

Abstract
The myogenic response of cerebral resistance arterial smooth muscle to intraluminal pressure elevation is a key physiological mechanism regulating blood flow to the brain. Rho-associated kinase plays a critical role in the myogenic response by activating Ca2+ sensitization mechanisms: (i) Rho-associated kinase inhibits myosin light chain phosphatase by phosphorylating its targeting subunit myosin phosphatase targeting subunit 1 (at T855), augmenting 20 kDa myosin regulatory light chain (LC20) phosphorylation and force generation; and (ii) Rho-associated kinase stimulates cytoskeletal actin polymerization, enhancing force transmission to the cell membrane. Here, we tested the hypothesis that abnormal Rho-associated kinase-mediated myosin light chain phosphatase regulation underlies the dysfunctional cerebral myogenic response of the Goto-Kakizaki rat model of type 2 diabetes. Basal levels of myogenic tone, LC20, and MYPT1-T855 phosphorylation were elevated and G-actin content was reduced in arteries of pre-diabetic 8–10 weeks Goto-Kakizaki rats with normal serum insulin and glucose levels. Pressure-dependent myogenic constriction, LC20, and myosin phosphatase targeting subunit 1 phosphorylation and actin polymerization were suppressed in both pre-diabetic Goto-Kakizaki and diabetic (18–20 weeks) Goto-Kakizaki rats, whereas RhoA, ROK2, and MYPT1 expression were unaffected. We conclude that abnormal Rho-associated kinase-mediated Ca2+ sensitization contributes to the dysfunctional cerebral myogenic response in the Goto-Kakizaki model of type 2 diabetes.
Keywords: Ca2+ sensitization, cerebral arteries, Goto-Kakizaki, Rho-associated kinase, type 2 diabetes
Introduction
Cerebral blood flow is matched to neuronal metabolic demand in dynamic physiological conditions through multiple mechanisms that influence force generation by vascular smooth muscle cells (VSMCs) within the walls of cerebral resistance arterioles and small arteries.1 The myogenic response of these vessels permits constriction and relaxation in response to intraluminal pressure elevation and reduction, respectively, and thereby maintain flow relatively constant during changes in perfusion pressure within the physiological range, i.e. blood flow autoregulation. This fundamental mechanism also determines capillary perfusion pressure within the downstream arterial tree and establishes a state of partial constriction from which vessels can dilate or further constrict. The latter permits local control of flow by vasoactive agonists and retrograde propagating vasodilation arising from neurovascular coupling to accommodate temporal changes in oxygen and nutrient demand.2,3 Myogenic dilation is key to the maintenance of blood flow at low perfusion pressure, preventing vascular insufficiency, and ischemic injury. In contrast, myogenic constriction evoked by pressure elevation protects downstream arterioles, capillaries, and the blood–brain barrier against damage and rupture resulting from unrestricted, excessive blood flow.1,3 That the cerebrovascular myogenic response is crucial for the structural and functional integrity of the brain is indicated by a direct link between myogenic dysfunction, brain injury, and cognitive impairment in aging and disease.4,5
Accumulating evidence indicates that impairment of the myogenic response is a defect that is common to multiple disorders: increased basal myogenic tone development and/or loss of myogenic constriction and proportional dilation to pressure elevation were reported for animal models and humans with stroke, hypertension, Alzheimer disease, and type 2 diabetes.3,5–9 For example, enhanced myogenic tone was observed in mesenteric arteries of db/db mice,10 cerebral arterioles of BBZDR/WOR rats,11 and cerebral and mesenteric arteries of Goto-Kakizaki (GK) rats,12–14 but a loss of myogenic constriction was reported for cerebral and coronary arteries of older GK rats12,15 and gluteal arterioles of diabetic patients.16 The varied findings for cerebral vessels of GK rats12 imply that vascular bed, species or type 2 diabetes model cannot account for these differences in myogenic behavior. Alternatively, it is possible that myogenic dysfunction changes with the progression of type 2 diabetes and severity of insulin resistance, hyperinsulinemia, and hyperglycemia.
The specific molecular defects responsible for the impaired myogenic regulation of cerebral arterial diameter are not known with certainty. The sensitivity of resistance arteries and arterioles to intraluminal pressure derives from cellular mechanisms of force generation that are inherent to VSMCs, i.e. intrinsic myogenic mechanisms. Contractile force development in response to pressure elevation is postulated to require: (i) Ca2+-calmodulin-dependent activation of myosin light chain kinase (MLCK) in response to increased cytosolic free [Ca2+] ([Ca2+]i); (ii) inhibition of MLCP via Rho-associated kinase (ROK)-mediated phosphorylation of its targeting/regulatory subunit MYPT1; and (iii) ROK and protein kinase C (PKC)-stimulated actin polymerization involving reduced globular (G)-actin and increased filamentous (F)-actin within the cortical cytoskeleton.2,17 Specifically, pressure elevation favors increased myosin regulatory light chain (LC20) phosphorylation required for cross-bridge cycling, and actin polymerization provides more efficient transmission of contractile force over the cell surface and to the extracellular matrix.17–20 Whether a defect(s) in these mechanisms contributes to the dysfunctional myogenic regulation of cerebral blood flow in type 2 diabetes is unknown, but available evidence suggests that defective ROK signaling may be involved.15,21,22
Here, we tested the hypothesis that abnormal regulation of MLCP and actin polymerization contributes to the dysfunctional myogenic response of cerebral arteries (CAs) in the GK rat model of type 2 diabetes. Arterial pressure myography was used to assess myogenic dysfunction, and ultra-high-sensitivity western blotting to quantify MYPT1 and LC20 phosphorylation ( ± ROK inhibitor H1152) and G-actin content at varied intraluminal pressures in middle and posterior CAs of 8–10 and 18–20 week old GK rats. Our findings indicate the presence of progressive dysfunction in the cerebral myogenic response in the GK rat model involving a transient increase in basal ROK-mediated MYPT1 phosphorylation and actin polymerization, and an increasing impairment in the pressure-dependent regulation of MLCP and actin cytoskeletal remodeling with age.
Materials and methods
Animals
Male Wistar (WR) and GK rats (Charles River, Montréal, Canada) were studied at 8–10 and 18–20 weeks of age. Animals were maintained and euthanized by halothane inhalation and exsanguination according to the standards of the Canadian Council on Animal Care, and a protocol reviewed by the Animal Care Committee of the Cumming School of Medicine, University of Calgary and the ARRIVE guidelines. Serum glucose and insulin levels were determined by IDEXX Laboratories (Calgary, Canada) and a commercial ELISA kit (Millipore, Bellerica, USA); values of serum glucose and insulin in the 18–20 week GK animals employed here were previously published.23
Arterial pressure myography
Rat brains were removed and transferred to ice-cold Krebs' solution (composition in mmol/L): NaCl 120, NaHCO3 25, KCl 4.8, NaH2PO4 1.2, MgSO4 1.2, glucose 11, CaCl2 1.8 (pH 7.4 when aerated with 95% air-5% CO2). CAs were removed, cut into segments of 2–3 mm length, cannulated, denuded of endothelium (unless indicated otherwise) prior to vessel diameter analysis by videomicroscopy.24,25 Vessel diameter ± 0.5 μmol/L ROK inhibitor H115226 was evaluated with pressure steps from 10 to 140 mmHg in 20 mmHg increments prior to exposure to zero Ca2+-containing Krebs' saline (i.e. no added Ca2+ and 2 mmol/L EGTA) to determine passive diameter.24,25 Phosphoprotein and G-actin content was quantified in CAs pressurized to 10, 60, 120 mmHg ± H1152 (0.5 μmol/L) and flash-frozen or treated with F-actin stabilization buffer once stable myogenic constriction was achieved.
RT-PCR and real-time qPCR
Reverse transcriptase polymerase chain reaction (PCR) (RT-PCR) and real-time quantitative PCR (qPCR) were conducted as previously described27 using appropriate primer pairs to quantify transcript levels of RhoA (108 bp; F: CAGCAAGGACCAGTTCCCAGA; R: AGCTGTGTCCATAAAGCCAACTC), ROK2 (85 bp; F: CTAACAGTCCGTGGGTGGTTCA; R: TCCACCTGGCATGTACTCCATC) and MYPT1 (103 bp; F: AGGAAGCAATGGAAGAGCTA; R: CCTCGCGTCTCTAAGCATTA).
Western blotting
Proteins were extracted by heating flash-frozen vessels at 95 ℃ for 10 min in sample buffer (4% SDS, 100 mmol/L dithiothreitol (DTT), 10% glycerol, 0.01% bromophenol blue, 60 mmol/L Tris-HCl, pH 6.8) and rotated overnight at 4 ℃. A three-step western blotting protocol was used to quantify phospho-MYPT1 (ROK sites T697 and T855), -LC20, -FAK-Y397, and ROK2 content, as described.24,26,28 Phospho-MYPT1-T697, -MYPT1-T855, -FAK-Y397, and ROK2 levels were normalized to actin content in each sample determined by conventional two-step western blotting. Phosphorylated and unphosphorylated LC20 were separated by phosphate-affinity tag SDS-PAGE (Phos-Tag™ SDS-PAGE) and quantified as a percentage of total LC20.24,26,28
G-actin determination
G-actin content was detected by immunoblotting and normalized to SM22 as previously described.29,30 F-actin content was not determined based on previous analysis showing contamination by G-actin from intact cells and cell fragments within the high speed pellet obtained by differential centrifugation and that normalization to SM22 provided an accurate quantification of the change in G-actin content with increased pressurization.26,29,30
Chemicals
All chemicals were purchased from Sigma (Oakville, Canada) unless indicated otherwise. H1152 was obtained from Calbiochem (San Diego, CA, USA), polyclonal rabbit anti-LC20 and GAPDH from Santa Cruz Biotechnology (Santa Cruz, CA, USA), polyclonal rabbit phosphospecific anti-MYPT1, ROK2, and FAK-Y397 from Millipore (Temecula, CA, USA), polyclonal goat anti-SM22 from Novus Biologicals (Littleton, MA, USA), biotin-conjugated goat anti-rabbit secondary antibody from EMD Millipore (Bellerica, MA, USA), horseradish peroxidase-conjugated streptavidin from Pierce Biotechnology (Rockford, IL, USA) and Phos-tag™ acrylamide from NARD Institute Ltd (Amajasaki City, Japan)
Statistical analysis
All values are presented as means ± SEM with n values indicative of the number of vessels analyzed, i.e. only one vessel per rat was analyzed in each experimental group, but multiple vessels from each rat were used in different treatment groups (e.g. untreated vs. H1152-treated) or in different types of experiments (e.g. myography vs. western blotting). Statistical difference was determined using unpaired Student's t-test or repeated measures ANOVA followed by Bonferroni's post hoc test as indicated. A value of P < 0.05 was considered statistically significant.
Results
Serum glucose and insulin levels in GK and WR
Serum insulin (ng/ml) and glucose (mmol/L) levels were not statistically different in GK and WR at 8–10 weeks (1.2 ± 0.1 and 1.05 ± 0.1 for insulin; 8.4 ± 0.4 and 8.2 ± 0.3 for glucose; n = 6 each), but were increased at 10–12 weeks in GK rats (1.58 ± 0.1 vs. 1.07 ± 0.1 (P < 0.05) in WR for insulin; 14.4 ± 1.7 vs. 8.5 ± 0.2 (P < 0.05) in WR for glucose; n = 6 each). Insulin and glucose levels of the 18–20 week GK rats and WR used here were previously reported23, i.e. 2.8 ± 0.5 versus 1.28 ± 0.1 (P < 0.05) for insulin; 19.0 ± 1.3 versus 11.4 ± 0.9 (P < 0.05) for glucose (n = 6 each). These measurements are consistent with previous data for GK rats indicating the presence of insulin resistance after ∼5 weeks, and overt diabetes with marked hyperglycemia and hyperinsulinemia after ∼10 weeks.31,32 Although the levels of insulin and glucose were not different in the 8–10 week GK rats, we found that the vessels displayed a reduced response to insulin challenge. We applied insulin at 50 nmol/L (a submaximal concentration) for inhibition of myogenic tone in age-matched WR) to pressurized, endothelium-denuded CAs from 8 to 10 week GK rats and WR. Insulin-evoked dilation at 80 mmHg (calculated as % of maximal dilation evoked by Ca2+-free conditions) was significantly reduced in 8–10 week GK compared to age-matched WR: 7.9 ± 1.9% for GK compared to 22.9 ± 1.8% for WR (P < 0.05, n = 4 vessels each). This result is consistent with the presence of insulin resistance in the 8–10 week GK rats employed in this study. Based on these data, we assessed the myogenic behavior of posterior CAs from age-matched WR and 8–10 week GK which demonstrated reduced insulin-evoked dilation but normal insulin and glucose levels, as well as, 18–20 week WR versus GK rats that demonstrated evidence of elevated insulin and glucose levels. Whether the observed changes in the myogenic autoregulation of cerebral arterial diameter in the GK vessels were due to abnormal phosphorylation of MYPT1 and LC20, and/or to differences in actin polymerization were determined by ultra-high-sensitivity western blotting.
Abnormal pressure-evoked cerebral myogenic constriction in GK rat
Standard pressure myography was used to assess the myogenic response of endothelium-denuded posterior CAs evoked by step increases in intraluminal pressure between 10 and 140 mmHg. Arteries of 8–10 and 18–20 week age-matched control WR exhibited myogenic behavior as described previously for CAs of SD rats2,24,33, i.e. vessel diameter increased proportionally with pressure until ∼40 mmHg, active myogenic constriction was detected between ∼40 and ∼140 mmHg with evidence of a negative slope in the pressure–diameter relation between ∼40 and ∼80 mmHg (Figure 1(a) and (e)). In contrast, arteries from 8 to 10 week GKs exhibited a significant increase in basal myogenic tone at 10 and 40 mmHg, maintenance of diameter and no indication of a negative slope in the pressure–diameter relation between 40 and 80 mmHg, and slight dilation with pressurization to >80 mmHg (Figure 1(b) and (c)). The myogenic response of endothelium-intact vessels from 8 to 10 week WR and GK rats was also assessed (the presence of a functional endothelium was assessed with bradykinin at 30 nmol/L). No difference in the extent of myogenic constriction was detected between endothelium-denuded and -intact WR vessels (Figure 1(c)). Increased basal tone was not detected in CAs of 18–20 week GKs, and diameter increased proportionally with pressure elevation over the entire pressure range between 20 and 140 mmHg (Figure 1(f) and (g)). The extent of active myogenic constriction following pressure elevation was identical in 8–10 and 18–20 week WRs, but it was depressed at > 80 mmHg in GK vessels, with a significantly greater reduction in force evident in vessels from the 18 to 20 week animals (Figure 1(d) and (h)). There was no significant difference in passive diameter between vessels from WR and GKs in the absence of extracellular Ca2+ indicating that vascular remodeling may be excluded as a factor in the dysfunctional myogenic response of 8–10 and 18–20 week GK rats.
Figure 1.

Effect of ROK inhibition on the myogenic response of CAs from GK and WR. Panels (a), (b), and (c): Representative recordings (a) and (b) and mean values ± SEM (c) of CA diameter in response to step increases in pressure between 10 and 140 mmHg in control conditions (2.5 mmol/L Ca2+), in ROK inhibitor (H1152; 0.5 μmol/L), and in zero external Ca2+ (Ca2+-free) for 8–10 week WR (WR8, panel (a) and black lines in panel (c)) and GK (GK8, panel (b) and red lines in panel (c)) rats (n = 8 each). Open squares show mean diameter ± SEM of endothelium-intact CAs from 8-10 week WR (black) and GK rats (red) (n = 5 and 8, respectively). *Significantly different (P < 0.05) from corresponding value in WR8. Panel (d): Mean active constriction versus pressure ± SEM between 10 and 140 mmHg of CAs from WR8 (black lines) to GK8 (red lines) rats in control conditions (open symbols) and in the presence of H1152 (0.5 μmol/L; closed symbols) (n = 8).* Significantly different (P < 0.05) from value in WR8. Panels (e), (f), and (g): Representative traces (e and f) and mean values ± SEM (g) of CA diameter in response to step increases in pressure between 10 and 140 mmHg in control conditions (2.5 mmol/L Ca2+), in ROK inhibitor (H1152; 0.5 μmol/L), and in zero external Ca2+ (Ca2+-free) for 18–20 week WR (WR18, panel (e) and black lines in panel (g)) and GK rats (GK18, panel (f) and blue lines in panel (g)) (n = 8). Significantly different (P < 0.05) from corresponding value in WR18. Panel (h): Mean active constriction versus pressure ± SEM between 10 and 140 mmHg of CAs from WR18 (black lines) and GK18 (blue lines) rats in control conditions and in the presence of H1152 (0.5 μmol/L) (n = 8).* Significantly different (P < 0.05) from value in WR18.
H1152 (0.5 µmol/L) was used to assess the contribution of ROK to the abnormal myogenic behavior of GK vessels. H1152 abolished myogenic constriction of WR CAs as previously reported.24 Significantly, H1152 completely reversed the elevated basal myogenic tone in CAs of 8–10 week GKs, as well as the residual pressure-dependent tone observed in 8–10 and 18–20 week GK vessels (Figure 1).
Loss of pressure-evoked LC20 and MYPT1-T855 phosphorylation in GK CAs
Previous studies identified an H1152-sensitive, pressure-dependent increase in LC20 and MYPT1-T855 phosphorylation in CAs.24 Here, we determined if the elevated basal tone and proportional dilation with increased pressure in the 8–10 and 18–20 week GK rats, respectively, were due to inappropriate LC20 and MYPT1 phosphorylation by flash-freezing CAs at 10, 60, and 120 mmHg. The selected pressures represent the unpressurized, basal condition (10 mmHg), and approximate the lower and upper limits of the physiological pressure range experienced by rat posterior CAs.2,3 Figure 2(a) and (c) illustrates that pressure elevation from 10 to 60 or 120 mmHg evoked increased LC20 phosphorylation in WR arteries as detected in previous studies.24,26 Consistent with our hypothesis, phospho-LC20 content was significantly elevated at 10 mmHg in 8–10 week GK compared to WR arteries (Figure 2(c)). Also, in contrast to the pressure-dependent increase at 60 and 120 compared to 10 mmHg in WR vessels, phospho-LC20 content in 8–10 week GK arteries at 60 mmHg was not different from the level at 10 mmHg (but identical to the level in WR vessels at 60 mmHg), and at 120 mmHg, the level was significantly lower than the value in WR vessels and not different from that at 10 mmHg (Figure 2(a) and (c)). Basal phosphorylation of LC20 at 10 mmHg was not different in arteries of 18–20 week GK and WRs, but the levels at 60 and 120 mmHg in GK vessels were significantly lower than those in WR at the same pressures and not different from that at 10 mmHg (Figure 2(d) and (e)). Taken together, these data indicate an absence of pressure-dependent regulation of LC20 phosphorylation in the 8–10 and 18–20 week GK vessels.
Figure 2.

Effect of ROK inhibition on pressure-evoked LC20 phosphorylation in CAs of GK and age-matched WR. Panels (a) and (b): Representative western blot for CAs of 8–10 week WR (WR8) and GK (GK8) rats at 10, 60, 120 mmHg (a), and for WR8 and GK8 vessels at 10 and 120 mmHg ± ROK inhibitor H1152 (0.5 μmol/L) (b). Monophosphorylated and unphosphorylated LC20 were separated by Phos-tag™ SDS-PAGE: upper band is monophosphorylated LC20 (1-P) and the lower band is unphosphorylated protein (0-P). Panel (c): Mean values ± SEM of LC20 phosphorylation as % of total LC20 at 10, 60, and 120 mmHg for CAs of WR8 (white bars) and GK8 (black bars) (n = 7), and at 10 and 120 mmHg for WR8 + H1152 (rising hatched bars) and GK8 + H1152 (falling hatched bars) (n = 3 and 7, respectively). * and # indicate significant difference (P < 0.05) from WR8 GK8 values at 10 mmHg, respectively. ** indicates significant difference (P < 0.05) from WR8 at 120 mmHg. Panels (d) and (e): Representative western blot (d) and mean values ± SEM (e) of LC20 phosphoprotein as % of total LC20 at 10, 60, 120 mmHg for CAs of 18–20 week WR (WR18, white bars) and GK rats (GK18, black bars) (n = 8). * and # indicate significantly different (P < 0.05) from WR18 value at 10 mmHg and WR18 value at the identical pressure, respectively. Note: All densitometric comparisons here and in subsequent figures were performed using single western blots; blots subsequently cut to remove lanes containing data not pertinent to the figure is denoted by a vertical bar, as in panel (a) between the WR8 and GK8 lanes.
To determine whether the abnormal levels of LC20 phosphorylation resulted from a dysregulation of MLCP activity by ROK, we quantified the levels of phosphorylation of the two major ROK phosphorylation sites on MYPT1, T855 and T697,3,24 at 10, 60, and 120 mmHg in WR and GK CAs. Figure 3 shows the normal pressure-dependent increase in phospho-MYPT1-T855 content previously reported24 was detected in both WR age-groups. Notably, a significant elevation in phospho-MYPT1-T855 content was detected at 10 mmHg in 8–10 week GK compared to WR CAs, but no pressure-dependent change was apparent at 60 and 120 mmHg, and the levels of phospho-MYPT1 were significantly lower in 8–10 week GK compared to WR CAs at 120 mmHg (Figure 3(a) and (c). The levels of phospho-MYPT1-T855 at 10 mmHg were identical in CAs of 18–20 week GK and WR, but there was a complete lack of any pressure-evoked increase in phosphorylation of MYPT1-T855 in the 18–20 week GK vessels (Figure 3(d) and (e)). This indicates that a lack of pressure-dependent regulation of MLCP and resulting impairment in the control of LC20 phosphorylation and force generation underlies the impaired myogenic response of CAs from 18 to 20 week GK rats with established diabetes.
