetic data from 29 different ADCs, with multiple iterations of SNP data,50–52 which were analyzed together with neuropathological and clinical data gathered through NACC.53 The three alleles identified were analyzed according to ADGC SNP nomenclature and were GRN.rs5848 (A/G), TMEM106B.rs1990622 (A/G; note that other reports have used T/C for this allele: the “A” allele is analogous to “T” allele in other reports, whereas the “G” allele we report is analogous to “C” allele), and ABCC9.rs704180 (A/G).24,25 APOE ɛ4 genotype information from NACC was also used in the analysis because APOE alleles are known to be associated with cerebral amyloid angiopathy (CAA), which could lead to vascular wall distortions.54
Neuroimaging and genetic parameters in ADNI data set
T1-weighted brain MRI scans were acquired using a sagittal MP-RAGE sequence following the ADNI MRI protocol.47,55 Arterial spin labeling (ASL) images were obtained from ADNI data set. Data from 15 Caucasian individuals were acquired from six different American research centers using a standardized pulsed arterial spin labeling (pASL) protocol: Field Strength= 3.0 tesla; Flip angle = 90.0 degree; Manufacturer = SIEMENS; Matrix X = 320.0 pixels; Matrix Y = 320.0 pixels; Pixel Spacing X = 4.0 mm; Pixel Spacing Y = 4.0 mm; Pulse Sequence = EP; Slice Thickness =4.0 mm; TE = 12.0 ms; TR = 3400.0 ms. Control and label images were subtracted, and quantitative CBF (mL/100 g/min units) was calculated using in-house Matlab software using the following equation56,57 and then correlated with genotyping
where λ was the brain/blood partition coefficient in mL/g, SIcontrol and SIlabel were the time-averaged signal intensities in the control and label images, respectively, T1,blood was the longitudinal relaxation time of blood in seconds, α was the labeling efficiency, M0 was the equilibrium brain tissue magnetization, TI1 was post-labeling delay, and TI2 was the label duration. SNP rs704180 in ABCC9 and APOE ε4 status information came from ADNI.
Statistical analyses
Exploratory bivariate analyses and regression modeling were used to assess the association between clinical vascular risk factors and B-ASC pathology. Initially, a Chi-square test, a Mann-Whitney U test, or a Kruskal–Wallis test were used to determine possible risk factors for B-ASC severity in the two age at death groups. A Chi-square test was applied for categorical variables, whereas a Mann-Whitney U test or Kruskal-Wallis test was applied for continuous non-normally distributed variables. Subsequently, clinical variables that yielded a p < 0.05 in these analyses were included as independent variables in an ordinal logistic regression to further elucidate the association between clinical variables and B-ASC pathology while controlling for confounding effects. The variables in this logistic regression model included age at death, sex, hypertension, diabetes, smoking pack years, and hypercholesterolemia.
Logistic regression modeling was used to determine the association between the ABCC9 HS-Aging risk genotype and B-ASC pathology. Age at death, sex, hypertension, diabetes, pack years, and hypercholesterolemia were used as covariates. Mild, moderate and severe B-ASC pathologies were collapsed into one category and treated as a dependent variable.
A linear regression model was used to assess the association between B-ASC pathology with MMSE and CDRSUM scores. B-ASC pathology was treated as the main independent variable. MMSE or CDRSUM scores were treated as dependent variables while adjusting for age at death, sex, Braak neurofibrillary tangles (NFT) stage, presence of any microinfarcts, presence of neocortical Lewy bodies, and presence of HS-Aging pathology. Adjusted mean MMSE and CDRSUM scores derived from the linear regression analyses were reported for each B-ASC severity category and compared using the least squares method.
In order to assess the relationship between the ABCC9 HS-Aging risk genotype and CBF, a possible manifestation of B-ASC pathology, a Welch's two sample t-test was used to compare CBF measurements between individuals with the ABCC9 HS-Aging homozygous non-risk and risk genotypes. All statistical analyses were performed using IBM SPSS Statistics 22 Properties and PC-SAS 9.34 (SAS Institute, Inc.; Cary, NC, USA).
Results
The inclusion/exclusion criteria applied to individuals used in our analyses from the NACC data set are shown in Figure 1(a). Individuals used in these analyses were predominantly (>90%) Caucasian (data not shown), and median year at death was 2010 (range: 2005–2013). In order to convey examples of blood vessel profiles representing the spectrum of B-ASC pathology, images were obtained from four human brain sections stained with hematoxylin and eosin (H&E) (Figure 2). The percentage of individuals with B-ASC pathology trended upward with increasing age at death. (Figure 1(b)). When stratifying by age at death, the percentage of individuals with moderate or severe B-ASC pathology was higher in older age at death groups (p < 0.0001) (Figure 1(c)). These results indicate that B-ASC is a common pathology in the NACC data set, becoming more severe with increasing age at death.
Figure 1.
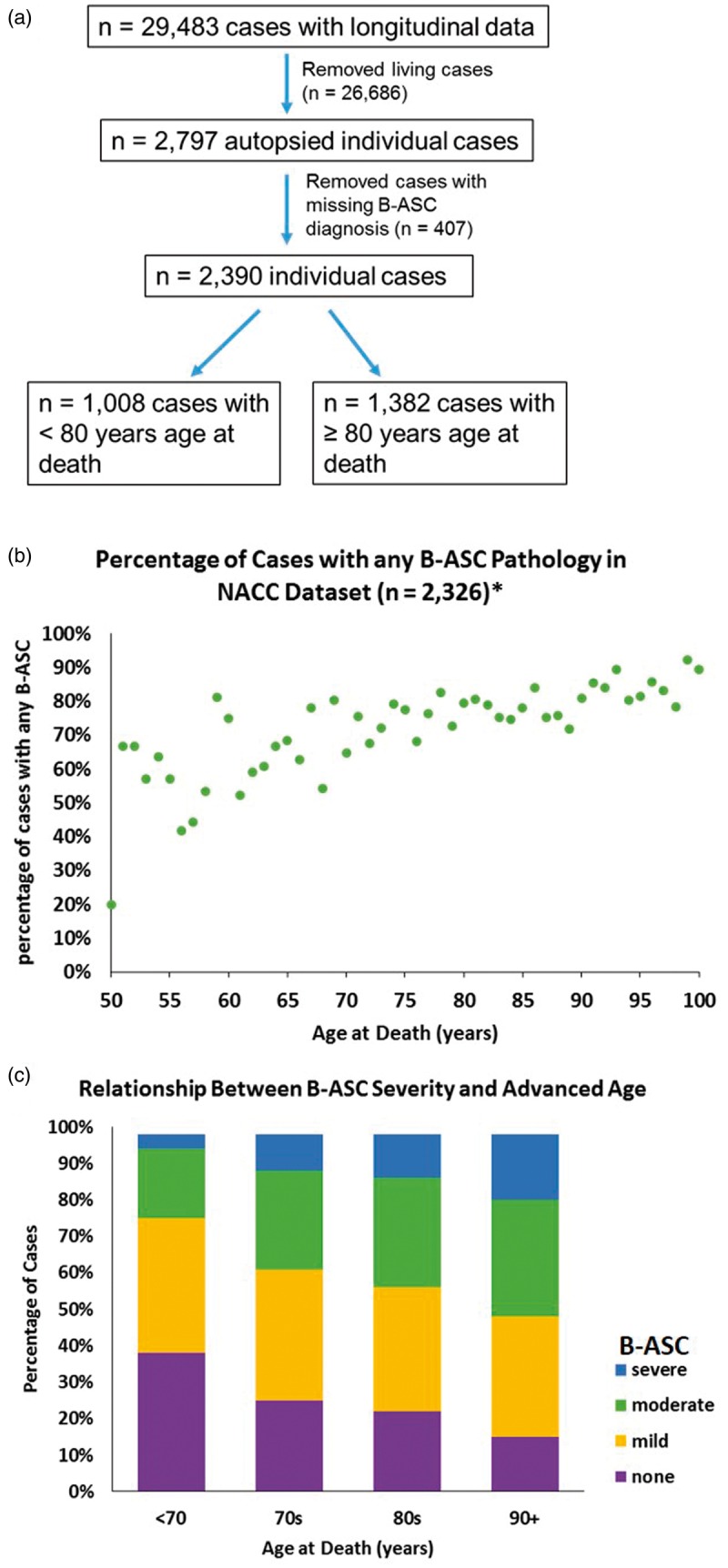
Exclusion/inclusion criteria and frequency of brain arteriolosclerosis (B-ASC) pathology in autopsied cases from the National Alzheimer's Coordinating Center (NACC) data set. (a) Living cases and autopsied cases with missing information which would preclude making an assessment of B-ASC status were excluded from analysis. (b) The percentage of cases with any B-ASC pathology (mild, moderate or severe) in the NACC data set. Asterisk (*) indicates that 64 cases with age at death <50 years or > 100 years were not plotted. (c) A stacked bar graph showing that B-ASC severity increases with age at death, Chi-square p-value < 0.0001.
Figure 2.

Semi-quantitative severity grading of B-ASC pathology and its non-association with APOE ɛ4 allele in the NACC data set. (a, b, c, d) Photomicrographs of hematoxylin- and eosin-stained blood vessels. (a) What we assume to be a normal arteriole (green arrow). (b) mild B-ASC with relatively mild thickening of vessel wall. (c) moderate B-ASC severity grade 2 with increased thickening of vessel wall. (d) severe B-ASC with prominent thickening of vessel wall and partly occluded vessel lumen. Scale bars = 100 µm. (e) The association between APOE ɛ4 genotypes and any degree of CAA (mild, moderate, severe) combining both age at death groups: *Chi-square p-value < 0.0001 among 1883 individuals for whom APOE genotype data and CAA diagnosis were available. Those with ɛ2/ɛ3 and ɛ2/ɛ2 genotypes were combined into one category. There were no statistically significant associations that could be determined between B-ASC severity and APOE genotype.
B-ASC: brain arteriolosclerosis; CAA: cerebral amyloid angiopathy.
As a result of prior studies showing different outcomes and clinical-pathological correlations in the “oldest old,”35–37 we analyzed the overall cohort in two separate age groups. More specifically, the cutoff of 80 years was chosen because it has been used before to help highlight neurodegenerative disease and/or neuropathologic features that differ – often quite dramatically – among the “oldest-old”.39–41,58–60 Furthermore, this cutoff was close to the overall cohort mean (80.0 years) and median (82.0 years) age at death. Comparing the two age at death groups in the NACC data set, individuals in the ≥80 years age at death group were more often female, hypertensive, and less often impaired cognitively when compared to individuals in the <80 years age at death group (Table 1). In addition, individuals in the ≥80 years age at death group were less likely to show “high” levels of AD pathology, but more likely to show B-ASC and HS-Aging pathologies at autopsy, compared to individuals in the <80 years age at death group. These data, in addition to the prior precedents in the literature, confirmed that the two age groups show differing clinical risk factors, cognitive profiles, and neuropathological autopsy results.
Table 1.
Age at death group comparison on clinical, cognitive, neuropathologic, and genetic variables.
| <80 years age of death group | ≥80 years age of death group | Missing cases, n | p a | ||
|---|---|---|---|---|---|
| n = 1008 | n = 1382 | ||||
| Demographic variables | |||||
| Age of death, median (25th, 75th quartile) | 70.0 (64.0, 76.0) | 88.00 (84.0, 92.0) | <0.0001b,c | ||
| Gender (%) | <0.000b,c | ||||
| Male | 61.7 | 48.3 | |||
| Female | 38.3 | 51.7 | |||
| Clinical variables | |||||
| Hypertension (%)d | 46.1 | 64.6 | 8 | <0.0001b,c | |
| Diabetes (%)d | 9.6 | 13.2 | 2 | 0.008c | |
| Hypercholesterolemia (%)d | 47.7 | 49.8 | 28 | 0.315 | |
| Pack years median (25th, 75th quartile)d | 0.0 (0.0, 13.1) | 0.0 (0.0, 15.0) | 181 | 0.496 | |
| BMI median (25th, 75th quartile)e | 25.4 (22.8, 28.5) | 24.9 (22.2, 27.6) | 980 | 0.004c | |
| Cognitive variables | |||||
| MMSE median (25th, 75th quartile) | 16.0 (6.0, 24.0) | 21.0 (14.0, 27.0) | 697 | <0.0001b,c | |
| CDRSUM median (25th, 75th quartile) | 13.0 (7.0, 18.0) | 9.0 (2.0, 15.0) | <0.0001b,c | ||
| Neuropathological variables | |||||
| AD pathology “High”f | 47.3 | 42.5 | 36 | <0.0001b,c | |
| HS-Aging (%)e | 7.1 | 14.2 | 113 | <0.0001b,c | |
| B-ASC (%) | <0.0001a,b,c | ||||
| None | 31.3 | 19.9 | |||
| Mild | 37.0 | 33.9 | |||
| Moderate | 23.8 | 31 | |||
| Severe | 7.8 | 15.2 | |||
| Genetic variables | |||||
| ABCC9.rs704180 genotype A_A (%) | 23.8 | 25.8 | 1465 | 0.508 | |
| APOE ɛ4 allele (%) | 48.1 | 39.9 | 467 | <0.0001b,c |
In comparing the two age at death groups, p-values for age at death, pack years, and BMI are from Mann-Whitney U analyses. p-Values for gender, hypertension, diabetes, hypercholesterolemia, B-ASC, ABCC9 HS-Aging risk genotype (rs704180.A_A), and APOE ɛ4 allele are determined from Chi-square tests. Percentages were recorded after excluding missing cases for each variable.
