

Abstract
Oncolytic herpes simplex virus (oHSV) type I constructs are investigational anti-neoplastic agents for a variety of malignancies, including malignant glioma. Clinical trials to date have supported the safety of these agents even when directly administered in the CNS. Traditional pre-clinical US Food and Drug Administration (FDA) toxicity studies for these agents have included the use of two species, generally including murine and primate studies. Recently, the FDA has decreased its requirement of non-human primates as an animal model for ethical reasons, especially for established viral systems where there are good alternative model systems. Here we present data demonstrating the safety of C134, a chimeric oHSV construct, in CBA mice as well as in a limited number of the HSV-sensitive non-human primate Aotus nancymaae as a proposed agent for clinical trials. These data, along with the previously conducted clinical trials of oHSV constructs, support the use of the CBA mouse model as sufficient for the pre-clinical toxicity studies of this agent. We summarize our experience with different HSV recombinants and differences between them using multiple assays to assess neurovirulence, as well as our experience with C134 in a limited number of A. nancymaae.
Keywords: oncolytic virus, HSV, Δ, γ134.5, PKR, IRF3, interferon, neurotoxicology, alpha US11, glioma, IRS1
Introduction
The unresponsiveness of malignant gliomas (MGs) to traditional therapies has led to the investigation of novel therapies, including oncolytic viruses (OVs), to treat this disorder. Oncolytic herpes simplex virus (oHSV) has been studied in human trials; while oHSV is effective in some patients, it has not uniformly produced extended survival or cures, likely due to over-attenuation of the virus for safety purposes. We previously reported the construction of a chimeric HSV-1, C134, which is a neurovirulent and is deleted for the γ134.5 gene but expresses the human cytomegalovirus (HCMV) IRS1 gene. The IRS1 gene improves Δγ134.5 replication within tumors and improves its anti-tumor activity in murine tumor models, yet C134 remains as safe as its parent Δγ134.5 virus in lethal dose 50% (LD50) and virus recovery studies.1, 2
Immunohistochemistry (IHC) of the infected murine CNS provides an additional method for evaluating sublethal viral encephalitic potential and complements viral recovery and LD50 studies. Unlike other viruses developed for oncolytic therapy (e.g., measles, adenovirus, poliovirus), HSV replicates in non-human animal cells and there are well established animal models for evaluating neuroinvasion and neurovirulence of the virus.3, 4, 5, 6 The commonly accepted method for evaluating the encephalitic potential of HSVs is by direct intracerebral injection in mice and has been used for over 40 years.3, 6 The CBA/J mouse strain (4- to 6-week-old mice) is our preferred model because it is more sensitive to herpes simplex encephalitis (HSE) than other murine strains (e.g., C57/BL6).7, 8, 9, 10 By stereotactically injecting virus into the caudate putamen nucleus and recording neurologic dysfunction and mouse deaths over a 28-day period, an objective and quantitative measure of neurovirulence (LD50 values) can be calculated using the Spearman-Karber calculation.3, 6, 11, 12 Wild-type (WT) HSV replicates more efficiently, generates greater viral progeny, and efficiently undergoes cell-to-cell spread, leading to death of the animal.1, 2, 13, 14, 15, 16
The studies described summarize the C134 neurotoxicology data from two animal models (murine and limited data from the highly HSV-sensitive non-human primate [NHP] model Aotus nancymaae). These studies extend our prior published studies and show that despite similar objective viral recovery and LD50 studies, subtle histologic differences exist between C134 and Δγ134.5 oHSVs in the murine model. In addition to the obvious difference in HCMV IRS1 gene expression in HSV antigen-positive regions of the brain, mice injected with C134 showed greater parenchymal HSV antigen staining than do those inoculated with the Δγ134.5 parent virus. However, consistent with the first-generation Δγ134.5 oHSVs, C134 did not show evidence of efficient cell-to-cell spread. In contrast, a more virulent protein kinase R (PKR)-evasion virus (Δγ134.5, HCMV IE US11) showed lower LD50 values and enhanced cell-to-cell spread. We also appreciated increased IRF3 IHC staining in previously HSV antigen-positive regions of the CNS. IRF3 expression increases correlated with a decrease in HSV antigen expression in these regions. IRF3 and HSV antigen were less prominent in the parenchyma of Δγ134.5-treated mice. This is consistent with our previous findings showing that C134, like its parent Δγ134.5 virus, induces early IRF3 and interferon (IFN) signaling and supports our hypothesis that this enhanced IFN response is linked to C134 aneurovirulence.17, 18
In addition to the murine biotoxicology studies, two non-human primates were administered 1 × 107 plaque forming units (PFU) of C134 (equivalent to a human dose of 1 × 109 PFU on a PFU/brain mass basis). One animal (T975) underwent MRI immediately prior to C134 inoculation and then again on days D15 and D33 post-inoculation. Both animals were analyzed for evidence of HSV infection (infectious virus and qPCR analysis). Infectious virus was not recovered from any site (brain periphery or shedding) from either animal. However, in both animals, viral DNA was detected by qPCR studies in multiple locations in the CNS (suggestive of trans-synaptic spread), similar to that reported for Δγ134.5 in previous murine studies1 and other oHSVs in NHP-based studies (G207 and M032).19, 20, 21, 22 Similar to our past analysis of another Δγ134.5 oHSV (M032) in NHPs, infectious virus was not recovered from the periphery on either D15 or D34; however, low levels of HSV DNA (101–103) were detectable at select sites (spleen, ureter), suggestive of viral clearance at 1 month post-injection.20
Finally, we also investigated two highly unlikely but theoretically possible events that could lead to patient harm: spontaneous mutations and recombination events. To identify whether C134 was likely to develop secondary mutations that would render it virulent and limit its clinical utility, we performed serial infection studies in mice. After six serial infections in the mouse CNS, the recovered virus (C134CNS passage 6) had no increase in virulence, with an LD50 value that was identical to the un-passaged C134 virus (>1 × 107 PFU). Next, to identify whether a C134 and WT-HSV recombination event could produce a more virulent virus, we created a WT HSV encoding HCMV IRS1 and tested it in murine neurotoxicity studies. The results show that IRS1 expression does not increase WT HSV-1 virulence and suggest that the resulting IFN response limits C134 replication spread, consistent with our past studies.17, 18 Together, these data support advancing C134 for production of a clinical-grade product for phase I clinical trials.
Results
Mouse Neurotoxicity Studies: IHC Analysis
We previously showed that (1) IRS1 selectively improved oHSV late viral protein synthesis and replication in tumors and (2) both C134 and a Δγ134.5 recombinant had equivalent LD50 neurotoxicity profiles.5 To determine whether C134 was capable of producing a subclinical encephalitis not detected by LD50 testing, We examined the mouse CNS by IHC on D2, D4, D6, and D8 after HSV infection (Figures S1 and S2). The D6 mouse CNS samples showed HSV antigen staining in both Δγ134.5- and C134-injected mice. C101 (Δγ134.5)-inoculated mice demonstrated HSV antigen staining in the ventricular ependymal cells on D2 and D4 post-injection but only rare antigen-positive cells were detectable by D6. The C134-inoculated mice demonstrated a similar temporal pattern for HSV staining as the Δγ134.5-inoculated mice. However, C134 produced greater HSV parenchymal staining around the injection site in contrast to the C101-administered mouse samples. The C134 brain sections were also examined for IRS1 gene expression. We took advantage of the C-terminal V5 epitope tag in the C134-expressed IRS1 protein. The results show IRS1 gene staining in multiple HSV antigen-positive regions of the brain (Figure S3A). Despite this improved protein expression and antigen staining, viral recovery remained similar between C134 and the Δγ134.5 in the non-malignant CNS (Figure S3B).
To identify whether this parenchymal staining was unique to C134, we compared C134 (Δγ134.5, IE IRS1) with another Δγ134.5-based PKR-evasion virus C122 (Δγ134.5, IE US11) by IHC staining. C122 is a more virulent PKR-evasion virus that expresses the HSV US11 gene also under control of the HCMV IE promoter Consequently, US11 is expressed earlier in infection, similar to our previously reported studies.23, 24 IHC analysis comparing C122, C134, and Δγ134.5 was performed at a dose approaching the LD50 of C122 (8.3 × 105 PFU). The results show differences in Δγ134.5, C134, and C122 infection. Consistent with our high-dose staining results, Δγ134.5 gene expression was limited to the ventricular ependyma (Figure 1). C134 infected ependymal and parenchymal cells near the injection site, although fewer antigen-positive cells were detected than with the high-dose injection. In contrast to Δγ134.5 and C134, the more virulent C122 (Δγ134.5, IE US11) injected at a dose approaching its LD50 value demonstrated extensive HSV spread through the parenchyma, beyond the injection site outside of the cortex, suggestive of efficient viral cell-to-cell spread. Due to these safety concerns, we did not further pursue CNS-based studies using C122 (Δγ134.5 IE US11), and we instead focused on C134 (Δγ134.5 IE IRS1) for clinical development for CNS tumors.
Figure 1.

HSV IHC Staining of CBA Mouse Brains 3 and 4 Days after Being Injected with Doses of R3616, C134, and C122 at the LD50 Level of C122: 8 × 105 PFU
Consistent with the high-dose results (shown in Figures S1 and S2), the Δγ134.5-inoculated mice demonstrate HSV in ventricular ependymal cells. C134-injected mice demonstrate HSV parenchymal staining (shown magnified in the inset) as well as ventricular staining (not shown). Mice inoculated with C122 virus (LD50 8 × 105 PFU) show extensive HSV parenchymal staining, suggesting viral cell-to-cell spread.
We previously demonstrated that the C134 virus induces an IRF3-mediated anti-viral response and that type I IFN limits C134 replication and cell-to-cell spread.17, 18 We hypothesized that this IFN response limited C134 replication and spread in the CNS.17, 18 To examine the kinetics of the IFN-mediated anti-viral activity elicited by C134 replication and spread in the CNS, we performed two studies. First, TaqMan qPCR studies were performed on RNA extracted from the brains of CBA/J mice injected stereotactically with saline and C134. The results showed that C134 infection upregulates an IRF3-inducible transcript, CXCL10, early after injection (peak expression, 24 hr) but gene expression levels returns to baseline by D3 post-injection (Figure 2). Mice injected stereotactically and administered saline had no change in their CXCL10 gene expression. After identifying the timing for this gene expression response, we next performed IHC studies to evaluate IFN-stimulated gene expression in the HSV-administered mice. We chose to examine IRF3 based on its performance in paraffin-embedded tissue samples. IRF3 staining was detected in the D4 samples but became readily apparent by D6 and D8 post-infection in C134-inoculated mice. This IRF3 increase correlated temporally with HSV clearance in the C134- and C101-inoculated brain sections. The IRF3 staining appeared earlier in the C134-inoculated samples (D4 for C134 versus D6 for C101) and localized to regions of HSV infection in the CNS. There was more prominent IRF3 parenchymal staining present in the sections from C134-inoculated animals when compared with those from C101 (Δγ134.5)-injected mice (Figure 3). In summary, we determined that transcriptional upregulation of the IRF3-inducible gene CXCL10 occurs the day following oHSV treatment but that ISG antigen staining, as shown by IRF3 staining in the CNS, was identified later at D4–D8 post-infection. We also identified that there was greater parenchymal HSV and IRF3 antigen detection in C134-inoculated mice when compared to Δγ134.5-treated controls and that they were found in shared locations in the CNS. Finally, the results show that IRF3 staining inversely correlated with HSV antigen expression, providing further evidence that the ISG response is integrally related to control of the Δγ134.5 and C134 infection in the CNS.
Figure 2.

CXCL10 Gene Expression from CBA Mice Stereotactically Injected with 1 × 107 PFU C134 or with Saline
qPCR shows a >100× increase in the IRF3-inducible transcript (CXCL10) the day after C134 injection when compared to saline-injected mice. The CXCL10 levels return to baseline by D3 post-C134 injection.
Figure 3.
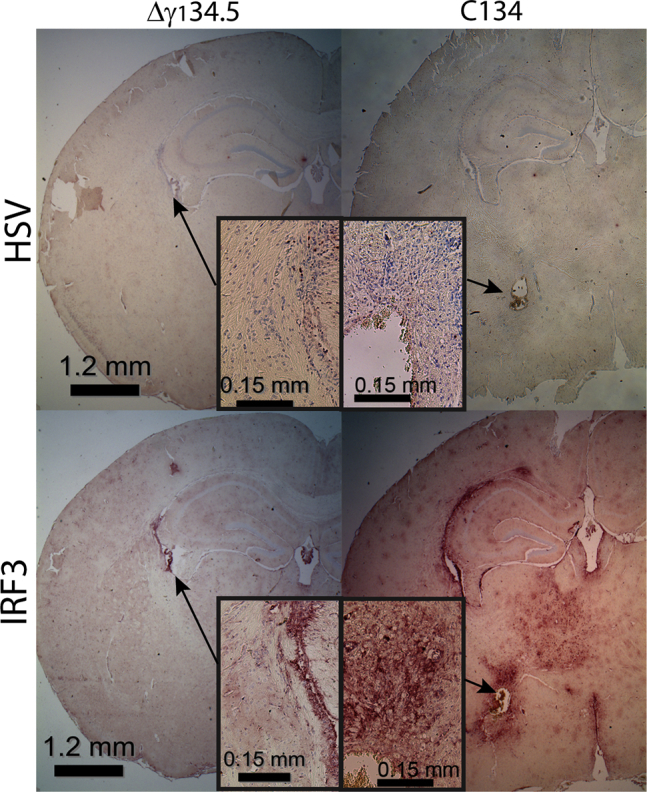
HSV and IRF3 IHC Staining in the CNS of CBA Mice Injected with 1 × 107 PFU C101 or C134
Low-magnification and corresponding high-magnification views of the CNS 6 days post-injection show limited HSV antigen detection in both the C101- and C134-inoculated mice. IRF3 staining is readily apparent in the C134-administered mouse CNS parenchyma and ependyma on D6 and coincides with the area of prior HSV detection in the mouse CNS. Composite representative images of the IHC-stained HSV-infected brains at D2, D4, D6, and D8 are provided in Figures S1 and S2 (low and high magnification, respectively).
NHP-Based Studies
Clinical Evaluation of C134 in NHPs and Autopsy Results
In addition to the mouse studies, we sought to verify safety in the highly HSV-sensitive non-human primate model A. nancymaae before advancing C134 for current good manufacturing practice (cGMP) virus production. Two animals were inoculated with 1 × 107 PFU of C134 (equivalent to a human dose of 1 × 109 PFU on a PFU/brain mass ratio). Neither of the animals developed clinical evidence of encephalitis, although one animal was euthanized on D15 post-inoculation. The first NHP tested (T970) displayed typical clinically normal behavior and activities during the initial 2-week period of twice-daily observation by veterinary staff and staff veterinarians. On D15, although the animal was still capable of nibbling on an occasional orange, it was unable to eat solids and required intravenous (i.v.) resuscitation. The animal was euthanized at the request of veterinarians. A sterile necropsy revealed that the animal had developed an overwhelming maxillary periodontal abscess secondary to a carious broken incisor. At the time of euthanasia, the animal had no fever and showed no focal neurologic changes, seizures, or concerns indicative of encephalitis. Laboratory abnormalities consisted of a mild leukocytosis with neutrophil predominance (8,770/μL with 81.2% polymorphonuclear leukocytes or neutrophils [PMNs]), an elevated amylase (1,026 mg/dL) consistent with the periodontal abscess, and electrolyte and blood chemistry changes consistent with dehydration (albumin, 5 g/dL; total protein, 7.6 g/dL; sodium [Na], 161 mmol/L; potassium [K], 6.0 mmol/L; hematocrit [Hct], 53%; blood urea nitrogen [BUN], 97 mg/dL; and creatinine, 0.5 mg/dL). Viral recovery, HSV qPCR, and metabolic studies including unremarkable liver function tests, liver appearance, and normal spleen size showed no evidence of HSV dissemination.
While autopsy results on the first animal (T970) suggested that C134 was safe, a second animal (T975) was injected with good laboratory practice (GLP)-like OptiPrep C134 (1 × 107 PFU) and received scheduled MRI scans immediately prior to injection and then again on D15 and D33 post-injection to ensure that there was no evidence of encephalitis. Because of the experience with the earlier NHP’s periodontal infection, the second animal had three carious teeth removed while anesthetized for the pre-inoculation MRI 1 day prior to C134 inoculation. T975, like the earlier monkey, did not show any clinical evidence of toxicity, as assessed by significant temperature, neurologic performance, feeding, weight loss, or change in social behavior and was euthanized on D34 for a scheduled necropsy.
MRI
Scheduled MRI on D15 and D33 post-injection did not show changes indicative of subclinical encephalitis. As demonstrated in Figures 4A and 4B, photomicrographs of MRI (coronal and axial planes) on D15 and D33 (respectively) show evidence of the needle tract for injecting virus but no evidence of enhancement, blood, or edema on T2-weighted images.
Figure 4.
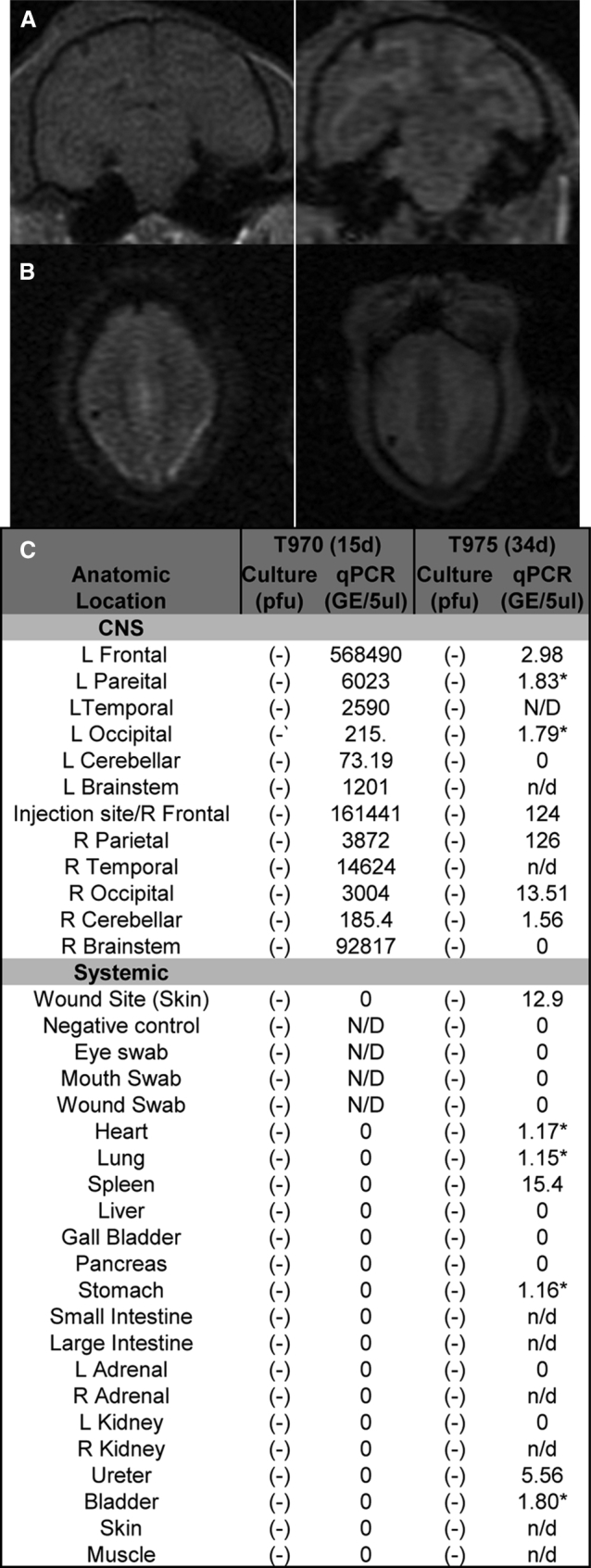
Composite of NHP Results
(A and B) MRI images from T975 at 2 weeks (A) (coronal view, upper panels) and D33 (B) (axial view, lower panels) post-injection show evidence of the needle tract but no radiographic evidence of HSV encephalitis. (C) No infectious virus was detected from the eye, nares, mouth, or any tissue but HSV DNA was detected distant from the site in both animals consistent with results from G207. Greater HSV DNA levels were detected in the CNS at D15 post-infection but were not detected outside the CNS. By D33, HSV DNA copies declined in the CNS but were detectable at low levels systemically in the spleen, ureter, and bladder. The asterisk indicates only one of three samples (+) by qPCR for HSV DNA. L, left; R, right.
Infectious Virus Recovery and Pathology
Gross examination of both primates (T970 and T975) showed no evidence of encephalitis or hemorrhagic necrosis. Eye, mouth, nares, and wound swabs were collected on D15 and D33 post-inoculation from T970 and T975, respectively. In addition to HSV shedding, brain and organs from both animals were also collected at necropsy for viral culture. No infectious HSV was recovered from any of the 29 brain and organ samples collected from either monkey (Figure 4C).
HSV DNA Is Detected in the C134-Infected NHPs
Previous experiences with M032 and G207 demonstrated that HSV DNA is detectable by PCR in the CNS beyond the injection site and even in the periphery weeks to months after HSV administration.19, 20, 22 We examined the C134-administered NHPs on D15 and D34 post-injection for differences in the relative distribution of HSV DNA in the CNS or periphery. Consistent with prior experiences with M032 and G207, C134 was detected in multiple sites within the brain of both animals euthanized on D15 and D34. The quantity of HSV DNA in the brain was lower in the animal euthanized on D34 (∼101–103 copies) when compared to the animal euthanized on D15 (104–106 copies). As described in the Materials and Methods, the 50-mg CNS and peripheral tissue samples were divided, with half being devoted to viral recovery studies and the other half to qPCR; none of the q-PCR-positive organs yielded infectious HSV by a viral recovery assay.
Lower quantities of HSV DNA were detected in the CNS of the monkey euthanized on D34 than in the animal euthanized on D15. HSV DNA was detected at the highest levels in the right brain (right frontal and right parietal) at or adjacent to where the virus had been inoculated at D34 post-injection. Lower HSV DNA copy numbers were detected distant from the injection site (right occipital > right cerebellum). Similar to the D15 NHP, HSV DNA was also detected in the left brain, albeit at lower levels than in the right hemisphere. Unlike in the D15 animal, HSV DNA was also detectable outside of the CNS in the D34 animal (Figure 4C). Low levels of HSV DNA (<50 copies/mg tissue) were detected in the wound site (skin from the site of inoculation), spleen, and ureter in at least two of three samples tested. Only one of three samples was positive for HSV DNA at four peripheral sites (heart, 3.5 genome equivalents [GEs]; lungs, 3.43 GEs; bladder, 5.4 GEs; and stomach, 3.49 GEs). None of the other organ or brain samples tested positive for HSV DNA in the animal euthanized on D34 (Figure 4C). There was no evidence of viral shedding by viral culture or qPCR (Figure 4C). HSV detection in the periphery is consistent with our past published experience with other Δγ134.5 HSVs 1 month after inoculation.20 The presence of HSV DNA at the limit of detection in the spleen and other vascular organs at D34 suggests immune clearance of viral DNA from the site of infection.
Theoretical Concerns: Spontaneous Mutations and Recombination Events
Intracranial Serial Passage
We and others have shown in previous studies that Δγ134.5 recombinants can develop compensatory mutations during serial infection that increase Δγ134.5 neurovirulence.4, 25, 26 We also showed that different compensatory mutations are elicited during in vivo passage in flank tumors that attenuate and improve anti-tumor activity.26 Spontaneous mutations and re-established virulence are theoretical concerns for attenuated viral therapeutics. A particular worry is that pathogenic mutations could develop during infection in non-malignant cells. To identify whether C134 could develop pathogenic mutations that may prohibit its use as a therapeutic, we chose in vivo IC serial passage. After six serial passages in CBA/J mouse brains, isolated progeny virus (C134CNS passage6) was as safe (LD50 > 1 × 107) as the starting (passage 0) C134 recombinant (Figure 5A).
Figure 5.

Modeling Spontaneous Mutations or a Recombination Event
(A) LD50 values for serially passaged and engineered viruses. A theoretical concern with a recombinant is that secondary mutations could develop during replication, thus limiting its safety and clinical utility. To assess this, we serially passaged C134 in vivo and found that after six passages in the CNS of mice, the virus retained it pre-passage LD50 profile. Another theoretical concern with an oHSV vector containing a foreign gene is that a recombination event could produce an HSV with greater virulence or encephalitic potential than the WT species. To test this hypothesis, we mimicked such a recombination event and created C124 (WT+HCMV IRS1). LD50 studies show that insertion of HCMV IRS1 into the UL3/UL4 intergenic region of WT HSV does not increase HSV-1 (LD50 of 86 PFU versus 18 PFU) virulence. (B and C) Furthermore, using dose-matched groups, 330 PFU (B) and 1,000 PFU (C) HSV-1 was more virulent than C124 and led to earlier death in the IC-administered mice in survival studies. A full summary of animal numbers and timing of deaths for all dose groups is provided in Figure S4.
Recombination Events
A theoretical concern with biologics is the potential for a recombination event. To test whether a recombination event could produce a more virulent virus in a patient, we created a WT HSV encoding HCMV IRS1 (C124) and tested it in murine neurotoxicity studies. The results showed that adding the HCMV IRS1 gene to a WT HSV did not increase its virulence (18 PFU for HSV-1 [F] versus 86 PFU for C124). We also examined the rate of death in the LD50 sample cohorts (330-PFU and 1,000-PFU cohorts) using Kaplan-Meier survival estimates. When cohorts were inoculated with 330 PFU or 1,000 PFU of WT or C124, WT HSV killed a greater proportion of mice, and they died earlier than in the C124-inoculated cohort. To date, a known recombination event has not occurred in any patient in oHSV trials in the United States or Europe. Nonetheless, our results indicate that should such an event occur, the HCMV IRS1 gene would not increase the encephalitic potential of WT HSV. As an added margin of safety, HSV recombinants intended for clinical use should retain sensitivity to acyclovir, the drug of choice for treating HSV encephalitis. Plaque reduction assays were performed and showed that C134 was as susceptible to acyclovir (ACV) (ED50 < 0.5 μg/dL) as other thymidine kinase (TK)-containing viruses (F, C101, and G207) tested (data not shown).
Discussion
Previous studies by our group indicate that the chimeric HSV C134 is an attractive oncolytic therapeutic. It is safe like first-generation Δγ134.5 oHSVs in murine neurovirulence LD50 studies. However, unlike Δγ134.5 recombinants, C134 evades PKR-mediated translational arrest and synthesizes late viral proteins. This improves C134 replication in tumor cells. Pre-clinical models show that C134 is superior to Δγ134.5 viral therapy in brain tumor models.5 For previous oHSVs that advanced to phase I clinical trials, the FDA requested testing on at least two species (including a NHP model) prior to their use in phase I trials. Based on its advantageous anti-tumor and biotoxicology profile, C134 has been manufactured as a cGMP product, has undergone biotoxicology testing, and has received RAC approval.
We first sought to investigate whether subclinical replication or pathologic changes (not reflected in LD50 assays) occurred in mice infected with the C134 virus. Our results were consistent with those reported by Markovitz et al.1 and show that like other Δγ134.5 viruses, C134 is cleared by D6–D8 post-infection. We also show that there are some notable differences between the Δγ134.5 and chimeric viruses. First, there was greater parenchymal HSV antigen expression in the C134-infected animals than in the first-generation Δγ134.5-infected animals. We hypothesize that this may be a function of improved HSV protein expression by the C134 recombinant. Virus recovery studies (shown in Figure S3B) indicate that C134 and C101 replicate similarly in the CNS. We suspect that C134, with its ability to evade PKR-mediated translational arrest, produces more HSV proteins in the infected cells and is more readily detectable in the CNS parenchyma immediately adjacent to the injection site. Second, C134, like the first-generation oHSV, infects the ependymal cells. Third, the duration of HSV antigen detection is longer than that observed with the Δγ134.5 virus-infected animals. The other significant finding was that C134 antigen staining differed from that of another PKR-evasion virus (C122). C122 expresses the HSV US11 gene from the HCMV IE promoter and is more virulent than C134 (LD50 of 8.3 × 105 PFU for C122 versus > 1 × 107 PFU for C134). This enhanced virulence was also reflected in the IHC studies that show diffuse CNS parenchymal staining suggestive of efficient viral spread 3 days after C122 inoculation at 8 × 105 PFU.
The speed with which the virus undergoes cell-to-cell spread was the focus of studies by Markovitz et al.1, 2 They showed that Δγ134.5 virus is capable of viral replication including synaptic spread but that this is slow and inefficient and that by D6–D8 post-infection, intrinsic and immune-mediated anti-viral responses limit Δγ134.5 replication and clear the infection.1 While our results are observational, our recent published experience18 and those of Markovitz et al. suggest that C134 behaves like a Δγ134.5 virus, with respect to replication and limited cell-to-cell spread in cells with intact IFN signaling response. We previously showed that C134 induces IRF3-mediated gene expression. To further investigate whether IRF3 staining (as a marker of ISG expression and IFN response) correlated with C134 replication, we performed further IHC studies and found that IRF3 staining increased by D4–D8. In addition, this increase in staining occurred at anatomic loci of a prior C134 infection in the CNS, and IRF3 expression correlated temporally with loss of HSV antigen expression. These studies show that while an IRF-inducible gene (CXCL10) peaks 1 day after oHSV injection, it returns to baseline by D3. Protein detection of another ISG (IRF3) occurs much later and was readily apparent by D4–D6 post-infection.
Another potential concern for a biologic therapeutic is the potential to develop virulence mutations. Our results using serial-passage and recombination experiments were reassuring. While negative results cannot eliminate the possibility of virulent mutations, the results from our intracerebral (IC) studies suggest that a Δγ134.5 containing the IRS1 gene was not any more likely than the Δγ134.5 to develop secondary pathogenic mutations. Thus far, no known spontaneous mutations have occurred during any of the phase I clinical trials in Europe and the United States using Δγ134.5-based recombinants. Likewise, based on LD50 studies, transfer of the immediate early (IE)1 promoter-driven IRS1 gene to WT HSV did not increase WT virulence; in survival studies, WT HSV led to more rapid death in mice than did equivalent doses of the C124 (WT, IRS1) virus.
In these limited NHP studies, C134 was also safe. There were no clinical or radiographic changes indicative of encephalitis and no infectious virus was recovered from the brain or peripheral tissue from either animal at D15 or D34 post-inoculation. HSV DNA was recovered from the brain on D15 and D34 post-inoculation, similar to that described for G207 and M032.20, 22 High levels of HSV DNA were detected throughout the CNS at D15 post-inoculation (102–105 GEs). By D34 post-infection, lower levels of viral DNA (≤102) were detectable and predominantly near the site of injection in the right cortex (right frontal, right parietal > right occipital). Similar to the M032 studies, we detected C134 DNA at low levels (∼5.5–15.4 GEs) in the periphery (spleen, wound site, and ureter) at ∼1 ms post-inoculation. We do not believe that this is a cross-contamination of HSV DNA from other sites or during extraction, because other samples remained free of HSV DNA and a negative control sample included during the extraction process was also negative (zero of three samples tested). Instead, we postulate that this low-level HSV detection represents HSV DNA at the threshold of qPCR detection from circulating immune cells in highly vascular organs. Taken together, the NHP results also provide insight into clearance of viral CNS infection in the primate model and are consistent with our past experience in Aotus with another Δγ134.5 virus and with murine Δγ134.5-based studies reported by Broberg et al.,27, 28 who demonstrated systemic immune response changes following CNS infection with attenuated HSVs. Based on the long history of murine-based neurotoxicity studies for oHSV and ethical concerns with further NHP-based toxicity studies, the decision was made to limit the toxicology analysis to the two NHPs.
Materials and Methods
Viruses, Cells, and Animals Used
Vero cell lines were obtained from American Type Culture Collection. Vero cells were propagated in DMEM supplemented with 5% newborn calf serum (Vero). HSV-1 (F) and R3616 (kindly provided by B. Roizman, University of Chicago) are the prototypical HSV-1 and Δγ134.5 recombinant, respectively, used in these studies. R3616, C101, and C134 have been described previously3, 5, 29 and are summarized in Table 1. C122 is another C101-based PKR-evasion virus and encodes the HSV-1 US11 gene under control of the CMV IE promoter in the same locus (UL3/UL4 intergenic region). C124 is a WT HSV that expresses HCMV IRS1 and was created using the same targeting plasmid used for C134.
Table 1.
Summary of Recombinants and Genetic Modifications
| Virus | γ134.5 | UL3–UL4 Gene Insertion |
|---|---|---|
| R3616 | deleted | none |
| C101 | deleted | IE EGFP |
| C134 | deleted | IE HCMV IRS1 |
| C122 | deleted | IE HSV US11 |
| HSV-1 (F) | intact | none |
| M2001 | intact | IE EGFP |
| C124 | intact | IE HCMV IRS1 |
IE, HCMV IE promoter.
Preparation of OptiPrep Clinical-Grade Virus
HSV virus stocks were obtained after purification on OptiPrep gradients (Axis Shield) similar to that described in Shah et al.26 In brief, Vero cells were infected at low MOI (0.01). Both culture supernatants and cells were collected separately once infection had reached a 100% cytopathic effect (CPE). Supernatants underwent three centrifugation steps: step 1, 1,811 × g (15 min); step 2, transfer of supernatants to Nalgene tubes and centrifugation at 3,568 × g (5 min, JA14 rotor); and step 3, transfer of the final supernatants to Beckman Coulter Ultra-Clear tubes and centrifugation at 53,000 × g (1 hr, SW28 rotor). Viral pellets were resuspended in PBS containing 10% glycerol. Cells were scraped, lysed in 0.4 M NaCl, and subjected to multiple centrifugations as described above prior to suspension in PBS/10% glycerol. Virus pellets were purified on OptiPrep gradients as follows. Equal volumes of OptiPrep were added to NaCl-extracted or culture supernatant virus pellets, mixed, and transferred to Quick-Seal polyallomer tubes. Tubes were then filled with 25% OptiPrep and centrifuged for 3 hr at 354,000 × g in an NVT90 rotor. After centrifugation, virus fractions were collected, diluted in PBS, and centrifuged. Virus pellets from supernatants and cell lysates were combined, and the titer was determined on Vero cells as described previously.
Intracranial Serial Passage
Similar to the methods described in Shah et al.,26 mice were inoculated with C134 GLP-like OptiPrep virus NC070516 (1 × 106 PFU) and were euthanized at D3 post-inoculation (based on our earlier studies showing that virus could still be recovered from the CNS at D3 post-inoculation: see Figure S3B). The brains were homogenized in 1 mL DMEM with 1% FBS serum using a sterile Dounce homogenizer. The homogenized passage virus recovered from the brain was then amplified in Vero cells and a viral stock was produced, resuspended in DMEM 10% glycerol, and titered and another in vivo passage was performed (using 1 × 106 PFU of virus in 10% glycerol). This procedure was repeated until we completed six serial infections in CBA/J mouse brains. The amplified sixth-passage virus was then analyzed in a murine LD50 neurotoxicity assay.
All animal studies were conducted in accordance with guidelines for animal use and care established by the University of Alabama at Birmingham Animal Resource Program and the Institutional Animal Care and Use Committee. Mice (CBA/J) were obtained from the National Cancer Institute (NCI). A. nancymaae were obtained from the US Department of Agriculture (USDA) and housed according to the University of Alabama at Birmingham animal husbandry conditions, allowing social interaction between animals. In keeping with the other members of the colony, animals were kept in a reverse light-cycle environment with special caging. In addition to the regular feeds, the primates received supplemental fresh fruit and “fruity gems.” For environmental enrichment, the animals had appropriate-sized perches and cubbies and were allowed social interaction.
Stereotactic Murine Intracerebral Inoculation
Murine studies were performed using the same method described in Cassady et al.4 and Shah et al.5 In brief, mice were anesthetized (ketamine and xylazine), prepped sterilely, slowly injected with OptiPrep GLP-like virus (5–10 μL) using a stereotactic system through a pre-drilled burr hole into the right subcortical frontal region, and allowed to equilibrate for 2 min prior to removal of the syringe.
Stereotactic NHP Intracerebral Inoculation and MRI Analysis
All intracranial inoculations and MRI of the primates was done under conditions of general anesthesia. Stereotactic injection was performed similar to that previously described.20 In brief, monkeys were sedated and intubated. Anesthesia was induced using isoflurane gas (1.7%–2.5%) and the animals were surgically prepped and mounted in a stereotactic head frame. Following scalp incision, a small burr hole was made with a sterile drill with a 1.5-mm burr, the dura was incised, a sterile needle was lowered into the right frontal lobe to a depth of 5 mm and OptiPrep GLP-like C134 (1 × 107 PFU) was infused, and post-operative analgesia was maintained with 0.008–0.01 mg/kg buprenorphine over the 24-hr post-operative period. Monkeys were observed daily and were examined for evidence of clinical toxicity; one monkey was also evaluated using a 1.5T MRI system (GE Healthcare) for radiographic evidence of C134 neurotoxicity at 2 weeks and 1 month after inoculation. Monkeys were anesthetized with veterinary assistance, immobilized in a custom plexiglas cradle, and stabilized with towels, blankets, and i.v. bags of warm saline and placed supine in a custom coil in the 1.5T MRI machine. In addition to clinical and radiologic evaluations, NHPs also underwent gross pathologic and laboratory testing for viral recovery.
Preparation of NHP Tissue from Necropsy and NHP Virus Recovery
Organs and brain sections from the infected NHPs were harvested, weighed, and divided during scheduled necropsy under sterile conditions in the operating room. Samples were stored in individually marked containers as follows: (1) tissue for frozen sections were snap-frozen in OTC using a liquid nitrogen bath; (2) archival specimens were snap-frozen in liquid nitrogen, placed on dry ice during transport, and stored in a liquid nitrogen storage system; and (3) samples for viral culture and qPCR were divided, weighed, and placed in separate vials. Viral culture samples (25 mg) were placed in MR5 transport media vials and immediately frozen (−80°C). The NHP culture samples were subjected to three freeze-thaw cycles, with tissue disruption during each freeze-thaw cycle with vigorous vortexing (using beads present in the MR5 transport media). Vero cells in 3.5-cm2 culture dishes were infected with 250 μL NHP homogenate in a humidified incubator with gentle rocking. After 2 hr, the lysate was removed and replaced with 500 μL 5% newborn calf serum (NBCS) (containing antibiotics and antifungals). Vero cells were incubated at 37°C for 1 week and checked on D2, D4, and D7 for evidence of cytopathic effect.
NHP DNA Recovery and qPCR
NHP samples for qPCR analysis (25 mg) were placed in sterile microcentrifuge tubes in proteinase K containing digestion buffer and digested overnight at 56°C. DNA was extracted into 200 μL elution buffer from NHP samples using the QIAamp DNA Blood and Tissue Mini Kit (QIAGEN) per the manufacturer’s instructions. The tissue preparation, DNA extraction, and TaqMan preparation steps were performed in a laminar flow hood in a laboratory that does not work with HSVs. Extracted DNA samples were incubated with the following HSV-specific primers and probes for HSV polymerase (sequences kindly provided by Dr. Fred Lakeman): PolF (forward), 5′-ACC GCC GAA CTG AGC AGA C-3′; and PolR (reverse), 5′-TGA GCT TGT AAT ACA CCG TCA GGT-3′. The fluorescent-labeled probe sequence was 5′-6FAM-CGC GTA CAC CAA CAA GCG CCT G-TAMRA-3′. HSV GEs of the amplified product were measured from triplicate samples using an ABI 7300 TaqMan machine (Applied Biosystems ) and compared against logarithmic dilutions of a positive control DNA sequence (106 – 101 copies). Descriptive statistical analyses (mean and SD) were used to compare differences in DNA copy numbers between samples using Prism 5.0 statistical software (GraphPad).
Neurotoxicity Studies
As described previously, independent cohorts of CBA/J mice were injected stereotactically using graded 1/2 log increments of OptiPrep virus (1 × 105, 3.3 × 105, 1 × 106, 3.3 × 106, and 1 × 107 PFU) and were followed for 4 weeks for neurologic dysfunction.5 For each virus (C122, C134, C101, or C134CNS passage 6-1 and C134CNS passage 6-2), an LD50 value was calculated based on the Spearman-Karber statistical method. HSV-1 (F) was used as the positive control and the results were repeated to verify results. In addition to traditional LD50 studies, Kaplan-Meier survival analysis from matched dose cohorts was also compared with the C124 LD50 studies to assess the effect of IRS1 on WT HSV-1 virulence.
RNA qPCR Analysis
For RNA qPCR studies, CBA mice injected stereotactically with saline or with 1 × 107 PFU of C134 were euthanized on D1, D2, and D3 post-injection mice (4–5/cohort), and the brain injection site was dissected and placed in RNAlater. The brain samples (50 mg/sample) were subject to RNA extraction using an RNeasy total RNA isolation kit (QIAGEN) with DNase digestion while silica bound as per the manufacturer’s instructions. Equivalent mass samples of RNA (1 μg/sample) were then subjected to first-strand synthesis using SuperScript III reverse transcriptase (Invitrogen) and oligo(dT) 21 primers from the saline- and C134-treated brain samples. CXCL10 gene expression was measured by using TaqMan primer sets (ABI Mm99999072_m1) and StepOne Plus (Applied Biosystems) compared against a standard curve prepared from logarithmic serial dilutions of a plasmid encoding murine CXCL10.
IHC
In separate studies, CBA/J mice were inoculated stereotactically with 8 × 105 PFU of C122, C134, or R3616 and euthanized on D3 and D4 post-inoculation or were inoculated with 1 × 107 PFU of C101 or C134 and euthanized on D2, D4, D6, and D8 post-injection. The virus-inoculated mouse brains were isolated, formalin fixed, paraffin embedded, and analyzed by IHC, as described previously.30 In brief, 6-μm-thick sections were prepared for IHC staining by deparaffinization, antigen retrieval using sodium citrate buffer (pH 6.0), hydrogen peroxide incubation to quench endogenous peroxidases, and blockade with Fc block and with either horse or goat serum (Vector Laboratories) depending on the antibody to be used. Antibodies used for these studies included anti-HSV rabbit polyclonal antibody (diluted 1:400; BioGenex Laboratories), mouse monoclonal antibody against V5 (clone SV5-Pk1 diluted 1:100; Pierce), and rabbit anti-IRF3 (EPR2418Y diluted 1:100; Epitomics). Both rabbit and mouse polyclonal IgGs were used as negative controls. Secondary antibodies specific for rabbit or mouse IgG were supplied as Vectastain ABC kits (Vector Laboratories). Slides were developed using a Vector VIP (Vector Laboratories) peroxidase substrate detection kit.
Author Contributions
K.A.C., scientific design, execution and manuscript preparation of the C134 studies; D.F.B. and M.R.C., aotus surgery; J.R., assisted with manuscript and experiments (viral recovery studies); T.S., veterinary care for NHPs; J.C., assisted with C134 lab studies (IHC); M.P., assisted with C134 lab studies (ACV resistance assays and qPCR studies); G.Y.G., assisted with C134 lab studies (Mouse LD50 Neurotox studies); J.M.M., manuscript preparation and scientific discussions.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the NIH (grants P20-CA151129 and X01-NCI DTP-RAID [Rapid Access to Intervention Development]), Alex’s Lemonade Stand Foundation for Childhood Cancer, and Hyundai Wheelin’ for Hope. The authors especially thank Dr. Steven Lloyd for providing the Aotus digital imaging and communications in medicine (DICOM) images from archival storage.
Footnotes
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.omto.2017.02.001.
Supplemental Information
References
- 1.Markovitz N.S., Baunoch D., Roizman B. The range and distribution of murine central nervous system cells infected with the gamma(1)34.5- mutant of herpes simplex virus 1. J. Virol. 1997;71:5560–5569. doi: 10.1128/jvi.71.7.5560-5569.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehta H., Muller J., Markovitz N.S. Ultrastructural analysis of ICP34.5- herpes simplex virus 1 replication in mouse brain cells in vivo. J. Virol. 2010;84:10982–10990. doi: 10.1128/JVI.00337-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chou J., Kern E.R., Whitley R.J., Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 4.Cassady K.A., Gross M., Gillespie G.Y., Roizman B. Second-site mutation outside of the U(S)10-12 domain of deltagamma(1)34.5 herpes simplex virus 1 recombinant blocks the shutoff of protein synthesis induced by activated protein kinase R and partially restores neurovirulence. J. Virol. 2002;76:942–949. doi: 10.1128/JVI.76.3.942-949.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah A.C., Parker J.N., Gillespie G.Y., Lakeman F.D., Meleth S., Markert J.M., Cassady K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007;14:1045–1054. doi: 10.1038/sj.gt.3302942. [DOI] [PubMed] [Google Scholar]
- 6.Hill T.J., Field H.J., Blyth W.A. Acute and recurrent infection with herpes simplex virus in the mouse: a model for studying latency and recurrent disease. J. Gen. Virol. 1975;28:341–353. doi: 10.1099/0022-1317-28-3-341. [DOI] [PubMed] [Google Scholar]
- 7.Halford W.P., Balliet J.W., Gebhardt B.M. Re-evaluating natural resistance to herpes simplex virus type 1. J. Virol. 2004;78:10086–10095. doi: 10.1128/JVI.78.18.10086-10095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kastrukoff L.F., Lau A.S., Thomas E.E. The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS) Herpesviridae. 2012;3:4. doi: 10.1186/2042-4280-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan W.L., Ziltener H.J., Liew F.Y. Interleukin-3 protects mice from acute herpes simplex virus infection. Immunology. 1990;71:358–363. [PMC free article] [PubMed] [Google Scholar]
- 10.Ito T., Jagus R., May W.S. Interleukin 3 stimulates protein synthesis by regulating double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA. 1994;91:7455–7459. doi: 10.1073/pnas.91.16.7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards J.T., Kern E.R., Overall J.C., Jr., Glasgow L.A. Differences in neurovirulence among isolates of Herpes simplex virus types 1 and 2 in mice using four routes of infection. J. Infect. Dis. 1981;144:464–471. doi: 10.1093/infdis/144.5.464. [DOI] [PubMed] [Google Scholar]
- 12.Whitley R.J., Kern E.R., Chatterjee S., Chou J., Roizman B. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Invest. 1993;91:2837–2843. doi: 10.1172/JCI116527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson R.L., Wagner E.K., Stevens J.G. Physical location of a herpes simplex virus type-1 gene function(s) specifically associated with a 10 million-fold increase in HSV neurovirulence. Virology. 1983;131:180–192. doi: 10.1016/0042-6822(83)90544-5. [DOI] [PubMed] [Google Scholar]
- 14.Thompson R.L., Stevens J.G. Biological characterization of a herpes simplex virus intertypic recombinant which is completely and specifically non-neurovirulent. Virology. 1983;131:171–179. doi: 10.1016/0042-6822(83)90543-3. [DOI] [PubMed] [Google Scholar]
- 15.Thompson R.L., Devi-Rao G.V., Stevens J.G., Wagner E.K. Rescue of a herpes simplex virus type 1 neurovirulence function with a cloned DNA fragment. J. Virol. 1985;55:504–508. doi: 10.1128/jvi.55.2.504-508.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Javier R.T., Thompson R.L., Stevens J.G. Genetic and biological analyses of a herpes simplex virus intertypic recombinant reduced specifically for neurovirulence. J. Virol. 1987;61:1978–1984. doi: 10.1128/jvi.61.6.1978-1984.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassady K.A., Saunders U., Shimamura M. Δγ1134.5 herpes simplex viruses encoding human cytomegalovirus IRS1 or TRS1 induce interferon regulatory factor 3 phosphorylation and an interferon-stimulated gene response. J. Virol. 2012;86:610–614. doi: 10.1128/JVI.05099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson J.D., Markert J.M., Li L., Carroll S.L., Cassady K.A. STAT1 and NF-κB inhibitors diminish basal interferon-stimulated gene expression and improve the productive infection of oncolytic HSV in MPNST cells. Mol. Cancer Res. 2016;14:482–492. doi: 10.1158/1541-7786.MCR-15-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markert J.M., Cody J.J., Parker J.N., Coleman J.M., Price K.H., Kern E.R., Quenelle D.C., Lakeman A.D., Schoeb T.R., Palmer C.A. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J. Virol. 2012;86:5304–5313. doi: 10.1128/JVI.06998-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth J.C., Cassady K.A., Cody J.J., Parker J.N., Price K.H., Coleman J.M., Peggins J.O., Noker P.E., Powers N.W., Grimes S.D. Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to Aotus nonhuman primates. Hum. Gene Ther. Clin. Dev. 2014;25:16–27. doi: 10.1089/humc.2013.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter W.D., Martuza R.L., Feigenbaum F., Todo T., Mineta T., Yazaki T., Toda M., Newsome J.T., Platenberg R.C., Manz H.J., Rabkin S.D. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J. Virol. 1999;73:6319–6326. doi: 10.1128/jvi.73.8.6319-6326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Todo T., Feigenbaum F., Rabkin S.D., Lakeman F., Newsome J.T., Johnson P.A., Mitchell E., Belliveau D., Ostrove J.M., Martuza R.L. Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in aotus. Mol. Ther. 2000;2:588–595. doi: 10.1006/mthe.2000.0200. [DOI] [PubMed] [Google Scholar]
- 23.Cassady K.A., Gross M., Roizman B. The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2. J. Virol. 1998;72:8620–8626. doi: 10.1128/jvi.72.11.8620-8626.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cassady K.A., Gross M., Roizman B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J. Virol. 1998;72:7005–7011. doi: 10.1128/jvi.72.9.7005-7011.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohr I., Gluzman Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 1996;15:4759–4766. [PMC free article] [PubMed] [Google Scholar]
- 26.Shah A.C., Price K.H., Parker J.N., Samuel S.L., Meleth S., Cassady K.A., Gillespie G.Y., Whitley R.J., Markert J.M. Serial passage through human glioma xenografts selects for a deltagamma134.5 herpes simplex virus type 1 mutant that exhibits decreased neurotoxicity and prolongs survival of mice with experimental brain tumors. J. Virol. 2006;80:7308–7315. doi: 10.1128/JVI.00725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broberg E.K., Peltoniemi J., Nygårdas M., Vahlberg T., Röyttä M., Hukkanen V. Spread and replication of and immune response to gamma134.5-negative herpes simplex virus type 1 vectors in BALB/c mice. J. Virol. 2004;78:13139–13152. doi: 10.1128/JVI.78.23.13139-13152.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broberg E.K., Nygårdas M., Salmi A.A., Hukkanen V. Low copy number detection of herpes simplex virus type 1 mRNA and mouse Th1 type cytokine mRNAs by Light Cycler quantitative real-time PCR. J. Virol. Methods. 2003;112:53–65. doi: 10.1016/s0166-0934(03)00191-5. [DOI] [PubMed] [Google Scholar]
- 29.Cassady K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005;79:8707–8715. doi: 10.1128/JVI.79.14.8707-8715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker J.N., