

Abstract
A hallmark of stroke is water accumulation (edema) resulting from dysregulation of osmotic homeostasis. Brain edema contributes to tissue demise and may lead to increased intracranial pressure and lethal herniation. Currently, there are only limited treatments to prevent edema formation following stroke. Aquaporin 4 (AQP4), a brain water channel, has become a focus of interest for therapeutic approaches targeting edema. At present, there are no pharmacological tools to block AQP4. The role of AQP4 in edema after brain injury remains unclear with conflicting results from studies using AQP4−/− mice and of AQP4 expression following stroke. Here, we studied AQP4 and its role in edema formation by testing AQP4−/− mice in a model of middle cerebral artery occlusion using novel quantitative MRI water content measurements, histology and behavioral changes as outcome measures. Absence of AQP4 was associated with decreased mortality and increased motor recovery 3 to 14 days after stroke. Behavioral improvement was associated with decreased lesion volume, neuronal cell death and neuroinflammation in AQP4−/− compared to wild type mice. Our data suggest that the lack of AQP4 confers an overall beneficial role at long term with improved neuronal survival and reduced neuroinflammation, but without a direct effect on edema formation.
Keywords: Aquaporin, astrocyte, blood-brain barrier, stroke, transient middle cerebral artery occlusion
Introduction
The role of aquaporins in cerebral edema
Edema is a hallmark of stroke as well as other brain disorders (e.g. traumatic brain injury, tumors, inflammation). Cerebral edema appears as a result of water accumulation from dysregulation of brain osmotic homeostatic mechanisms and leads to brain swelling with decreased brain perfusion and aggravation of secondary injuries. Although edema has been observed for many years, there are only limited treatment options including administration of mannitol or hypertonic saline to prevent or attenuate edema formation or expansion after stroke, but their benefit is not entirely clarified and the molecular and cellular events in edema formation/resolution are still poorly understood.
The discovery of the brain aquaporins (AQPs) raised new hope for the development of new therapies to treat edema. AQP1, 4 and 9 show changes in their levels of expression in stroke models.1 AQP4 is expressed on astrocytic endfeet surrounding blood vessels in cortex and striatum, and its expression increases after ischemia and correlates with the time course of edema formation and resolution.2,3 AQP4 knockout (AQP4−/−) mice are powerful tools to study the role of AQP4 after stroke. AQP4−/− mice do not exhibit any major structural and physiological changes with the exception of an extracellular space expansion by about 20% in AQP4−/− mice compared to wild-type (WT).4–7 Despite the absence of obvious structural or physiological changes, it cannot be excluded that some astrocyte properties could be altered in AQP4−/− mice, such as wider extracellular space resulting in a reduced potassium clearance4,8 or an impaired glutamate reuptake due to a decreased expression of the glutamate transporter GLT-1.9 Such changes could influence the outcome and need to be considered when interpreting the opposite outcomes seen with in AQP4−/− experimental rodent brain injury models, including stroke.10,11 WT fared better or worse compared to AQP4−/− (for review see Badaut et al., BBA 201428). Previous results from Verkman and colleagues lead to the hypothesis of a dual role for AQP4 in the development of edema: deleterious during edema formation and beneficial during the edema resolution phase.13 However, the absence of available drugs to acutely and specifically block AQP4 has not allowed direct testing of the dual role hypothesis after brain injury.
AQP4 and edema build-up versus edema resolution
We previously showed in a model of transient middle cerebral artery occlusion that AQP4 expression is rapidly up-regulated in perivascular astrocyte endfeet, peaking at 1 h after stroke onset.2,3 This increase was observed at the future lesion site and in the peri-infarct region in a mouse stroke model, and the degree of its increase temporally correlated with the degree of brain swelling.2,3 However, increased AQP4 expression is not observed in more severe stroke models,14 prompting the suggestion that under great tissue duress, the brain is not able to synthesize sufficient new AQP4 proteins during the early phase of reperfusion. The variability of AQP4 expression, which seems to depend on the degree of severity and the model, highlights the complexity of the role of AQP4 after ischemia. This confusion is definitely supported by the contradictory results obtained on edema and lesion outcomes with AQP4−/− after stroke.10,11 Overall, AQP4 has been proposed to have dual role: it is deleterious in the development of edema10,11 and beneficial for water clearance during edema resolution based on the finding that infusion of artificial cerebral spinal fluid (ACSF) in brain parenchyma induced a significant increase in intracranial pressure in AQP4−/− mice compared to the WT.15,16 Despite the lack of a clear answer regarding the role of AQP4 in edema resolution, several results support this hypothesis: increased AQP4 was observed to be associated with edema resolution measured over time using MRI,1,17 and increased AQP4 is observed near the lesion site in perivascular astrocyte endfeet, astrocyte processes and the glia limitans.2,18 These changes may indicate that excess AQP4 could facilitate edematous fluid elimination through the subarachnoid space.19
To address the role of AQP4 in the outcome and edema formation and resolution after ischemia/reperfusion, we subjected AQP4−/− and WT mice to 30 min transient MCAo followed by serial MRI (T2WI and DWI) determinations as well as behavioral testing and long-term outcome assessment using histology/immunohistochemistry 14 days post-stroke.
Material and methods
Animals
The manuscript was written in accordance with the ARRIVE guidelines. Experiments and animal care were conducted according to the principles and procedures of the Guidelines for Care and Use of Experimental Animals and were approved by the Loma Linda University Institutional Review Board. Adult (two months old) wild type ICR-CD1 mice and AQP4−/− mice were housed in a temperature-controlled (22 ℃ to 25 ℃) animal facility on a 12-h light/dark cycle seven days before surgery. Animal groups were randomized. Experimenters (LH, AF) were blinded to the groups during surgery and other tests (behavior, neuroimaging and histology).
Middle cerebral artery occlusion surgery
Transient focal ischemia was induced in male AQP4−/− (30.6 ± 1.3 g, n = 8) and matched ICR-CD1 wild type (WT) mice (weight 34.8 ± 0.7 g, n = 10) as previously described.2 The mice were anesthetized with 2.5% isoflurane (induction) and maintained at 1% isoflurane in 30% oxygen and 70% nitrous oxide with a facemask. In all animals, regional cerebral blood flow (rCBF) was measured by laser-Doppler flowmetry with a flexible probe fixed on the skull (1 mm posteriorly and 6 mm laterally from the bregma) starting before onset of ischemia until 10 min after reperfusion. Focal cerebral ischemia was induced by MCAO by introducing a silicone-coated 8–0 filament from the common carotid artery into the internal carotid artery and advancing it until blood flow was reduced (rCBF 14% ± 1% of baseline). The filament was removed after 30 min to allow reperfusion (rCBF 98% ± 8%, no difference between the groups). Rectal temperature was controlled and maintained at 36.5 ℃ with a temperature control unit and a heating pad during the anesthesia period (37.1 ℃ ± 0.1 ℃). The WT (n = 10) and AQP4−/− (n = 8) were randomized during the experiments and experimenters (LH/AF) were blinded to the group until the final analysis. In our study, we applied the following exclusion criteria: the loss of laser flow signals during surgery and the absence of correct reperfusion (more than 50% of the baseline CBF). Therefore, one WT mouse and three AQP4−/− mice were withdrawn (Table 1). MRI and behavioral evaluation was performed on 9 WT and 5 AQP4−/−. Automated Hierarchical Region Splitting (HRS) analysis for the lesions on MRI scans failed in one WT mouse and was excluded resulting in n = 8 WT and 5 AQP4−/−.
Table 1.
Number of mice for each experimental group at the beginning of the study, after surgery and at each time point.
| After surgery | Outcome | 1 day | 3 days | 6 days | 7 days | 14 days | |
|---|---|---|---|---|---|---|---|
| WT | |||||||
| n = 10 | n = 9a | MRI | 8 | 8 | n.a. | 4 | 4 |
| Behavior | 9 | 9 | 4 | n.a. | 4 | ||
| Histology | n.a. | n.a. | n.a. | n.a. | 4 | ||
| AQP4−/− | |||||||
| n = 8 | n = 5b | MRI | 5 | 5 | n.a. | 4 | 4 |
| Behavior | 5 | 5 | 4 | n.a. | 4 | ||
| Histology | n.a. | n.a. | n.a. | n.a. | 4 | ||
n.a., not applicable.
Loss of CBF signal in one animal.
Absence of reperfusion in three animals.
The animals were prepared for histology at 14 days (n = 8) after MCAO. Mice were deeply anesthetized with an overdose of pentobarbital and perfused transcardially with 50 ml of paraformaldehyde 4% in phosphate-buffered saline (PBS) 0.1 M, pH 7.4, at 4 ℃. Fixed brains were removed from the skull and immersed overnight in fixative solution. Brains were then stored in PBS containing 30% sucrose for 48 h at 4 ℃ for cryoprotection. Serial coronal sections (50 μm) were cut on a cryostat at −22 ℃ (Leica, Richmond, IL) for immunohistochemical analysis.3,17
Neuroimaging and lesion volume analysis
Mice underwent MRI imaging at 1, 3, 7 and 14 days post-MCAO induction as previously reported.20 Briefly, mice were lightly anesthetized using isoflurane (1.0%) and then imaged on a Bruker Avance 11.7 T MRI (Bruker Biospin, Billerica, MA, USA) and T2 weighted-imaging (T2WI; TR/TE=2357.9 ms/10.2 ms, 20 × 1 mm slices) and diffusion weighted-imaging (DWI; TR/TE:3000 ms/25 ms, 20 × 1 mm slices, b-values = 0.72, 1,855.64 s/mm2) data were acquired. All MRI data used a matrix = 128 × 128, field of view of 2–3 cm and total imaging time was 45 min for both T2 and DWI. T2 and DWI data were post-processed for quantitative T2 maps and apparent diffusion coefficients (ADC) using in-house software written in Matlab (Mathworks, Natick, MA, USA) as described previously.21 Lesion, total brain, and hemispheric volumes were calculated using our recently developed automated Hierarchical Region Splitting (HRS) software.22 Our previous studies established that automatically derived core and penumbral lesion volumes co-localized with those determined by immunohistochemistry or diffusion-perfusion mismatch. All quantitative data were summarized in Microsoft Excel. Additional corrections for lesion volume due to hemispheric swelling and evaluation of hemispheric space-occupying effect (%HSE) for edema were determined using the protocol developed by Gerriets et al.23
Behavioral testing
Foot-fault and rotarod testing was performed at 1, 3, 6, 8, 12, and 14 days, where the foot-fault test was used to evaluate sensorimotor and proprioception and the rotarod test evaluated sensorimotor coordination as previously reported in our earlier studies.21 All tests at each time-point were carried out on WT and AQP4−/− mice within a 3-h morning time-block (8–11 am) and WT and AQP4−/− were interleaved during the testing sequence.
Foot-fault testing was carried out on an elevated platform (50 cm × 155 cm, ClosetMaid, Ocala, FL, USA) with parallel wire bars 1.5 cm apart and raised 100 cm above the floor. Mice were placed in the middle of the platform and allowed to freely move for 60 s in two separate trials, 30 min apart. When a mouse's paw (fore- or hindlimb) slipped completely through the wire mesh, it was considered as an individual fault. The average foot-fault score was calculated from the total number of faults from two 60 s trials.
Active time was calculated on foot-fault tests, counting the time spent exploring on the 60 s test on elevated platform described above.
Rotarod evaluation was performed on all the animals at 1, 3, 6, 8, 12, and 14 days (SD Instruments, San Diego, CA, USA). A rotating 7 cm wide spindle with a continuous speed (20 r/min) was used to evaluate performance during two trials (15 min apart). Latency to fall was recorded as a measure of motor coordination and balance. The maximum time spent on the test was 60 s, if the mouse did not fall. The two fall latencies were summed and expressed in total time (s) for two trials.
Tissue processing and immunohistochemistry
For immunoglobulin (IgG) extravasation immunohistochemistry, sections were washed with PBS, blocked with 1% BSA in PBS, and then incubated for 2h at room temperature with IRDye 800 conjugated affinity purified goat-anti-mouse IgG (1:500, Rockland, Gilbertsville, PA, USA) in PBS containing 0.1% Triton X-100 and 1% bovine serum albumin. After washing, sections were scanned on an infra-red (IR) scanner (Odyssey) to quantify fluorescence and the area of IgG extravasation for the different ROIs as previously described.3,17
Immunostaining was done for AQP4 with AQP4-rabbit polyclonal antibodies (1:300, Alpha Diagnostic, Owings Mill, MD, USA), for glial fibrillary acidic protein (GFAP, astrocyte marker) with GFAP-chicken polyclonal antibodies (1:500, Millipore, Billerica, MA, USA), Neuronal Nuclei (NeuN) with NeuN-mouse monoclonal antibodies (1:200, Abcam, Cambridge, MA, USA), for ionized calcium-binding adapter molecule (marker of microglia cells) with Iba-1-rabbit polyclonal antibodies (1:300, Wako, Richmond, VA, USA). Sections were washed with PBS, blocked with 1% BSA in PBS, incubated with the respective primary antibodies overnight, and then incubated with affinity purified secondaries conjugated to dyes of the desired wavelength to either be scanned on an InfraRed (IR) scanner or to be observed under a confocal microscope (Olympus). In detail, slides were incubated with secondary antibodies conjugated to dyes in the IR wavelength of either 680 or 800 nm. These slides were then scanned on an infrared scanner (Odyssey, Lincoln, NE, uSA), where images were saved with a resolution of 21 μm per pixel. Slides for confocal microscopy were incubated with secondary antibodies: goat anti-mouse secondary antibody coupled with Alexa-Fluor-488 or with Alexa-Fluor-594, goat anti-chicken coupled with Alexa-Fluor-568 and goat anti-rabbit secondary antibody coupled with Alexa-Fluor-594 (all secondaries from Invitrogen, Grand Island, NY, USA). After washes in PBS, sections for classical immunofluorescence were mounted on glass slides and coverslipped with vectashield antifading medium containing DAPI (Vector, Vector Laboratories, Burlingame, CA, USA). All confocal image acquisition parameters for the same proteins were kept constant for all of the animals for analysis and visualization purposes. All analyses were carried out in a non-biased, blinded manner. Negative control staining where either the primary or secondary antibody was omitted showed no detectable labeling (not shown).
AQP4, GFAP, and Iba-1 were quantified in a similar manner to IgG extravasation staining as published previously.24 Three identical circular regions of interests (ROIs) were drawn in the perilesional cortex (Figure 3(a)) at three different bregma levels (−1.40 mm, −2.56 mm, and −3.80 mm): the bregma level where the lesion area was largest, one slice anterior and one posterior, for a total of nine regions of interest per animal. The infrared scanner software measures the intensity of the fluorescence expressed in fluorescent integrated intensity with arbitrary units (A.U). The average fluorescence of these ROIs was calculated.
Figure 3.
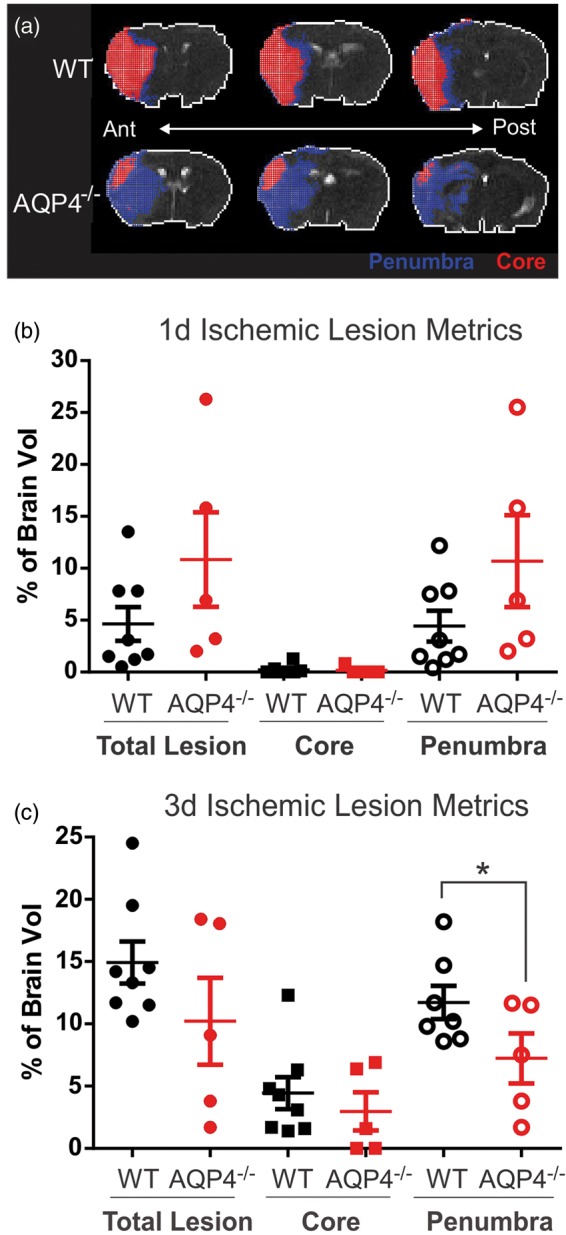
Evaluation of the lesion composition at three days post-ischemia. (a) Hierarchical region splitting (HRS) was performed to determine lesion composition, specifically core (red) and penumbra (blue) from T2-weighted MRI at three days post-ischemia. (b) There is a large heterogeneity between in the AQP4−/− mice, predominately in the total lesion (core+penumbra). There was a trend towards reduced total lesion (p = 0.078) and penumbra (p = 0.069) volumes in AQP4−/− mice. (c) At three days post-stroke, the WT mice had elevated total lesion volumes, predominately a reflection of dramatic increases in the penumbra volume. AQP4−/− mice had significantly smaller penumbra volumes at three days post-injury (p < 0.040). (n = 8 for WT and n = 5 for AQP4−/−).
NeuN staining was quantified using the Mercator software (Explora-Nova, La Rochelle, France). NeuN positive cells were counted on images acquired using constant parameters: i.e. the same settings of the contrast, gain, and brightness were used. The tissue section with the largest lesion was selected and the NeuN positive cells were counted in an automated fashion via the Mercator software and the density was determined.21
Statistics
MRI and behavioral data were analyzed by two-way repeated measures analysis of variance with a post hoc Bonferroni test. One-way ANOVA was used for immunohistochemistry analysis. All data are expressed as the mean ± SEM.
Results
Part 1: AQP4 deficient (AQP4−/−) mice develop a less severe neurological deficit than WT mice after transient MCAO
Mortality was decreased and survival increased in AQP4−/− mice consistent with decreased vulnerability to transient focal cerebral ischemia with loss of AQP4 (Figure 1(a)). Motor performance was tested using the rotarod test (Figure 1(b)) with both groups showing poor performance on day 1. However, there was a striking recovery in AQP4−/− mice that performed significantly better up to day 14 post-injury (Figure 1(b)). Activity was recorded in 1-min bins during the foot fault test and we observed an early decrease in the duration of the active periods on days 1 and 3 in WT mice and but not in AQP4−/− (Figure 1(c)). The length of the active periods of WT mice increased thereafter and no significant differences were observed from day 6 onwards. Sensorimotor skills were evaluated by counting foot-faults of paretic fore and hind limbs on an elevated platform with wire bars. Performance was impaired on days 1 and 3 wherein we observed a 2- to 4-fold increase in the number of foot-faults in WT compared with AQP4−/− mice. Again, no additional significant differences were observed between groups from day 6 onwards. Taken together, these results indicate that the lack of AQP4 improves survival and attenuates the neurological deficits after MCAO in mice.
Figure 1.

(a). Survival curve: significantly higher mortality in WT compared to AQP4−/− mice. The survival of the animals is illustrated in a Kaplan–Meier survival curve (a). There was mortality in both groups starting from day 3. The mortality was significantly higher in WT than in AQP4−/− mice. (b) Significant increase of the time spent on the rotarod treadmill for the AQ4−/− mice compared to the WT up to 14d after stroke onset. Mice were tested on a rotarod treadmill with constant acceleration and the time spent on the treadmill was recorded (b). The performance on the treadmill was poor in both groups on the day after surgery. Starting from day 3, there was a rapid improvement of the performance of AQP4−/− mice (n = 9 for WT and n = 5 for AQP4−/−), which remained significantly better than WT until day 14 (n = 4 for WT and n=4 for AQP4−/−). (c) Evaluation of the active time shows a significant difference between AQP4−/− mice compared to WT during the edema formation. Active time was assessed using a video recording system. On days 1 and 3, the length of an active time period was greater AQP4−/− than in WT mice (n = 9 for WT and n = 5 for AQP4−/−). Starting from day 6, there was no longer a significant difference between groups (n = 4 for WT and n = 4 for AQP4−/−). (d) Significant decrease of number of foot fault in AQP4−/− compared to WT. Foot faults were tested using an elevated platform with wire bars. On days 1 and 3, the recorded number of foot faults was significantly greater in WT than AQP4−/− mice (D) (n = 9 for WT and n = 5 for AQP4−/−). Starting from day 6, there was no suppress difference between groups (n = 4 for WT and n = 4 for AQP4−/−).
Part 2: AQP4−/− mice develop a smaller lesion and larger edema formation than WT mice after transient MCAO
Lesion volumes and hemispheric swelling
WT and AQP4−/− mice underwent serial MR imaging from 1 to 14 days post-MCAo induction (Figure 2(a)) with lesion volumes derived from T2 values for each animal that had a complete MR data set at each time point (Figure 2(b)). Lesion volume in WT mice was 16.05 ± 0.74%, 18.16 ± 0.74, 10.49 ± 0.26 and 3.54 ± 0.09% (% of total brain volume) at 1, 3, 7, and 14 days, respectively. At all-time points AQP4−/− mice had smaller lesion volumes 8.67 ± 0.90%, 10.47 ± 2.32, 5.48 ± 1.67 and 1.16 ± 0.50%, 1, 3, 7, and 14 days, respectively (Figure 2(a) and (b)). Our results demonstrate an ∼50% smaller lesion volume in AQP4−/− mice compared to WT at all-time points except at 14 days when lesion volumes were only 32% smaller than WT. Similar trends were observed in lesion volumes when all animals were analyzed, including those that did not have serial MR imaging (Supplementary Figure 1 using the hemispheric corrections of Gerriets and collaborators,23 in WT at one day 11.47 ± 3.87%, at three days 27.08 ± 5.81 and in AQP4−/− at one day 17.67 ± 8.06%, at three days 11.66 ± 5.87). Diffusion weighted imaging (DWI) and its quantitative apparent diffusion coefficient maps (ADC) exhibited similar evolution in lesion volumes. Thus, lack of AQP4 results in an overall decrease in lesion volume. Similar trends were observed in lesion volumes after correction of hemispheric space-occupying effects (%HSE) due to edema formation and calculated from MRI using the approach of Gerriets and collaborators.23 After correction for %HSE, AQP4−/− mice had significantly smaller lesion volumes (p < 0.04) (Supplement Figure 1A), and this difference was still present when all the MCAO mice in the study were included in the analysis (p < 0.05), specifically the 3-day time point ((p < 0.02), Supplement Figure 1B).
Figure 2.

Temporal evolution of the ischemic lesion in AQP4−/− mice is reduced. (a) Representative MR images from wildtype and AQP4−/− mice at 1–14 days post-ischemia. Hyperintense regions on the T2-weighted images highlight ischemic brain regions. (b) Quantitative analysis of manually derived lesion volumes from T2 images reveals that AQP4−/− mice had significantly reduced lesion volumes compared to wild type mice at every time point (n = 8 for WT and n = 5 for AQP4−/−). (c) Hemispheric swelling was also assessed by left/right (L/R) hemispheric asymmetry. Left panel illustrates sample L/R measures. Graphical analysis of the temporal evolution of brain swelling illustrates that there was increased swelling at three days (n = 8 for WT and n = 5 for AQP4−/−) and seven days post-stroke in the KO mice compared to WT, but this did not reach statistical significance (n = 4 for WT and n = 4 for AQP4−/−).
As mentioned earlier, MCAO is well known to significantly induce edema within brain tissues particularly at early time points. When hemispheric swelling was estimated using hemispheric asymmetry measures, we observed striking differences in right/left asymmetries (Figure 2(c)). Surprisingly, while we found decreased lesion volumes in AQP4−/− mice, there was increased hemispheric swelling, particularly at 3 and 7 days. This observation was confirmed by an increase of the %HSE (edema) in AQP4−/− compared to the WT in particularly at 3–7 days (Supplemental Figure 1(C) and (D)). There was a significant difference between groups over time (p<0.02, Two way ANOVA) but no significant difference was observed in post hoc tests for AQP4−/− versus WT. Moreover, at 3 and 7 days, these AQP4−/− mice had larger hemispheric swelling that exceeded those of the control WT mice (Figure 2(c)). This might suggest that lack of AQP4 leads to a reduced lesion volume, but edematous fluids are not adequately transported out from the injured tissues during the clearance of the water from ischemic hemisphere. This induces a delay in water clearance. At 14d, these AQP4−/− mice had reductions in hemispheric swelling that exceeded those of the control WT mice.
Lesion composition was then determined using a computational analysis method, Hierarchical Region Splitting (HRS), that has been previously validated in neonatal ischemic brain injury22 focusing on tissue volumes that comprised ischemic core and penumbra tissues (Figure 3(a)). Based on T2 values, we determined the relative composition of core/penumbra and that of total lesion volumes (Figure 3(a)). At one day post-MCAO, we found no significant differences in total, core or penumbral tissue volumes (Figure 3(c)), but penumbral lesion volumes tended to be larger in AQP4−/− than WT. Interestingly, the variance of penumbral volumes in the AQP4−/− was 2.5 times larger than WT (4.03% vs 1.61%, respectively). At three days post-MCAO, the lesion volumes and in particular the penumbral volumes increased significantly p < 0.04) in WT mice (Figure 3(b)). Interestingly, there was no significant increase in core volumes in either group at three days post-MCAO. One potential conclusion is a change in temporal profile with lesion and penumbral volumes progressing faster in the AQP4−/− mice than WT.
We then further investigated if there was a particular anatomical location that was perhaps more vulnerable to stroke damage. Analysis of lesion volumes in an anterior to posterior fashion demonstrated that the ischemic lesion was globally reduced throughout the entire brain of AQP4−/− relative to WT mice (Figure 4(a) and (b)). Similar to whole brain lesion volumes, global reductions in core or penumbral tissues in the anterior–posterior direction were observed for the AQP4−/− mice (Figure 4(b)). Thus, AQP4−/− mice had lesion volumes (including core/penumbra) that were globally reduced relative to WT mice.
Figure 4.

Anterior–posterior distribution of the lesion. (a) The ischemic injury, rostro-caudally, is different between AQP4−/− and wildtype mice. In this example, T2 and diffusion-weighted (DWI) images at three days post-ischemia differences in the distribution of the ischemic lesion are notable, particularly within the striatum. (b) Lesion characteristics (total lesion, core, penumbra) between WT and AQP4−/− were similar along the anterior posterior axis, but overall AQP4−/− mice had reduced volumes in every measure. (n = 8 for WT and n=5 for AQP4−/−).
Quantitative lesion analysis
Quantitative T2 and diffusion ADC values can provide relative information on the physiological processes underlying tissue injury.26 Again, HRS was used to derive tissue level values for total lesion, core and penumbral volumes. There was a significant increase in global T2 values between AQP4−/− and WT mice over time within the ischemic lesion (p < 0.0001) but post hoc testing revealed that no time point (1–14 days post-MCAO) was significantly different between the groups (Figure 5(a)). A similar finding was observed when ADC values were compared (Figure 5(b)) wherein there was a large two-fold increase in ADC at 14 days. This would suggest that at 14 days, while the volume of the ischemic tissue is reduced in AQP4−/− compared to WT mice, the lesion volume itself is not different between groups and becoming equally cystic (see Figure 2(a)).
Figure 5.

Quantitative T2 and ADC analysis. (a) T2 values were significantly different between WT and AQP4−/− mice (p < 0.0001), but post hoc testing did not identify a critical time window. (b) Similarly, ADC values were also found to be significantly different (p < 0.0001) without a within time point significance. (c) When HRS derived total lesion T2 values were assessed there was also a significant difference between WT and AQP4−/− mice (p < 0.03). (d) While there were no significant differences in penumbral T2 values, a significant between time and group (p < 0.05 was found in core T2 values between WT and AQP4−/− mice. Post hoc testing identified a significant increase in AQP4−/− core T2 values (p < 0.04). (n = 4 for WT and n = 4 for AQP4−/−).
Segmentation of the ischemic lesion found that only T2 values within the ischemic core were significantly increased in the AQP4−/− mice compared to WT (159.0 ± 27.94 vs. 115.25 ± 7.76 ms, respectively) (Figure 5(d)). This increase was not found in the penumbral volumes (data not shown). Similarly, when total lesion T2 values (core+penumbra) were assessed, they were significantly increased throughout the lesion (p < 0.03), but no significance was found in post hoc testing for temporal changes (Figure 5(c)). Again, these findings would suggest that the AQP4−/− mice have accelerated lesion characteristics, based on cystic T2 values within the core tissues (Figure 5(b)).
Part 3: AQP4 deficiency attenuates neuronal death, blood brain barrier breakdown and neuroinflammation markers after MCAO
Neuronal nuclei were labeled with an anti-NeuN antibody in both groups (Figure 6(a) to (c)). The number of NeuN positive nuclei per mm2 was significantly (2.6 fold) higher in mice lacking AQP4 (with about 1600 positive cells/mm2 compared to 600 positive cells/mm2 for WT), suggesting that the absence of AQP4 promotes neuronal survival after MCAO. AQP4 is expressed on astrocytic endfeet on capillaries participating in the blood brain barrier. Blood brain barrier leakage was evaluated by staining for extravasated IgG. Interestingly, IgG labeling was reduced by four times in the absence of AQP4 (with about 5% for AQP4−/− versus 20% IgG total brain area in WT, Figure 6(d) and (e)), indicating that BBB breakdown for large molecular weights (<150 kDa) is largely prevented in the absence of AQP4 up to 14 days post-stroke.
Figure 6.

(a–c) NeuN staining in WT and AQP4−/− mice in perilesion/penumbra: significant decrease of the number of NeuN positive cells in WT compared to AQP4−/− at 14 days post-stroke. NeuN staining (a to c) was performed to show viable neurons in the perilesion area. Neuronal nuclei are stained in green in perilesional cortex (a, b) in WT (a) and AQP4−/− (b). The number of NeuN positive neuronal nuclei per square mm was significantly smaller (p < 0.01) in WT (a, c) than in AQP4−/− mice (b, c) attesting a less pronounced neuronal death in AQP4−/− mice (n = 4 for WT and n = 4 for AQP4−/−). (d, e) BBB status: IgG extravasation at 14 days post-stroke significantly higher at the cortical and striatal site of the injury in WT mice. Blood brain barrier integrity was evaluated by staining for IgG extravasation (d, e). The staining was significantly stronger in WT (p < 0.01) than in AQP4−/− mice indicating a preserved BBB integrity for large molecular weights (<150 kDa) in the absence of AQP4 (n = 4 for WT and n = 4 for AQP4−/−). Scale bars: a, b = 100 μm.
Reactive astrocytes were labeled with an anti-GFAP antibody in the perilesional region (Figure 7(a) to (e′)). The labeling pattern was different between groups, with decreased infrared signal intensity by about 40% in the absence of AQP4 (Figure 7(a) to (c)). However, an increased labeling area in AQP4−/− was measured compared to WT mice (Figure 7(a) to (c)). In addition, the morphology of astrocytes with GFAP staining is altered between the experimental groups with more processes in AQP4−/− mice compared to WT in the cortex adjacent to the site of the lesion (Figure 7(d), (d′), (e), and (e′)). Microglia were labeled with an anti-Iba1 antibody (Figure 7(f)). The labeling was significantly stronger in the absence of AQP4, suggesting that microglial activation is attenuated in the absence of AQP4 (Figure 7(f)).
Figure 7.

GFAP and IBA1 staining patterns in the WT and AQP4−/− animals. (a, b, c, d, d′ e, e′) GFAP staining was carried out to characterize the astroglial reaction in the perilesion area at day 14. There is a perilesional increase of the GFAP staining intensity in WT compared to AQP4−/− however the spread of the reactive astrocytes is higher in the AQP4−/− compared to the WT (a, b and c). The intensity of staining was stronger in WT (a, c) than in AQP4−/− mice (b, c). However, the spread of the labeling, expressed in percent of staining of the total surface, was higher in AQP4−/− (n = 4, b, c) than WT mice (n = 4, a, c). Similarly, the shape of the reactive astrocytes differs between WT and AQP4−/− with more swollen astrocytes in AQP4−/− compared to WT (arrows, d, d′, e e′). Inset black white pictures are representing zoom on GFAP staining in WT (d′) and AQP4−/− (e′). Sections were double labeled for AQP4 and GFAP ((d) and (e)) with perivascular AQP4 labeling in WT (arrowheads, d) and no labeling in AQP4−/− mice (e). (f) Iba-1 staining was carried out in the WT and AQP4−/− and showed a significant decrease (p < 0.01) of the Iba-1 staining in the peri-lesion tissue (n = 4 for WT and n = 4 for AQP4−/−). Scale bars d, e=100 μm; d′, e′=50 μm.
Discussion
Cerebral ischemia is a complex and multifactorial process triggered by an arterial occlusion leading to energy failure, depolarization and excitoxicity with ensuing neuronal death involving multiple other contributing phenomena such as the development of ionic and vasogenic edema, breakdown of the blood brain barrier and later onset development of inflammation. One of the earliest events after ischemia is the development of increased edema in and adjacent to the injury site. Water channels within the brain, such as AQPs, are well positioned to modulate water compartmentalization as needed particularly in response to injury. AQP4 is expressed on astrocytes, the most abundant cell type in the central nervous system playing an important role in brain metabolism, homeostasis, synaptic transmission, intercellular signaling and in neuroinflammation. In non-activated cortical and striatal astrocytes, AQP4 is preferentially expressed on astrocytic endfeet that surround the brain vasculature in the vicinity of the BBB, a very strategic localization in cerebral ischemia. Although, as a water channel, AQP4 is primarily passive to water movements through the astrocytic plasma membrane, we show here that its absence contributes to improving short- and long-term outcomes following ischemic stroke.
AQP4 expression on astrocytes allows water movements through the astrocytic plasma membrane driven by osmotic gradients. Depending on these gradients, the presence of AQP may facilitate astrocytic swelling or shrinking which can be observed in MRI-derived DWI signals (ADC) and hemispheric volumes. Surprisingly, AQP4 absence is associated with greater hemispheric enlargement at three and seven days compared to wild type animals (Figure 2). This strongly suggests that the benefits of AQP4 absence on behavioral outcomes and lesion volume do not reduce edema formation as a principal mechanism. In support of this possibility, we observed that the ADC values, reflecting water diffusion/mobility, were globally different between WT and AQP4−/− mice at all time-points. However, differences were not observed at any specific individual time-point. This indicates that the absence of AQP4 has a subtle influence on astrocyte swelling after MCAO, over a longer time period. The T2 signal, reflecting water content, also differed overall between groups, but did not reach significance at individual time-points. There are two phases of hemispheric enlargement after transient MCAO with peaks at 1 and 48h after ischemia onset.2 We have shown previously, using a preconditioning paradigm, that upregulating AQP4 on astrocytic end feet attenuated the early phase of hemispheric enlargement, 1 h after MCAO.3 We now show that hemisphere swelling three and seven days after MCAO is significantly enhanced in the absence of AQP4. This indicates an increased water accumulation in knockout mice. This result differs from the observation of an attenuated edema response in knock-out mice in a model of permanent MCAO27 and, more recently, in a 1-h MCAO model with evaluation at 23 h.11 Absence of AQP4 is correlated with less hemispheric enlargement at 23 h post-injury.11,27 The difference between the permanent MCAO results27 and the current results showing increased water accumulation in AQP4−/− mice at three and seven days strongly indicates that reperfusion causes vasogenic edema and impaired clearance of this vasogenic water accounts for the greater degree of hemispheric swelling. Commonly, larger lesions are associated with increased hemisphere swelling. In our experiments, however, these two features evolved differently and larger swelling was associated with smaller ischemic lesions in AQP4−/− mice. While at early time-points (1–4 days) the hemispheric volume depends mainly on edema, at later time-points (i.e. several weeks and beyond) it reflects atrophy and cystic encephalomalacia. The 14-day time-point shows a greater degree of hemispheric shrinkage in knockouts. Due to the smaller lesions, a less pronounced atrophy was anticipated in AQP4−/− mice. A possible explanation is that this 14-day time-point is influenced both by the atrophy and the water content caused by decreased water clearance consequence of absence of AQP4. We have shown previously, using a preconditioning paradigm, that upregulating AQP4 on astrocytic endfeet prevented an early hemispheric enlargement at 1h after MCAO3.
Our experiments show that the absence of AQP4 has a beneficial effect with smaller lesions and improved behavioral outcomes at early time-points and long term. Our work extends the previous observations made on early improvement of the lesions.11,27 We observed higher mortality in WT compared to AQP4−/−, which is in contrast with an earlier observation in Nanjing AQP4−/− mice.10 This difference between both studies can be due to the different background strains of mice used (ICR-CD1 vs CD1) and the different anesthesia protocol (isoflurane vs choral hydrate). Nevertheless, our results strongly suggest that the neuroprotective effect of removing AQP4 reaches beyond simple regulation of brain volume/edema process and has the potential to influence other astrocyte functions involved in brain tissue survival and function. Under physiological conditions, AQP4 is located strategically on astrocyte endfeet surrounding brain vasculature.28 We show here that the absence of AQP4, beyond the regulation of water movements, affects the permeability of the BBB, with an attenuated leakage of immunoglobulins into the extracellular space in the absence of AQP4. However, we can not exclude that BBB dysfunction is still present in the AQP4−/−, as it has been described at late time points after stroke in wild type rodents.29 This raises the question of additional roles of AQP4, e.g. in astrocyte endfeet and interaction with the neighboring BBB. Our observation suggests that the astrocytes can take part in opening the BBB after transient focal cerebral ischemia. Recently, it has been proposed that AQP4 may be involved in intracellular Ca2+ signaling by activation of purinergic receptors after ATP release. Ca2+ changes could very likely affect some basic functions of the astrocyte in BBB maintenance.30 In addition, release of ATP and activation of purinergic receptors modulate the neuroinflammatory process.31 Neuroinflammation, the activation of resident inflammatory cells in the brain, contributes to the evolution of the ischemic lesion and to tissue remodeling. Our results show that astrocyte activation is modified in the absence of AQP4, with less intense labeling in the lesion border but a more widespread distribution of GFAP labeling. The morphology of the reactive astrocyte is different between the groups with moderate astrogliosis after deletion of AQP4 compared to severe astrogliosis in wild type mice. The difference in the glial scar may also influence the ability in post-injury neuroplasticity32 with a better recovery for the AQP4−/− mice (Figure 1(b) to (d)). Although microglia do not express AQP4, microglial activation was also reduced in the knockout mice, possibly by altered release of ATP and activation of their purinergic receptors. These observations also point to further functions of normal AQP4-expressing astrocytes in the regulation of neuroinflammation.
Summary/Conclusions
Astrocytes express the water channel AQP4 on their end feet surrounding cerebral capillaries. We show that cerebral ischemia, in animals lacking AQP4, has a long-term improved outcome in terms of lesion volumes and behavioral deficits. Furthermore, despite enhanced edema, neuroinflammation is reduced and BBB disruption partly prevented. Our observations demonstrate a complex role of AQP4 and astrocytes in pathophysiological cascades initiated by ischemia. Our observations continue to support the concept of investigating AQP4 as a target for pharmacological treatment of cerebral ischemia.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Institute of child disorders (NICHD) R01HD061946 and the Swiss Science Foundation FN 310030_135617. JB was supported by a grant from the French Agence Nationale de la recherche within the context of the investments for the Future Program, referenced ANR-10-LABX-57 (Translational Research and Advanced Imaging Laboratory, TRAIL). A portion of this material was performed in the Loma Linda University School of Medicine Advanced Imaging and Microscopy Core (LLUSM AIM) that is supported by the National Science Foundation under Major Research Instrumentation, Division of Biological Infrastructure (grant no. 0923559) (Sean M Wilson) and the Loma Linda University School of Medicine.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Contribution
LH, AMF, KA, and FR generated the data; LH, DB, AO, and JB contributed to the experimental design and the analysis; LH, AMF, DB, AV, SA, AO, JB took part in writing of the manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Badaut J, Ashwal S, Obenaus A. Aquaporins in cerebrovascular disease: A target for treatment of brain edema? Cerebrovasc Dis 2011; 31: 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Castro Ribeiro M, Hirt L, Bogousslavsky J, et al. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res 2006; 83: 1231–1240. [DOI] [PubMed] [Google Scholar]
- 3.Hirt L, Ternon B, Price M, et al. Protective role of early aquaporin 4 induction against postischemic edema formation. J Cereb Blood Flow Metab 2009; 29: 423–433. [DOI] [PubMed] [Google Scholar]
- 4.Binder DK, Papadopoulos MC, Haggie PM, et al. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci 2004; 24: 8049–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao X, Hrabetova S, Nicholson C, et al. Aquaporin-4-deficient mice have increased extracellular space without tortuosity change. J Neurosci 2008; 28: 5460–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zador Z, Magzoub M, Jin S, et al. Microfiberoptic fluorescence photobleaching reveals size-dependent macromolecule diffusion in extracellular space deep in brain. Faseb J 2008; 22: 870–879. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Adwanikar H, Werb Z, et al. Matrix metalloproteinases and neurotrauma: evolving roles in injury and reparative processes. Neuroscientist 2010; 16: 156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Binder DK, Oshio K, Ma T, et al. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 2004; 15: 259–262. [DOI] [PubMed] [Google Scholar]
- 9.Li YK, Wang F, Wang W, et al. Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: Involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology 2012; 37: 1867–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng XN, Xie LL, Liang R, et al. AQP4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci Therapeut 2012; 18: 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao X, Derugin N, Manley GT, et al. Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci Lett 2015; 584: 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badaut J, Bix GJ. Vascular neural network phenotypic transformation after traumatic injury: Potential role in long-term sequelae. Translat Stroke Res 2014; 5: 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binder DK, Yao X, Zador Z, et al. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006; 53: 631–636. [DOI] [PubMed] [Google Scholar]
- 14.Friedman B, Schachtrup C, Tsai PS, et al. Acute vascular disruption and aquaporin 4 loss after stroke. Stroke 2009; 40: 2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papadopoulos MC, Manley GT, Krishna S, et al. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. Faseb J 2004; 18: 1291–1293. [DOI] [PubMed] [Google Scholar]
- 16.Saadoun S, Papadopoulos MC. Aquaporin-4 in brain and spinal cord oedema. Neuroscience. 168: 1036–1046. [DOI] [PubMed] [Google Scholar]
- 17.Badaut J, Ashwal S, Adami A, et al. Brain water mobility decreases after astrocytic aquaporin-4 inhibition using RNA interference. J Cereb Blood Flow Metab 2011; 31: 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuda AM, Pop V, Spagnoli D, et al. Delayed increase of astrocytic aquaporin 4 after juvenile traumatic brain injury: Possible role in edema resolution? Neuroscience 2012; 222: 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tait MJ, Saadoun S, Bell BA, et al. Increased brain edema in aqp4-null mice in an experimental model of subarachnoid hemorrhage. Neuroscience. 167: 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badaut J, Ashwal S, Tone B, et al. Temporal and regional evolution of aquaporin-4 expression and magnetic resonance imaging in a rat pup model of neonatal stroke. Pediatr Res 2007; 62: 248–254. [DOI] [PubMed] [Google Scholar]
- 21.Fukuda AM, Adami A, Pop V, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab 2013; 33: 1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh N, Yuan X, Turenius CI, et al. Automated core-penumbra quantification in neonatal ischemic brain injury. J Cereb Blood Flow Metab 2012; 32: 2161–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerriets T, Stolz E, Walberer M, et al. Noninvasive quantification of brain edema and the space-occupying effect in rat stroke models using magnetic resonance imaging. Stroke 2004; 35: 566–571. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda AM, Hirt L, Ambadipudi K, et al. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. Stroke 2013; 44: ATP97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuda AM, Adami A, Pop V, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab 2013; 33: 1621–1632. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashwal S, Tone B, Tian HR, et al. Comparison of two neonatal ischemic injury models using magnetic resonance imaging. Pediatr Res 2007; 61: 9–14. [DOI] [PubMed] [Google Scholar]
- 27.Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 2000; 6: 159–163. [DOI] [PubMed] [Google Scholar]
- 28.Badaut J, Fukuda AM, Jullienne A, et al. Aquaporin and brain diseases. Biochim Biophys Acta 2014; 1840: 1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strbian D, Durukan A, Pitkonen M, et al. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience 2008; 153: 175–181. [DOI] [PubMed] [Google Scholar]
- 30.Thrane AS, Rappold PM, Fujita T, et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2 + signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A 2011; 108: 846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chrovian CC, Rech JC, Bhattacharya A, et al. P2X7 antagonists as potential therapeutic agents for the treatment of CNS disorders. Progress Med Chem 2014; 53: 65–100. [DOI] [PubMed] [Google Scholar]
- 32.Sofroniew MV, Vinters HV. Astrocytes: Biology and pathology. Acta Neuropathol 2010; 119: 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.