AD: Alzheimer's disease; B-ASC: brain arteriolosclerosis; BMI: body mass index; CDRSUM: clinical dementia rating sum of boxes; HS-Aging: hippocampal sclerosis of aging; MMSE: mini mental state examination.
Group comparisons exclude cases with missing data.
p-Value <0.05.
Significant p-value after Bonferroni correction for multiple comparisons.a,b,c,d,e,f
Self-reported vascular risk factors.
Derived variables from NACC variables.
NIA/Reagan Institute Criteria, 1997.
Global cognitive status associated with B-ASC pathology
To analyze the global cognitive status of individuals with B-ASC pathology, linear regression analyses were performed using B-ASC pathology as a predictor, other dementia-inducing pathologies as covariates, and MMSE or CDRSUM scores as outcome variables. After adjusting for age at death, cortical microinfarcts, AD pathology (Braak NFT stage and CERAD neuritic plaque rating), neocortical Lewy bodies, and HS-Aging pathologies, B-ASC was a significant pathological predictor in the MMSE model using cases from the <80 years age at death group and in the CDRSUM model using cases from both age groups (Supplementary Tables 3 and 4). In the < 80 age at death group, the adjusted MMSE and CDRSUM group means for cases with severe B-ASC were worse compared to that of cases with none, mild, or moderate B-ASC pathology (Table 2). In the ≥ 80 age at death group, the adjusted MMSE group mean score for cases with severe B-ASC was worse compared to that of cases with none or mild B-ASC pathology. The adjusted CDRSUM group mean score for cases with severe B-ASC pathology was worse compared to that of cases with none, mild, or moderate B-ASC pathology (Table 2). In addition, the adjusted CDRSUM group mean score for cases with moderate B-ASC pathology was worse compared to that of cases with mild B-ASC pathology. These results indicate that after adjusting for age and co-morbid brain pathologies, individuals with moderate and severe B-ASC pathology had worse global cognition compared to those with none or mild B-ASC pathology.
Table 2.
Adjusted final MMSE and CDRSUM Group means associated with brain arteriolosclerosis (B-ASC) pathology.a
| Age of death < 80 years | Age of death ≥ 80 years | |||
|---|---|---|---|---|
| B-ASC | MMSE | CDRSUM | MMSE | CDRSUM |
| None | 17.0 ± 0.7 | 10.5 ± 0.4 | 21.3 ± 0.6 | 6.8 ± 0.4 |
| Mild | 16.8 ± 0.6 | 10.8 ± 0.3 | 21.2 ± 0.4 | 6.8 ± 0.3 |
| Moderate | 17.1 ± 0.8 | 10.3 ± 0.4 | 20.4 ± 0.5 | 7.5 ± 0.3 b |
| Severe | 13.1 ± 1.3 c | 12.7 ± 0.7 c | 19.5 ± 0.7 d | 9.0 ± 0.4 c |
CDRSUM: Clinical Dementia Rating Sum of Boxes. MMSE: Mini Mental State Exam.
Means are adjusted for age at death (years), sex, Braak & Braak stage, semi-quantitative ratings of diffuse and neuritic plaques, and dummy indicators for microinfarcts, HS-aging, and Lewy body pathology.
p < 0.05 versus mild.
p < 0.01 versus none, mild, and moderate.
p < 0.05 versus none and mild. Bold values indicate significant finding.
Clinical vascular risk factors associated with B-ASC pathology
Bivariate analyses and regression modeling were used to assess the relationship between vascular risk factors and B-ASC pathology. Race and ethnicity were not adjusted for in the models because of the low sample size within each group (data not shown). In the < 80 age at death group, hypertension, diabetes, and hypercholesterolemia were associated with B-ASC severity (Supplementary Table 2). In the ≥80 age at death group, sex and smoking pack years were associated with B-ASC severity. With respect to clinically evident cerebrovascular disease, self-reported stroke history was associated with autopsied confirmed B-ASC pathology in the <80 years age at death group (p = 0.003) and the ≥ 80 years age at death group (p = 0.033) (data not shown). However, there was no association between atrial fibrillation and B-ASC pathology (data not shown). Five vascular risk factors, along with age at death, were used in an ordinal logistic regression, with B-ASC severity as the ordinal outcome measure, to determine risk factors associated with B-ASC severity. In the < 80 age at death group, age at death, female sex, and hypertension were associated with predicting B-ASC severity. In the ≥ 80 age at death group, only age at death and female sex remained to be significant variables in the model but not hypertension (Table 3). These findings suggest that age at death and sex are associated with autopsy-proven B-ASC in both younger and older-aged individuals. However, hypertension may only be a risk factor for B-ASC pathology only in young elderly individuals, raising the possibility of other B-ASC risk factors being important in more advanced old age.
Table 3.
Brain arteriolosclerosis and vascular risk factors.a
| Beta coefficient ± S.E. | Odds ratio | 95% C.I. | p | |
|---|---|---|---|---|
| <80 years age at death group (n = 922) | ||||
| Age at death (one-year increase) | 0.04 ± 0.01 | 1.0 | 1.0–1.1 | <0.0001b |
| Gender (male vs. female) | −0.3 ± 0.1 | 0.8 | 0.7–0.8 | 0.029b |
| Hypertension (vs. no hypertension) | 0.3 ± 0.1 | 1.4 | 1.1–1.8 | 0.017b |
| Diabetes (vs. no diabetes) | 0.3 ± 0.2 | 1.4 | 0.9–2.1 | 0.120 |
| Pack years (one-year increase) | 0.0 ± 0.003 | 1.0 | 1.0–1.006 | 0.962 |
| Hypercholesterolemia (vs. no hypercholesterolemia) | 0.2 ± 0.1 | 1.3 | 1.0–1.6 | 0.067 |
| ≥80 years age at death group (n = 1255) | ||||
| Age at death (one-year increase) | 0.02 ± 0.01 | 1.0 | 1.00–1.04 | 0.021b |
| Gender (male vs. female) | −0.2 ± 0.1 | 0.8 | 0.7–1.0 | 0.041b |
| Hypertension (vs. no hypertension) | 0.2 ± 0.1 | 1.2 | 1.0–1.5 | 0.134 |
| Diabetes (vs. no diabetes) | 0.001 ± 0.2 | 1.0 | 0.7–1.4 | 0.995 |
| Pack years (one-year increase) | 0.003 ± 0.002 | 1.0 | 1.0–1.0 | 0.102 |
| Hypercholesterolemia (vs. no hypercholesterolemia) | −0.1 ± 0.1 | 0.9 | 0.7–1.1 | 0.257 |
An ordinal logistic regression model was applied in both age at death groups using vascular risk factors identified from exploratory analysis (see Supplementary Table 2). In both age at death groups, the statistical models included cases with available data on all six variables.
p-Value < 0.05.
Novel B-ASC genetic risk factor: ABCC9
Genetic and pathological information from NACC were used to assess the association between the ABCC9 HS-Aging risk genotype and B-ASC pathology. Of the 2390 cases included in the analysis, a total of 925 persons had available ABCC9 SNP information. Individuals with ABCC9 genotype information were slightly more likely to have AD pathology (45% vs. 40%) than those lacking genotype data (data not shown). Among subjects with available ABCC9 SNP data, bivariate analysis showed that the ABCC9 HS-Aging risk genotype was associated with the presence of any B-ASC pathology (mild, moderate, and severe combined) in the ≥ 80 age at death group (p = 0.032) (Figure 3). By contrast, APOE genotype status was strongly associated with CAA pathology as expected but not with B-ASC pathology (Figure 2(e)).
Figure 3.

Relationship between any degree of brain arteriolosclerosis (B-ASC) pathology in NACC and HS-Aging risk genotypes. (a) The association between ABCC9 HS-Aging risk genotype (rs704180 A_A, as determined previously)24,25 and B-ASC pathology, stratifying by age of death. *Chi-square p-value = 0.032. By contrast, as shown in (b,c), neither the TMEM106B (rs1990622 A_A or A_G) nor GRN (rs5848 T_T) risk genotypes are associated with B-ASC pathology.
To account for relevant covariates, a logistic regression analysis was used, treating the presence of B-ASC pathology as a dependent variable, the ABCC9 HS-Aging risk genotype as an independent variable, and age at death, sex, smoking pack years, and history of hypertension, diabetes, and hypercholesterolemia as covariates. Results from this model showed that individuals in the ≥80 years at death group with the ABCC9 HS-Aging risk genotype were 1.9 times more likely to have a diagnosis any B-ASC pathology (mild, moderate, or severe) compared to individuals without the ABCC9 HS-Aging risk genotype (p = 0.04). There was no association between the ABCC9 HS-Aging risk genotype and B-ASC pathology in the <80 age at death group. In a sensitivity analysis adjusting for research center identifications as a fixed effect, the association between the ABCC9 HS-Aging risk genotype and B-ASC pathology was still observed (p = 0.04) in the ≥ 80 years at death group.
ABCC9 and cerebral blood flow
To provide further testing on the association between the ABCC9 HS-Aging risk genotype and B-ASC pathology, we assessed a neuroimaging modality that we hypothesize detects a clinical manifestation of B-ASC pathology. This was performed by measuring CBF in convenience sample of individuals (n = 15) from the ADNI data set comparing persons with A_A versus G_G ABCC9.rs704180 genotype. Individuals were on average 72 years of age at the time of scan and both groups of rs704180 homozygotes (A_A and G_G) were matched for cognitive status, APOE alleles, sex, and age (see Supplementary Table 5 for complete data on these parameters). Individuals with two ABCC9 HS-Aging non-risk alleles (n = 7 with rs704180 G_G) showed higher global CBF measurements compared to those with two ABCC9 HS-Aging risk alleles (n = 8 with rs704180 A_A) (Figure 4). The group level relative difference in CBF was 28%, p < 0.001 (Figure 4).
Figure 4.

Arterial Spin labeling (ASL) neuroimaging indicates that ABCC9 HS-Aging risk genotype is associated with decreased cerebral blood flow (CBF), compatible with a novel pathogenetic mechanism. (a) A representative scan of a 77-year-old female with the ABCC9 HS-Aging non-risk genotype: rs704180 G_G. (b) A representative scan from a 76-year-old male with rs704180 A_A. (c) Individuals with the rs704180 G_G genotype showed significantly higher global CBF compared to those with the rs704180 A_A genotype, group level relative difference in CBF is 28%, p < 0.001. Neither other scans nor SNPs were analyzed from the ADNI data set. (d) An interpretation of the results presented in this paper. We hypothesize that an ABCC9 gene variant increases risk for B-ASC and/or decreased CBF, which, in turn, may cause or exacerbate hippocampal TDP-43 pathology and HS-Aging. All of these pathologies have been associated with cognitive impairment. Other variants in GRN and TMEM106B genes are not associated with B-ASC pathology and thus may represent downstream factors, perhaps more directly related to risk of hippocampal TDP-43 pathology. Presumed non-cerebrovascular pathologies (e.g. frontotemporal lobar degeneration (FTLD), brain trauma, and Alzheimer's disease) also are associated with hippocampal TDP-43 pathology, indicating that hippocampal TDP-43 pathology is a common downstream pathological phenomenon.
Discussion
In this study, we describe the global cognitive status, in addition to both clinical and genetic putative risk factors of B-ASC, in autopsied cases from the NACC data set. The presence of severe B-ASC pathology was associated with global cognitive impairment. In addition, a neuroimaging CBF experiment further supports the association between the ABCC9 HS-Aging risk genotype and B-ASC. Potential risk factors for B-ASC included advanced age at death, hypertension, and sex. In younger individuals (age at death < 80 years), hypertension was associated with B-ASC. However, this association was not observed in older individuals (age at death ≥ 80 years). In the ≥ 80 years age at death group, the ABCC9 HS-Aging risk genotype was associated with B-ASC pathology. These findings suggest that B-ASC risk factors are age dependent. For example, hypertension appears to have a strong role in younger elderly individuals, while at least one genetic factor (ABCC9) may affect the B-ASC risk in the “oldest-old”.
There are potential limitations in this study. The NACC data set is not a population-based data set; it is better characterized as a clinical series of persons enrolled at ADCs, and in addition, carries known biases associated with autopsy cohorts.42,43,53,61 As a result, NACC participants are predominantly Caucasian, highly educated, and are drawn predominantly from dementia clinics.42,44 Due to the lack of socioeconomic information and low sampling of individuals from different ethnic/racial groups, race and ethnicity were not included in the regression models. The data on clinical disease risk factors are largely self-reported, which can lead to an underestimation of the true disease frequencies.62 In addition, duration of disease (i.e. hypertension) data was not available; therefore, it was not adjusted for in the regression models. We found that female sex was associated with B-ASC; this finding is potentially confounded by the increased longevity of women (age being a risk factor for B-ASC) and many other covariates that vary with sex. Although this association survived in a regression model that accounted for other factors including age at death, these data should be interpreted with caution and future work is merited in this area. The NACC neuropathology data set coding guidebook does not suggest optimal brain sections for B-ASC diagnosis. As a result, B-ASC diagnostic methods are inconsistent across ADCs. Although non-NACC guidelines exist for B-ASC diagnosis,63 different ADCs have reported using the basal ganglia64 or a “global”8 criteria in the diagnosis of B-ASC. In a prior, non-ADC autopsy study with 135 vascular dementia brains, B-ASC was seen in the frontal lobe (83.7% of cases), temporal lobe (80.7% of cases), and basal ganglia (89.6% of cases).65 Because the diagnosis of B-ASC is presently based on H&E staining, other pathologies that lead to a distortion of vascular walls (i.e. CAA) may mistakenly be diagnosed as B-ASC, leading to a biased estimation of B-ASC frequency. However, we saw a strong correlation between APOE status and CAA severity, but no correlation between APOE status and B-ASC severity in our sample, indicating that these pathologies are at least partly independent. In the future, improved methodologies and consensus for B-ASC pathologic diagnosis should improve the specificity of B-ASC operationalization. Until then, it can be argued that a multicenter approach, combining data from dozens of neuropathologists, each applying center-specific diagnostic rubrics, is the best way to achieve an outcome that is representative of what any given neuropathologist would define as “brain arteriolosclerosis.”
Despite the challenges inherent to a retrospective cross-sectional study, the NACC and ADNI databases provide relatively high-quality contexts to study clinical, genetic, neuroimaging, and/or pathological correlations. Detailed cognitive assessments, genetic, and neuropathological data have allowed us to study associations of B-ASC with cognitive status and both clinical and genetic variables. These include both “traditional” ASC risk factors (hypertension and diabetes), as well as novel genetic aspects (ABCC9 SNP). Mixed pathologies are frequently seen in the aged brain30,66,67 and detailed neuropathological data from NACC allowed us to adjust for other dementia-inducing pathologies in our analyses. B-ASC is a common cerebrovascular pathology that is often seen in a complex milieu along with other brain diseases30 and has been associated with motor impairment in advanced old age.12 In this NACC-ADC data set, the frequency of any B-ASC pathology (mild, moderate, and severe) was 75.3%, and became more severe with increasing age at death, which is consistent with findings from other autopsy cohorts.11,68,69
We showed that B-ASC pathology was associated with worse MMSE and CDRSUM scores after adjusting for co-morbid cognitive impairment-inducing pathologies. Prior studies reporting the cognitive profiles of B-ASC are limited, mostly focusing on patients diagnosed with vascular dementia (a very heterogeneous condition).5–7 Although both age at death groups had MMSE and CDRSUM scores indicative of cognitive impairment, individuals in <80 age at death group were more globally impaired compared to individuals in the ≥80 years age at death group. This finding may be attributed to the differing frequencies of AD and B-ASC pathologies present in the two age at death groups: higher number of individuals with “high” AD pathology in < 80 years age at death group and higher number of individuals with moderate or severe B-ASC pathology in ≥ 80 years age at death group. Individuals with AD pathology exhibit greater deficits in memory function and faster rates of information decay compared to individuals with cerebrovascular disease.70 Our study provided quantitative evidence to support the hypothesis that B-ASC is associated with worse cognitive status independent of other brain changes. Furthermore, it underscores the importance of identifying risk factors, specific neuroimaging abnormalities, and potential treatments of B-ASC, in order to prevent or reverse its development later in life.
Hypertension is a major risk factor for B-ASC.16,71 In an autopsy study, Moritz and Oldt16 observed that B-ASC pathology was more severe in the hypertensive group compared to the non-hypertensive group.16 In the < 80 age at death group, we found hypertension to be associated with B-ASC. However, in the ≥ 80 age at death group, there was no association detected between B-ASC and hypertension, and these results did not change after adjusting for anti-hypertensive medication use. Similarly, in an autopsy study consisting of 70 cases with B-ASC, 31% of cases were normotensive with 10 of these cases having an age at death ≥80 years.71 These results suggest that hypertension may not be the only risk factor for B-ASC pathology in older elderly individuals.
We hypothesize that the known strong impact(s) of diabetes on brain function may be mediated through a combination of vascular and metabolic etiologies.72 We did not find support for the direct impact of diabetes on B-ASC, but there was a trend between diabetes and B-ASC in the younger cohort. We note that there was not a distinction between Type 1 or Type II diabetes in the NACC data set, although the majority is presumed to be Type II diabetes. Evidently, beyond the “usual suspects”, there are additional, currently unknown risk factors for B-ASC in advanced old age.
One category of risk factors that may be relevant to B-ASC pathology in the “oldest-old” is genetics, and we here provide support for a specific candidate risk allele. The ABCC9 SNP rs704180 was previously associated with risk for HS-Aging,24,25 a hippocampal pathology seen in ∼10–25% of autopsied individuals beyond 80 years at death.26,73–75 Recently, we found an association between HS-Aging and B-ASC in three separate cohorts, including the NACC data set.8 Using digital image methods for analysis of arteriolar morphology, we found that HS-Aging cases had larger vessel areas, vessel perimeters, vascular areas, and vessel wall thicknesses compared to non HS-Aging cases.8 Research from a different cohort, using different neuropathological scoring, and statistical methods, reported that moderate B-ASC (but not severe or mild) was associated with hippocampal atrophy.76 Because of all these findings collectively, we hypothesized that the ABCC9 HS-Aging risk genotype is associated with B-ASC in advanced old age, possibly upstream of the risk for HS-Aging (Figure 4(d)). The present study showed an association between the ABCC9 HS-Aging risk genotype and B-ASC pathology in cases with an age at death ≥80 years. There was no evidence of that association among individuals with age at death <80 years.
In order to test the association between the ABCC9 HS-Aging risk genotype and B-ASC, we analyzed CBF in elderly individuals. The rationale for this experiment includes that (1) B-ASC is associated with white matter hyperintensities (WMHs) on MRI scans,77–81 and (2) WMHs have been correlated with decreases in CBF82–84 and cognitive impairment.85–89 We found the ABCC9 HS-Aging risk genotype to be associated with decreased CBF in elderly individuals. These findings support the hypothesis that the ABCC9 HS-Aging risk genotype promotes B-ASC in the oldest-old with decreases in CBF on neuroimaging. ABCC9 encodes a regulator of ATP-sensitive potassium channels that is expressed in vascular smooth muscle cells.90–93 The protein is important for vascular tone regulation and reactivity to metabolic factors and oxidative stress.90,94–96 We hypothesize that gene variants in ABCC9 could result in chronic perturbations of the neurovascular unit leading to decrease in CBF and B-ASC pathology. Therefore, the brain changes associated with ABCC9 gene variants may be part of a “brain wide” disease characterized by HS-Aging, TDP-43 pathology, and B-ASC in elderly individuals.97,98 Since TMEM106B and GRN SNPs were not associated with B-ASC pathology, we hypothesize that these are downstream in the pathological process of HS-Aging (Figure 4(b)). Further studies are warranted to test this hypothesis.
We conclude that B-ASC is a common vascular pathology with a deleterious impact on global cognition in elderly individuals. Risk factors for B-ASC include hypertension, which has long been considered to be a putative modifiable factor, as well as advanced age. Additional possibly targetable mechanisms involved in the B-ASC pathogenesis are mostly unknown, but the results of this study offer candidate pathways involving ABCC9 gene products. Furthermore, we provide evidence that ASL neuroimaging is a potential candidate biomarker to indicate ABCC9-related variations in CBF that could be useful in a clinical setting. These findings may serve to increase awareness about B-ASC, a common cerebrovascular pathology associated with cognitive impairment.
Funding
This work was supported by the following National Institute of Health grants: P30 AG028383, R01 AG038651, R01 AG042419, T32 AG000242, K25 AG043546. The National Alzheimer's Coordinating Center database from which clinical information was obtained was supported by the following National Institute of Health grants: P30 AG028383, R01 AG038651, R01 AG042419, T32 AG000242, K25 AG043546. The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI David Teplow, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), and P50 AG005681 (PI John Morris, MD). The Alzheimer's disease genetic consortium from which genetic information was obtained is funded by NIA/NIH Grant U01 AG032984 and RC2AG036528. Samples from the National Cell Repository for Alzheimer's Disease (NCRAD) which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the NIA Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the University of Southern California. Genetics data for this study were partially prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24-AG041689-01), funded by the NIA.
Supplementary Material
Supplementary Material
Acknowledgements
We are deeply grateful to all of the study participants who made this research possible. We thank Luke Broster, Holly Brothers, Kevin Foy, and Shaniya Maimaiti for editing versions of the manuscript. Peter Nelson had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. This research was conducted at the Department of Pathology and Laboratory Medicine, Division of Neuropathology, and Sanders-Brown Center on Aging, University of Kentucky, USA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
ETI and PTN both conceived the study, wrote the manuscript, and were involved in all aspects of the study design. ELA helped conceive the study, performed statistical analysis, and provided editorial input. AJ performed neuroimaging analysis and provided editorial input. SEM and WAK provided access to NACC data set and provided editorial input. FA provided assisted in statistical analysis and provided editorial input. DWF, YK, FAS, RJK, GAJ, JHN, SEM, WAK, FA, ADB, and LJV assisted in study design and/or clinical material, and provided editorial input.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Lammie GA. Hypertensive cerebral small vessel disease and stroke. Brain Pathol 2002; 12: 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roman GC and Benavent O. The neuropathology of vascular dementia. Seminars in Cerebrovascular Diseases and Stroke 2010; 4: 87–96.
- 3.Zheng L, Vinters HV, Mack WJ, et al. Cerebral atherosclerosis is associated with cystic infarcts and microinfarcts but not Alzheimer pathologic changes. Stroke 2013; 44: 2835–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esiri MM, Wilcock GK, Morris JH. Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 1997; 63: 749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jellinger KA. Pathology and pathogenesis of vascular cognitive impairment – a critical update. Front Aging Neurosci 2013; 5: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erkinjuntti T, Benavente O, Eliasziw M, et al. Diffuse vacuolization (spongiosis) and arteriolosclerosis in the frontal white matter occurs in vascular dementia. Arch Neurol 1996; 53: 325–332. [DOI] [PubMed] [Google Scholar]
- 7.Pantoni L, Garcia JH, Brown GG. Vascular pathology in three cases of progressive cognitive deterioration. J Neurol Sci 1996; 135: 131–139. [DOI] [PubMed] [Google Scholar]
- 8.Neltner JH, Abner EL, Baker S, et al. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 2014; 137(Pt 1): 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinters HV, Ellis WG, Zarow C, et al. Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 2000; 59: 931–945. [DOI] [PubMed] [Google Scholar]
- 10.Bridges LR, Andoh J, Lawrence AJ, et al. Blood-brain barrier dysfunction and cerebral small vessel disease (arteriolosclerosis) in brains of older people. J Neuropathol Exp Neurol 2014; 73: 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou SH, Shulman JM, Keenan BT, et al. Genetic susceptibility for ischemic infarction and arteriolosclerosis based on neuropathologic evaluations. Cerebrovasc Dis 2013; 36: 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchman AS, Yu L, Boyle PA, et al. Microvascular brain pathology and late-life motor impairment. Neurology 2013; 80: 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gavornik P, Galbavy S. Clinical picture of arteriolosclerosis. Bratisl Lek Listy 2001; 102: 326–331. [PubMed] [Google Scholar]
- 14.Grinberg LT, Thal DR. Vascular pathology in the aged human brain. Acta Neuropathol 2010; 119: 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker AB. Structure of the small cerebral arteries in hypertension. Am J Pathol 1941; 17: 39–46.1. [PMC free article] [PubMed] [Google Scholar]
- 16.Moritz AR, Oldt MR. Arteriolar sclerosis in hypertensive and non-hypertensive individuals. Am J Pathol 1937; 13: 679–728 7. [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson G. 1. On certain points in the anatomy and pathology of Bright's Disease of the kidney. 2. On the influence of the minute blood-vessels upon the circulation. Med Chir Trans 1868; 51: 57–78.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baumbach GL, Dobrin PB, Hart MN, et al. Mechanics of cerebral arterioles in hypertensive rats. Circulation Res 1988; 62: 56–64. [DOI] [PubMed] [Google Scholar]
- 19.Baumbach GL, Sigmund CD, Faraci FM. Cerebral arteriolar structure in mice overexpressing human renin and angiotensinogen. Hypertension 2003; 41: 50–55. [DOI] [PubMed] [Google Scholar]
- 20.Rizzoni D, Porteri E, Guelfi D, et al. Structural alterations in subcutaneous small arteries of normotensive and hypertensive patients with non-insulin-dependent diabetes mellitus. Circulation 2001; 103: 1238–1244. [DOI] [PubMed] [Google Scholar]
- 21.Schofield I, Malik R, Izzard A, et al. Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation 2002; 106: 3037–3043. [DOI] [PubMed] [Google Scholar]
- 22.Danis RP, Yang Y. Microvascular retinopathy in the Zucker diabetic fatty rat. Invest Ophthalmol Visual Sci 1993; 34: 2367–2371. [PubMed] [Google Scholar]
- 23.Like AA, Lavine RL, Poffenbarger PL, et al. Studies in the diabetic mutant mouse. VI. Evolution of glomerular lesions and associated proteinuria. Am J Pathol 1972; 66: 193–224. [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson PT, Estus S, Abner EL, et al. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathologica 2014; 127: 825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson PT, Wang WX, Partch AB, et al. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol 2015; 74: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson PT, Smith CD, Abner EL, et al. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta neuropathologica 2013; 126: 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Standen NB, Quayle JM, Davies NW, et al. Hyperpolarizing vasodilators activate ATP-sensitive K + channels in arterial smooth muscle. Science 1989; 245: 177–180. [DOI] [PubMed] [Google Scholar]
- 28.Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 2010; 20: 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thal DR, Ghebremedhin E, Orantes M, et al. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 2003; 62: 1287–1301. [DOI] [PubMed] [Google Scholar]
- 30.Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain 2013; 136(Pt 9): 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12(3): 189–98. [DOI] [PubMed] [Google Scholar]
- 32.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993; 43: 2412–2414. [DOI] [PubMed] [Google Scholar]
- 33.Monsell SE, Mock C, Roe CM, et al. Comparison of symptomatic and asymptomatic persons with Alzheimer disease neuropathology. Neurology 2013; 80: 2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch CA, Walsh C, Blanco A, et al. The clinical dementia rating sum of box score in mild dementia. Dementia Geriatr Cogn Disord 2006; 21: 40–43. [DOI] [PubMed] [Google Scholar]
- 35.Savva GM, Wharton SB, Ince PG, et al. Age, neuropathology, and dementia. N Engl J Med 2009; 360: 2302–2309. [DOI] [PubMed] [Google Scholar]
- 36.von Gunten A, Ebbing K, Imhof A, et al. Brain aging in the oldest-old. Curr Gerontol Geriatr Res 2010. DOI:10.1155/2010/358531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imhof A, Kovari E, von Gunten A, et al. Morphological substrates of cognitive decline in nonagenarians and centenarians: a new paradigm? J Neurol Sci 2007; 257: 72–79. [DOI] [PubMed] [Google Scholar]
- 38.Nelson PT, Head E, Schmitt FA, et al. Alzheimer's disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol 2011; 121: 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ubhi K, Peng K, Lessig S, et al. Neuropathology of dementia with Lewy bodies in advanced age: a comparison with Alzheimer disease. Neurosci Lett 2010; 485: 222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bancher C, Jellinger KA. Neurofibrillary tangle predominant form of senile dementia of Alzheimer type: a rare subtype in very old subjects. Acta neuropathologica 1994; 88: 565–570. [DOI] [PubMed] [Google Scholar]
- 41.Lorke DE, Wai MS, Liang Y, et al. TUNEL and growth factor expression in the prefrontal cortex of Alzheimer patients over 80 years old. Int J Immunopathol Pharmacol 2010; 23: 13–23. [DOI] [PubMed] [Google Scholar]
- 42.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 2006; 20: 210–216. [DOI] [PubMed] [Google Scholar]
- 43.Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord 2004; 18: 270–277. [PubMed] [Google Scholar]
- 44.Brenowitz WD, Monsell SE, Schmitt FA, et al. Hippocampal sclerosis of aging is a key Alzheimer's disease mimic: clinical-pathologic correlations and comparisons with both Alzheimer's disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimer's Dis 2014; 39: 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer's Disease Centers' Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord 2009; 23: 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mueller SG, Weiner MW, Thal LJ, et al. The Alzheimer's disease neuroimaging initiative. Neuroimaging Clin N Am 2005; 15: 869–877. xi-xii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mueller SG, Weiner MW, Thal LJ, et al. Ways toward an early diagnosis in Alzheimer's disease: the Alzheimer's Disease Neuroimaging Initiative (ADNI). Alzheimer's Dement 2005; 1: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006; 112: 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41: 479–486. [DOI] [PubMed] [Google Scholar]
- 50.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011; 43: 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011; 43: 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013; 45: 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the uniform data set. Alzheimer Dis Assoc Disord 2007; 21: 249–258. [DOI] [PubMed] [Google Scholar]
- 54.Nelson PT, Pious NM, Jicha GA, et al. APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol 2013; 72: 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leow AD, Klunder AD, Jack CR, Jr, et al. Longitudinal stability of MRI for mapping brain change using tensor-based morphometry. NeuroImage 2006; 31: 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Alsop DC, Song HK, et al. Arterial transit time imaging with flow encoding arterial spin tagging (FEAST). Magn Reson Med 2003; 50: 599–607. [DOI] [PubMed] [Google Scholar]
- 57.Ramage AE, Lin AL, Olvera RL, et al. Resting-state regional cerebral blood flow during adolescence: associations with initiation of substance use and prediction of future use disorders. Drug Alcohol Depend 2015; 149: 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brayne C, Richardson K, Matthews FE, et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimer's Dis 2009; 18: 645–658. [DOI] [PubMed] [Google Scholar]
- 59.Ohm TG, Scharnagl H, Marz W, et al. Apolipoprotein E isoforms and the development of low and high Braak stages of Alzheimer's disease-related lesions. Acta Neuropathol 1999; 98: 273–280. [DOI] [PubMed] [Google Scholar]
- 60.Dickson DW, Davies P, Bevona C, et al. Hippocampal sclerosis: a common pathological feature of dementia in very old ( > or = 80 years of age) humans. Acta Neuropathol 1994; 88: 212–221. [DOI] [PubMed] [Google Scholar]
- 61.Haneuse S, Schildcrout J, Crane P, et al. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology 2009; 32: 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnston DW, Propper C, Shields MA. Comparing subjective and objective measures of health: evidence from hypertension for the income/health gradient. J Health Econ 2009; 28: 540–552. [DOI] [PubMed] [Google Scholar]
- 63.Hachinski V, Iadecola C, Petersen RC, et al. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 2006; 37: 2220–2241. [DOI] [PubMed] [Google Scholar]
- 64.Buchman AS, Leurgans SE, Nag S, et al. Cerebrovascular disease pathology and parkinsonian signs in old age. Stroke 2011; 42: 3183–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deramecourt V, Slade JY, Oakley AE, et al. Staging and natural history of cerebrovascular pathology in dementia. Neurology 2012; 78: 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barker WW, Luis CA, Kashuba A, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord 2002; 16: 203–212. [DOI] [PubMed] [Google Scholar]
- 67.Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 2007; 66: 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Furuta A, Ishii N, Nishihara Y, et al. Medullary arteries in aging and dementia. Stroke 1991; 22: 442–446. [DOI] [PubMed] [Google Scholar]
- 69.Ferrer I, Bella R, Serrano MT, et al. Arteriolosclerotic leucoencephalopathy in the elderly and its relation to white matter lesions in Binswanger's disease, multi-infarct encephalopathy and Alzheimer's disease. J Neurol Sci 1990; 98: 37–50. [DOI] [PubMed] [Google Scholar]
- 70.Desmond DW. The neuropsychology of vascular cognitive impairment: is there a specific cognitive deficit? J Neurol Sci 2004; 226: 3–7. [DOI] [PubMed] [Google Scholar]
- 71.Lammie GA, Brannan F, Slattery J, et al. Nonhypertensive cerebral small-vessel disease. An autopsy study. Stroke 1997; 28: 2222–2229. [DOI] [PubMed] [Google Scholar]
- 72.Nelson PT, Smith CD, Abner EA, et al. Human cerebral neuropathology of Type 2 diabetes mellitus. Biochim Biophys Acta 2009; 1792: 454–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain 2011; 134(Pt 5): 1506–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zarow C, Weiner MW, Ellis WG, et al. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav 2012; 2: 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leverenz JB, Agustin CM, Tsuang D, et al. Clinical and neuropathological characteristics of hippocampal sclerosis: a community-based study. Arch Neurol 2002; 59: 1099–1106. [DOI] [PubMed] [Google Scholar]
- 76.Crystal HA, Schneider JA, Bennett DA, et al. Associations of cerebrovascular and Alzheimer's disease pathology with brain atrophy. Curr Alzheimer Res 2014; 11: 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fazekas F, Kleinert R, Offenbacher H, et al. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology 1993; 43: 1683–1689. [DOI] [PubMed] [Google Scholar]
- 78.Scheltens P, Barkhof F, Leys D, et al. Histopathologic correlates of white matter changes on MRI in Alzheimer's disease and normal aging. Neurology 1995; 45: 883–888. [DOI] [PubMed] [Google Scholar]
- 79.Shim YS, Yang DW, Roe CM, et al. Pathological correlates of white matter hyperintensities on magnetic resonance imaging. Dement Geriatr Cogn Disord 2015; 39: 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bal S, Goyal M, Smith E, et al. Central nervous system imaging in diabetic cerebrovascular diseases and white matter hyperintensities. Handb Clin Neurol 2014; 126: 291–315. [DOI] [PubMed] [Google Scholar]
- 81.Erten-Lyons D, Woltjer R, Kaye J, et al. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology 2013; 81: 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oishi M, Mochizuki Y. Differences in regional cerebral blood flow in two types of leuko-araiosis. J Neurol Sci 1999; 164: 129–133. [DOI] [PubMed] [Google Scholar]
- 83.Kobari M, Meyer JS, Ichijo M. Leuko-araiosis, cerebral atrophy, and cerebral perfusion in normal aging. Arch Neurol 1990; 47: 161–165. [DOI] [PubMed] [Google Scholar]
- 84.Promjunyakul N, Lahna D, Kaye JA, et al. Characterizing the white matter hyperintensity penumbra with cerebral blood flow measures. NeuroImage Clin 2015; 8: 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Albert M, Massaro J, DeCarli C, et al. Profiles by sex of brain MRI and cognitive function in the Framingham offspring study. Alzheimer Dis Assoc Disord 2010; 24: 190–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Au R, Massaro JM, Wolf PA, et al. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol 2006; 63: 246–250. [DOI] [PubMed] [Google Scholar]
- 87.Luchsinger JA, Brickman AM, Reitz C, et al. Subclinical cerebrovascular disease in mild cognitive impairment. Neurology 2009; 73: 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williams LR, Hutchinson CE, Jackson A, et al. Clinical correlates of cerebral white matter hyperintensities in cognitively normal older adults. Arch Gerontol Geriatr 2010; 50: 127–131. [DOI] [PubMed] [Google Scholar]
- 89.Silbert LC, Dodge HH, Perkins LG, et al. Trajectory of white matter hyperintensity burden preceding mild cognitive impairment. Neurology 2012; 79: 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res 2013; 112: 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang HL, Bolton TB. Two types of ATP-sensitive potassium channels in rat portal vein smooth muscle cells. Br J Pharmacol 1996; 118: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thorneloe KS, Maruyama Y, Malcolm AT, et al. Protein kinase C modulation of recombinant ATP-sensitive K(+) channels composed of Kir6.1 and/or Kir6.2 expressed with SUR2B. J Physiol 2002; 541(Pt 1): 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamada M, Isomoto S, Matsumoto S, et al. Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K + channel. J Physiol 1997; 499(Pt 3): 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flagg TP, Enkvetchakul D, Koster JC, et al. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev 2010; 90: 799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shi NQ, Ye B, Makielski JC. Function and distribution of the SUR isoforms and splice variants. J Mol Cell Cardiol 2005; 39: 51–60. [DOI] [PubMed] [Google Scholar]
- 96.Teramoto N. Physiological roles of ATP-sensitive K + channels in smooth muscle. J Physiol 2006; 572(Pt 3): 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ighodaro ET, Jicha GA, Schmitt FA, et al. Hippocampal sclerosis of aging can be segmental: two cases and review of the literature. J Neuropathol Exp Neurol 2015; 74: 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nelson PT, Jicha GA, Wang WX, et al. ABCC9/SUR2 in the brain: implications for hippocampal sclerosis of aging and a potential therapeutic target. Ageing Res Rev 2015; 24(Pt B): 111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.