Figure 3.

Effect of ROK inhibition on the pressure-evoked MYPT1-T855 phosphorylation in CAs from GK and age-matched WR. Panels (a) and (b): Representative blots of MYPT1-T855 phosphoprotein and corresponding levels of actin at 10, 60, and 120 mmHg for: (a) CAs of 8–10 week WR (WR8) and GK rats (GK8), and (b) CAs of WR8 (at 10 and 120 mmHg) and GK8 (at 10, 60, and 120 mmHg) ± H1152 (0.5 μmol/L). Panel (c): Mean values ± SEM of MYPT1-T855 phosphoprotein normalized to actin and expressed as a fraction of the WR8 value at 10 mmHg for CAs of WR8 (white bars), GK8 (black bars), and GK8 + H1152 (hatched bars) at 10, 60, and 120 mmHg (n = 7). * and # indicate significant difference (P < 0.05) from WR8 value at 10 mmHg and GK value at the identical pressure, respectively. Panels (d) and (e): Representative blots (d) of MYPT1-T855 phosphorylation and corresponding levels of actin in each lane and mean values ± SEM (e) of phosphoprotein normalized to actin and expressed as a fraction of the WR18 value at 10 mmHg for CAs of 18–20 week WR (WR18, white bars) and GK rats (GK18, black bars) at 10, 60, and 120 mmHg (n = 7). * and # indicate significant difference (P < 0.05) from WR18 value at 10 mmHg and WR18 value at the identical pressure, respectively.
H1152 (0.5 μmol/L) was employed to evaluate whether abnormal ROK activity was responsible for the elevated basal phosphoprotein content in 8–10 week GK arteries at 10 mmHg. ROK inhibition was previously shown to suppress LC20 and MYPT1-T855 phosphorylation at 10, 60, and 120 mmHg in CAs of Sprague-Dawley rats24 and this was confirmed in vessels of 8–10 week WR; H1152 (0.5 μmol/L) abolished the pressure-evoked increases in LC20 and MYPT1-T855 phosphorylation at 120 mmHg (Figures 2(b) and (c), and 3(b) and (c), respectively). Figure 2(b) and (c) shows that H1152 treatment suppressed the elevated basal level of LC20 phosphorylation in CAs of 8–10 week GKs at 10 mmHg, but had no effect on the reduced extent of LC20 phosphorylation at 120 mmHg. Figure 3(b) and (c) shows that the increased MYPT1-T855 phosphorylation at 10 mmHg was similarly blocked by H1152, as was phosphorylation at 60 and 120 mmHg. The elevated basal myogenic tone detected at 10 mmHg in 8–10 week GK arteries was, therefore, due to an H1152-sensitive increase in MYPT1-T855 and LC20 phosphorylation.
Phospho-MYPT1-T697 content of rat CAs was previously found to be unaffected by pressure elevation or H1152,24 and this was shown to be the case for MYPT1-T697 phosphorylation in 8–10 week GK and WR vessels (Figure 4(a) and (c)). A slight but significant increase in the level of MYPT1-T697 phosphorylation was evident at 10 and 60 mmHg in 8–10 week GK compared to WR vessels, but there was no difference in phosphoprotein content at 120 mmHg (Figure 4(a) and (c)). Figure 4 indicates that this slight elevation in MYPT-T697 phosphorylation at 10 mmHg in 8–10 week GK arteries was not sensitive to H1152; ROK phosphorylates this site in other vessels and smooth muscles,34 as well as zipper interacting protein kinase (ZIPK), myotonic dystrophy protein kinase (DMPK), integrin-linked kinase (ILK), and p21-activated protein kinase (reviewed in35), but their contribution in the GK rat is unknown.
Figure 4.
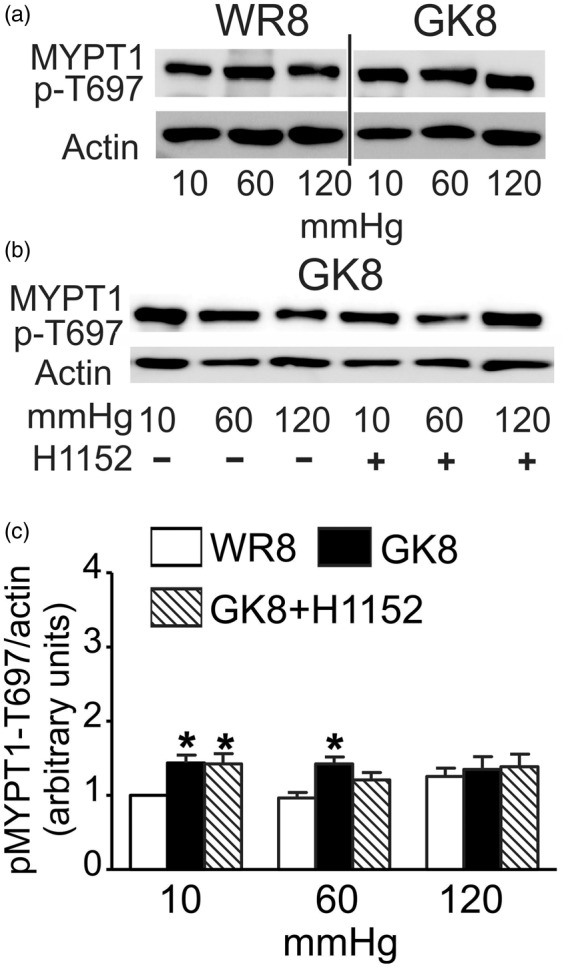
Effect of ROK inhibition on MYPT1-T697 phosphorylation in CAs from 8-10 week GK and WR. Panels (a) and (b): Representative blots of MYPT1-T697 phosphoprotein and corresponding levels of actin at 10, 60, and 120 mmHg for: (a) CAs of 8–10 week WR (WR8) and GK (GK8) rats; (b) CAs of GK8 rats ± H1152 (0.5 μmol/L) (n = 5). Panel (c): Mean values ± SEM of MYPT1-T697 phosphoprotein normalized to actin and expressed as a fraction of the WR value at 10 mmHg for CAs of WR8 (white bars), GK8 rats (black bars), and GK8 + H1152 (0.5 μmol/L) (hatched bars) at 10, 60, and 120 mmHg (n = 5). *Significant difference (P < 0.05) from WR8 value at 10 mmHg.
Loss of pressure-evoked actin polymerization in GK CAs
ROK activity was previously shown to elicit actin polymerization in the myogenic response.13,30 Based on the presence of H1152-sensitive basal tone and MYPT1-T855 phosphorylation in vessels of 8–10 week GK rats, and lack of pressure-dependent change in both at 18–20 weeks, we hypothesized that the regulation of actin dynamics in the myogenic response would also be defective. To evaluate whether abnormal pressure-evoked remodeling of the actin cytoskeleton contributes to the dysfunctional cerebral myogenic response of GK rats, G-actin content of CAs from WR and GK rats was determined at 10, 60, and 120 mmHg. Figure 5 shows that G-actin content decreased in a pressure-dependent manner at 60 and 120 compared to 10 mmHg in CAs from 8 to 10 and 18 to 20 week WR, as previously reported.26,30 However, no evidence of a pressure-dependent decline in G-actin content was obtained for GK vessels. G-actin was significantly lower at 10 mmHg, but there was no difference in content at 60 and 120 mmHg in 8–10 week GK compared to WR vessels (Figure 5). In contrast, the levels of G-actin at 10, 60, and 120 mmHg in 18–20 week GK arteries were not different from the level at 10 mmHg in WR vessels and there was no change in G-actin following pressurization (Figure 5).
Figure 5.

G-actin content in CAs from GK and age-matched WR. Panels (a) and (c): Representative western blots of G-actin and SM22 content (a) and mean values ± SEM (c) of G-actin normalized to SM22 and expressed as a fraction of the WR8 value at 10 mmHg in each age group for CAs of 8–10 week WR (WR8, white bars) and GK (GK8, black bars) at 10, 60, and 120 mmHg (n = 6). Panels (b) and (d): Representative western blots of G-actin and SM22 content (b) and mean values ± SEM (d) of G-actin normalized to SM22 and expressed as a fraction of the WR18 value at 10 mmHg in each age group for CAs of 18–20 week WR (WR18, white bars) and GK (GK18, black bars) at 10, 60, and 120 mmHg (n = 6). * and # indicate significant difference (P < 0.05) from WR value at 10 mmHg and WR value at the identical pressure, respectively, in the same panel.
Lack of pressure-dependent activation of focal adhesion kinase in GK CAs
Mechanical activation of integrin adhesions evokes an increase in tyrosine autophosphorylation of focal adhesion kinase (FAK) that is considered to be an essential initial event in integrin signaling.17,36 Recently, Colinas et al.37 provided direct evidence of a pressure-dependent increase in FAK-Y397 autophosphorylation and block of MYPT1-T855 and LC20 phosphorylation, as well as actin polymerization and myogenic constriction in rat middle and posterior CAs following treatment with the FAK inhibitor FI-14.37 Based on the observed loss of pressure-evoked phosphorylation of LC20 and MYPT1-T855, actin polymerization and active myogenic tone in arteries from 8 to 10 week GK rats, we investigated whether there was defective pressure-evoked FAK autophosphorylation in the GK vessels. The effect of pressure elevation on autophosphorylation of FAK-Y397 in CAs of 8–10 week WR and GK rats is shown in Figure 6. Pressurization from 10 to 80 mmHg was accompanied by an ∼3-fold (2.8 ± 0.3) increase in the autophosphorylation of FAK at Y397, but no further change was detected at 120 mmHg (2.3 ± 0.28), consistent with previous findings.37 Contrary to CAs of WR, no pressure-dependent change in the phosphorylation of FAK-Y397 was detected in cerebral vessels from GK rats.
Figure 6.
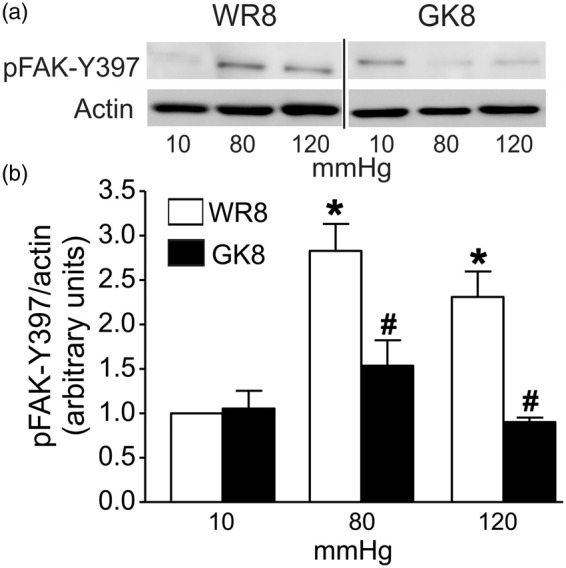
Pressure-evoked FAK-Y397 autophosphorylation in CAs from 8-10 week GK and WR. Panel (a): Representative western blots for phospho-FAK-Y397 and corresponding actin in each lane at 10, 80, and 120 mmHg for CAs of 8–10 week GK (GK8) and age-matched WR rats (WR8). Panel (b): Mean levels ± SEM of phospho-FAK-Y397 normalized to actin and expressed as a fraction of the WR value at 10 mmHg for CAs of WR8 (white bars), GK8 rats (black bars) at 10, 80, and 120 mmHg (n = 5). *Significantly different (P < 0.05) from WR value at 10 mmHg. #Significantly different (P < 0.05) from corresponding WR value at the same pressure.
MYPT1, RhoA, and ROK2 expression levels are not different in 8–10 week GK versus WR CAs
To determine if a change in the expression of RhoA, ROK2, and/or MYPT1 was responsible for the alteration in MYPT1 phosphorylation in CAs of 8–10 week GK rats, we quantified their transcript and/or protein levels in CAs of 8–10 week GK and WR. No difference in MYPT1 transcript levels was detected by real-time PCR (Figure 7(a)), and MYPT1 protein content was similar when normalized to two different housekeeping proteins, actin, and GAPDH (Figure 7(b) and (c). No difference in RhoA transcript levels was detected (Figure 7(d)), nor was a difference in expression of ROK2 (ubiquitously expressed in VSMC3) evident at the transcript or protein level (Figure 7(e) and (f)).
Figure 7.

MYPT1, RhoA, and ROK2 expression in CAs of GK and age-matched WR. Panel (a): Mean values ± SEM of MYPT1 transcript relative to ß-actin determined by real-time PCR using mRNA derived from CAs of 8–10 and 18–20 week GK (GK8, GK18; black bars) and WR (WR8, WR18; white bars). Relative transcript levels were determined by the 2−ΔΔCt method (n = 5). Panels (b) and (c): Representative western blots of MYPT1, actin, and GAPDH content in each lane (b) and mean values ± SEM (c) of MYPT1 protein normalized to actin (left) or GAPDH (right) and expressed as a fraction of the content in the age-matched WR value for CAs from 8 to 10 and 18 to 20 weeks GK (black bars) and WR (white bars) (n = 10). Panels (d) and (e): Mean values ± SEM of RhoA (d) and ROK2 (e) transcript expression relative to ß-actin determined by real-time PCR using mRNA derived from CAs of WR8 (white bars) and GK8 (black bars). Relative transcript levels were determined using the 2−ΔΔCt method (n = 5). Panel (f): Representative western blots of ROK2 and corresponding actin content and mean values ± SEM of ROK2 protein normalized to actin in CAs of GK8 (black bar) expressed as a fraction of the content in age-matched WR (white bar) (n = 4).
Discussion
Our findings provide novel mechanistic insights into the molecular basis of abnormal cerebrovascular myogenic autoregulation in the GK rat model of type 2 diabetes. Specifically, our data indicate that dysfunctional myogenic control of cerebral arterial diameter in the GK rat results from abnormal regulation of two mechanisms that underlie the phenomenon of Ca2+ sensitization in VSMCs. Ca2+ sensitization in the myogenic response of resistance arteries and arterioles has been attributed to: (i) ROK-mediated phosphorylation of MYPT1-T855 that inhibits MLCP resulting in increased LC20 phosphorylation and force generation, and (ii) ROK- and PKC-evoked actin dynamics that strengthens the cytoskeleton and increases the efficiency of force transmission to the cell membrane and extracellular matrix.13,17 Here, we show that (i) the presence of basal vasoconstriction and lack of myogenic dilation on pressure reduction at lower intraluminal pressures ( < 60 mmHg) in CAs of 8–10 week pre-diabetic GK rats resulted from increased H1152-sensitive, MYPT1-T855, and LC20 phosphorylation, as well as augmented actin polymerization; and (ii) loss of pressure-dependent myogenic constriction in 8–10 and 18–20 week GK CAs is due to the absence of pressure-dependent, ROK-mediated regulation of MYPT1-T855 and LC20 phosphorylation, as well as actin dynamics. Furthermore, we show that these defects are accompanied by defective pressure-dependent FAK-Y397 autophosphorylation, indicative of abnormal activation of integrin signaling in CAs of GK rats. Taken together, the data indicate a key role for abnormal regulation of two key processes, MLCP-dependent control of LC20 phosphorylation and cross-bridge cycling, as well as actin dynamics, in the dysfunctional myogenic response in the GK rat model of type 2 diabetes. These findings have important implications for the development of novel therapeutic approaches to correct defects in cerebral blood flow autoregulation in this disease.
We attribute the differences in myogenic behavior, phosphoprotein levels, and G-actin content of CAs from 8 to 10 and 18 to 20 week GK rats to an age-dependent progression from insulin resistance to overt diabetes marked by hyperinsulinemia and hyperglycemia. The diabetic condition in the non-obese GK rat model is spontaneous, polygenic in origin, and appropriate for determination of type 2 diabetes-related dysfunction in the absence of hyperlipidemia and/or obesity.38 No differences in serum insulin or glucose levels were detected in GK and WR prior to 10 weeks of age, but a progressive, age-dependent increase was apparent in GK rats after ∼12 weeks of age.23,31,32 A decreased magnitude of insulin-induced dilation was detected in vessels of GK compared to WR prior to 10 weeks of age indicative of resistance to the vascular actions of insulin in young GK rats before serum insulin or glucose levels were elevated at 10 to 12 weeks. Vascular remodeling was not evident in CAs from the GK rats used in this study as no significant change in passive diameter was detected. Previously, it was reported that passive diameter of 18–20 week GK CAs was either similar15 or reduced12 compared to age-matched WR. The reason(s) for these differences are not clear, but differences in the age-dependent progression/severity of the disease in GK rats from different colonies might be involved.
Previous studies identified abnormal myogenic behavior in resistance arteries of patients and animal models of type 2 diabetes, but the molecular mechanisms responsible for the abnormal contractility were not identified.10–12,14 We attribute the increased basal myogenic tone and vasoconstriction at low intraluminal pressures of CAs from 8 to 10 week GK rats to increased basal ROK2 activity and a resultant increase in Ca2+ sensitization. This view is substantiated by: (i) elevated levels of MYPT1-T855 and LC20 phosphorylation at 10 mmHg that were reversed, along with the increased basal tone, by ROK inhibition; and (ii) lower G-actin content indicating augmented actin polymerization. Increased basal Ca2+ sensitization is functionally significant as it prevents myogenic dilation on pressure reduction, i.e. no negative slope in the pressure–diameter curve between ∼80 and 20 mmHg. Myogenic dilation permits increased blood flow at lower perfusion pressures in situ and, when impaired, flow may not increase sufficiently upon pressure reduction resulting in inadequate cerebral perfusion and ischemia. As ROK2 mRNA and protein expression were similar in the GK and WR vessels, the elevated basal myogenic tone may be attributed to an increase in kinase activity rather than expression. This finding is consistent with previous reports of increased contractions of mesenteric arteries of db/db diabetic compared to control mice evoked by serotonin, angiotensin II, and phenylephrine due to enhanced ROK activity.39,40 Enhanced activation of RhoA/ROK signaling was cited as the cause of enhanced actin polymerization in cardiac myocytes from streptozotocin-diabetic rats,41 and in GK rats, diabetes-associated hypertension and enhanced contractile responses of mesenteric and carotid arteries to vasoconstrictor agonists was detected in the absence of altered RhoA or ROK2 expression.22,42,43 Enhanced myogenic tone was previously observed in CAs of 10 week GK rats12 and gracilis arteries of streptozotocin-treated rats,44 but at higher intraluminal pressures ( > 40 mmHg). The enhanced basal myogenic tone in the 8–10 week GK animals may preclude the vasodilatory response of CAs in periods of high metabolic activity and oxygen demand. Moreover, if the dysfunction is sufficiently severe, the reduced arterial diameter would be expected to restrict blood flow to the brain increasing the risk of ischemia.
We attribute the suppression of myogenic constriction in CAs of 18–20 week diabetic GK rats to a lack of pressure-dependent ROK-mediated MYTP1-T855 phosphorylation and actin dynamics. Maintenance and/or reduction in resistance arterial diameter in response to pressure elevation within the physiological range is due in part to pressure-dependent activation of RhoA and ROK.3,33 Similar to findings previously reported for Sprague-Dawley rats,24,26 pressure elevation from 10 to 60 or 120 mmHg enhanced MYPT1-T855 and LC20 phosphorylation and reduced G-actin content in WR vessels, indicative of pressure-dependent activation of these Ca2+ sensitization mechanisms. In contrast, there was complete lack of pressure-dependent increase in MYPT1-T855 and LC20 phosphorylation, or decline in G-actin content in vessels of 8–10 and 18–20 week GK rats. The greater basal ROK-mediated MYPT1-T855 phosphorylation and reduced G-actin content in 8–10 week GK vessels were apparently sufficient to offset the distending force of intraluminal pressure and permit the maintenance of CA diameter with only limited dilation upon pressure elevation to > 60 mmHg. However, the absence of increased basal, as well as pressure-evoked activation of mechanisms underlying Ca2+ sensitization in CAs of 18–20 week GK rats resulted in proportional dilation with increasing pressure. A similar loss of myogenic constriction was also reported for subcutaneous gluteal vessels of type 2 diabetes patients,16 and CAs and coronary arteries of 22 week GK rats.15 In the latter case, the impaired myogenic constriction was not due to a change in the regulation of cytosolic free [Ca2+] between 20 and 100 mmHg.15 Rather, dysfunction was speculated to result from the loss of Ca2+ sensitization due to defective ROK signaling.15 The present findings provide direct evidence that ROK-dependent mechanisms of Ca2+ sensitization are indeed defective in CAs in the GK model. A similar loss of myogenic constriction in stroke-prone spontaneously hypertensive rats was associated with inappropriate cerebral blood flow autoregulation and over-perfusion resulting in cerebral hemorrhage.45 The absence of ROK-dependent regulation of Ca2+ sensitization in GK rats is significant as chronic deficiency may be expected to disrupt cerebral arterial diameter autoregulation at high pressures resulting in unrestricted blood flow and damage to the blood–brain barrier, capillaries and/or neurovascular unit, and/or rupture owing to elevated intraluminal pressure in downstream vessels with implications for cognitive function. This view is consistent with MRI and autopsy findings indicating the presence of microinfarcts and increased white matter lesions in type 2 diabetes patients with clinical manifestations of dementia.7,8
Why basal ROK activity is enhanced and pressure-dependent ROK activity responsible for MYPT1 phosphorylation and actin dynamics is lost in GK rats remains to be determined. Several mechanisms have been postulated to account for an increase in ROK activity in cultured VSMCs from animal models of type 2 diabetes, including increased oxidative stress that stimulates ROK activity,21 loss of endothelium-derived NO that normally functions to suppress ROK activity,46 and/or defective insulin and insulin receptor signaling in VSMC.47. The interplay between insulin signaling and ROK activity in insulin-resistant VSMCs was previously studied using aortic VSMCs from 7 to 8 week GK rats47–49: insulin inhibited ROK activity, decreased MYPT1-T855 phosphorylation and actin polymerization, and induced vasorelaxation. We examined the myogenic response in endothelium-intact vessel segments from 8 to 10 week GK and WR, but no difference compared to denuded arteries was detected. This suggests that a difference in basal (unstimulated) NO release may not be the cause of the dysfunctional myogenic response in 8–10 week GK rats. On the other hand, the vasodilation evoked by 50 nmol/L insulin was significantly reduced in vessels of 8–10 week GK rats compared to age-matched WR. For this reason, future analysis should explore the relationship between insulin resistance and the increased basal phosphorylation of MYPT1-T855 and LC20, and actin polymerization detected in CAs of pre-diabetic 8–10 week GK rats. The defective pressure-evoked regulation of MLCP and actin dynamics was associated with the absence of a change in FAK autophosphorylation with pressurization in CAs of GK rats. This is similar to the suppression of phenylephrine-induced contraction of second order mesenteric arteries from ob/ob mice under hyperglycemic conditions owing to defective receptor-coupled RhoA activation and reduced ROK expression.50 The lack of pressure-dependent FAK autophosphorylation suggests a disruption of integrin signaling may contribute to the impairment of myogenic constriction. An interplay between integrin signaling and insulin action has been reported previously.51–53 Future studies should be directed at determining whether the loss of FAK-Y397 autophosphorylation is a direct result of the development of insulin resistance or the result of the increase in basal ROK activity.
In summary, this study provides novel biochemical evidence of a progressive dysfunction in the regulation of MLCP and actin dynamics by ROK in cerebral resistance arteries of the GK model of type 2 diabetes involving increased basal activity and a loss of pressure-dependent regulation of myogenic constriction. Novel strategies for therapeutic control of the vasculature must normalize both defects to reestablish physiological cerebral blood flow autoregulation in type 2 diabetes.
Funding
This study was supported by operating grants from the Canadian Institutes of Health Research to WCC (MOP-13505 and MOP-97988) and salary awards from the Alberta Innovates-Health Solutions (AI-HS) and Killam Foundation to KSA.
Acknowledgements
KSA is an Assistant Lecturer in the Department of Pharmacology & Toxicology, Faculty of Pharmacy, University of Alexandria, Egypt. WCC is the Andrew Family Professor in Cardiovascular Research and MPW is an AI-HS Scientist and recipient of a CRC (Tier 1) in Vascular Smooth Muscle Research.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
KSA, MPW, and WCC were responsible for the conception and design of all experiments and wrote the manuscript, KSA and EJW performed pressure myography experiments, KSA performed western blotting experiments and analyses, OC and HZ performed insulin experiments, CMC and EJW completed the RT-PCR and real-time PCR experiments and MPW contributed to the design, interpretation of the results and manuscript preparation. All authors have approved the submitted version of this manuscript.
References
- 1.Cipolla MJ. The cerebral circulation [2009] – PubMed – NCBI, www.ncbi.nlm.nih.gov/pubmed/21452434 (accessed 30 November 2015).
- 2.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 1999; 79: 387–423. [DOI] [PubMed] [Google Scholar]
- 3.Cole WC, Welsh DG. Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch Biochem Biophys 2011; 510: 160–173. [DOI] [PubMed] [Google Scholar]
- 4.Matsushita K, Kuriyama Y, Nagatsuka K, et al. Periventricular white matter lucency and cerebral blood flow autoregulation in hypertensive patients. Hypertension 1994; 23: 565–568. [DOI] [PubMed] [Google Scholar]
- 5.Iadecola C. The pathobiology of vascular dementia. Neuron 2013; 80: 844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schofield I. Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation 2002; 106: 3037–3043. [DOI] [PubMed] [Google Scholar]
- 7.Ryan JP, Fine DF, Rosano C. Type 2 diabetes and cognitive impairment: contributions from neuroimaging. J Geriatr Psychiatry Neurol 2014; 27: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luchsinger JA. Type 2 diabetes and cognitive impairment: linking mechanisms. J Alzheimers Dis 2012; 30: 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim Y-S, Davis SCAT, Truijen J, et al. Intensive blood pressure control affects cerebral blood flow in type 2 diabetes mellitus patients. Hypertension 2011; 57: 738–745. [DOI] [PubMed] [Google Scholar]
- 10.Su J, Lucchesi PA, Gonzalez-Villalobos RA, et al. Role of advanced glycation end products with oxidative stress in resistance artery dysfunction in type 2 diabetic mice. Arterioscler Thromb Vasc Biol 2008; 28: 1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarajapu YPR, Guberski DL, Grant MB, et al. Myogenic tone and reactivity of cerebral arteries in type II diabetic BBZDR/Wor rat. Eur J Pharmacol 2008; 579: 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly-Cobbs A, Elgebaly MM, Li W, et al. Pressure-independent cerebrovascular remodelling and changes in myogenic reactivity in diabetic Goto-Kakizaki rat in response to glycaemic control. Acta Physiol (Oxf) 2011; 203: 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abd-Elrahman KS, Walsh MP, Cole WC. Abnormal Rho-associated kinase activity contributes to the dysfunctional myogenic response of cerebral arteries in type 2 diabetes. Can J Physiol Pharmacol 2015; 93: 177–184. [DOI] [PubMed] [Google Scholar]
- 14.Sachidanandam K, Hutchinson JR, Elgebaly MM, et al. Glycemic control prevents microvascular remodeling and increased tone in type 2 diabetes: link to endothelin-1. Am J Physiol Regul Integr Comp Physiol 2009; 296: 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kold-Petersen H, Brøndum E, Nilsson H, et al. Impaired myogenic tone in isolated cerebral and coronary resistance arteries from the Goto-Kakizaki rat model of type 2 diabetes. J Vasc Res 2012; 49: 267–278. [DOI] [PubMed] [Google Scholar]
- 16.Schofield I, Malik R, Izzard A, et al. Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation 2002; 106: 3037–3043. [DOI] [PubMed] [Google Scholar]
- 17.Walsh MP, Cole WC. The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab 2013; 33: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J 2002; 16: 72–76. [DOI] [PubMed] [Google Scholar]
- 19.Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 2008; 295: 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamin R, Morgan KG. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J Physiol 2012; 590: 4145–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Didion SP, Lynch CM, Baumbach GL, et al. Impaired endothelium-dependent responses and enhanced influence of Rho-kinase in cerebral arterioles in type II diabetes. Stroke 2005; 36: 342–347. [DOI] [PubMed] [Google Scholar]
- 22.Rao MY, Soliman H, Bankar G, et al. Contribution of Rho kinase to blood pressure elevation and vasoconstrictor responsiveness in type 2 diabetic Goto-Kakizaki rats. J Hypertens 2013; 31: 1160–1169. [DOI] [PubMed] [Google Scholar]
- 23.Mishra RC, Wulff H, Cole WC, et al. A pharmacologic activator of endothelial KCa channels enhances coronary flow in the hearts of type 2 diabetic rats. J Mol Cell Cardiol 2014; 72: 364–373. [DOI] [PubMed] [Google Scholar]
- 24.Johnson RP, El-Yazbi AF, Takeya K, et al. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol 2009; 587: 2537–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Yazbi AF, Johnson RP, Walsh EJ, et al. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J Physiol 2010; 588: 1747–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno-Domínguez A, Colinas O, El-Yazbi A, et al. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol 2013; 591: 1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorneloe KS, Chen TT, Kerr PM, et al. Molecular composition of 4-aminopyridine-sensitive voltage-gated K(+) channels of vascular smooth muscle. Circ Res 2001; 89: 1030–1037. [DOI] [PubMed] [Google Scholar]
- 28.Takeya K, Loutzenhiser K, Shiraishi M, et al. A highly sensitive technique to measure myosin regulatory light chain phosphorylation: the first quantification in renal arterioles. Am J Physiol Renal Physiol 2008; 294: 1487–1492. [DOI] [PubMed] [Google Scholar]
- 29.Walsh MP, Thornbury K, Cole WC, et al. Rho-associated kinase plays a role in rabbit urethral smooth muscle contraction, but not via enhanced myosin light chain phosphorylation. Am J Physiol Renal Physiol 2011; 300: 73–85. [DOI] [PubMed] [Google Scholar]
- 30.Moreno-Dominguez A, El-Yazbi AF, Zhu H-L, et al. Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J Biol Chem 2014; 289: 20939–20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Rourke CM, Davis JA, Saltiel AR, et al. Metabolic effects of troglitazone in the Goto-Kakizaki rat, a non-obese and normolipidemic rodent model of non-insulin-dependent diabetes mellitus. Metabolism 1997; 46: 192–198. [DOI] [PubMed] [Google Scholar]
- 32.Gao W, Bihorel S, DuBois DC, et al. Mechanism-based disease progression modeling of type 2 diabetes in Goto-Kakizaki rats. J Pharmacokinet Pharmacodyn 2011; 38: 143–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osol G, Brekke JF, McElroy-Yaggy K, et al. Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am J Physiol Heart Circ Physiol 2002; 283: 2260–2267. [DOI] [PubMed] [Google Scholar]
- 34.Feng J, Ito M, Ichikawa K, et al. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem 1999; 274: 37385–37390. [DOI] [PubMed] [Google Scholar]
- 35.Ihara E, MacDonald JA. The regulation of smooth muscle contractility by zipper-interacting protein kinase. Can J Physiol Pharmacol 2007; 85: 79–87. [DOI] [PubMed] [Google Scholar]
- 36.Murphy TV, Spurrell BE, Hill MA. Cellular signalling in arteriolar myogenic constriction: involvement of tyrosine phosphorylation pathways. Clin Exp Pharmacol Physiol 2002; 29: 612–619. [DOI] [PubMed] [Google Scholar]
- 37.Colinas O, Moreno-Domínguez A, Zhu H, et al. 2015. α5-Integrin-mediated cellular signaling contributes to the myogenic response of cerebral resistance arteries. Biochem Pharmacol 2015; 97: 281–291. [DOI] [PubMed] [Google Scholar]
- 38.Nie J, Xue B, Sukumaran S, et al. Differential muscle gene expression as a function of disease progression in Goto-Kakizaki diabetic rats. Mol Cell Endocrinol 2011; 338: 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie Z, Gong MC, Su W, et al. Role of calcium-independent phospholipase A2beta in high glucose-induced activation of RhoA, Rho kinase, and CPI-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals. J Biol Chem 2010; 285: 8628–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Z, Su W, Allen S, et al. COX-2 up-regulation and vascular smooth muscle contractile hyperreactivity in spontaneous diabetic db/db mice. Cardiovasc Res 2005; 67: 723–735. [DOI] [PubMed] [Google Scholar]
- 41.Lin G, Craig GP, Zhang L, et al. Acute inhibition of Rho-kinase improves cardiac contractile function in streptozotocin-diabetic rats. Cardiovasc Res 2007; 75: 51–58. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto T, Watanabe S, Taguchi K, et al. Mechanisms underlying increased serotonin-induced contraction in carotid arteries from chronic type 2 diabetic Goto-Kakizaki rats. Pharmacol Res 2014; 87: 123–132. [DOI] [PubMed] [Google Scholar]
- 43.Matsumoto T, Ishida K, Taguchi K, et al. Short-term angiotensin-1 receptor antagonism in type 2 diabetic Goto-Kakizaki rats normalizes endothelin-1-induced mesenteric artery contraction. Peptides 2010; 31: 609–617. [DOI] [PubMed] [Google Scholar]
- 44.Ungvari Z, Pacher P, Kecskemeti V, et al. Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase C. Cardiovasc Res 1999; 43: 1018–1028. [DOI] [PubMed] [Google Scholar]
- 45.Smeda JS, Daneshtalab N. The effects of poststroke captopril and losartan treatment on cerebral blood flow autoregulation in SHRsp with hemorrhagic stroke. J Cereb Blood Flow Metab 2011; 31: 476–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sauzeau V, Le Jeune H, Cario-Toumaniantz C, et al. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem 2000; 275: 21722–21729. [DOI] [PubMed] [Google Scholar]
- 47.Begum N. Insulin signaling in the vasculature. Front Biosci 2003; 8: 796–804. [DOI] [PubMed] [Google Scholar]
- 48.Lee JH, Palaia T, Ragolia L. Impaired insulin-mediated vasorelaxation in diabetic Goto-Kakizaki rats is caused by impaired Akt phosphorylation. Am J Physiol Cell Physiol 2009; 296: 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sandu OA, Ragolia L, Begum N. Diabetes in the Goto-Kakizaki rat is accompanied by impaired insulin-mediated myosin-bound phosphatase activation and vascular smooth muscle cell relaxation. Diabetes 2000; 49: 2178–2189. [DOI] [PubMed] [Google Scholar]
- 50.Nobe K, Hashimoto T, Honda K. Two distinct dysfunctions in diabetic mouse mesenteric artery contraction are caused by changes in the Rho A-Rho kinase signaling pathway. Eur J Pharmacol 2012; 683: 217–225. [DOI] [PubMed] [Google Scholar]
- 51.Williams AS, Kang L, Wasserman DH. The extracellular matrix and insulin resistance. Trends Endocrinol Metab 2015; 26: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wertheimer E, Taylor SI, Tennenbaum T. Insulin receptor regulation of cell surface integrins: a possible mechanism contributing to the development of diabetic complications. Proc Assoc Am Physicians 110: 333–339. [PubMed] [Google Scholar]
- 53.Huang D, Khoe M, Ilic D, et al. Reduced expression of focal adhesion kinase disrupts insulin action in skeletal muscle cells. Endocrinology 2006; 147: 3333–3343. [DOI] [PubMed] [Google Scholar]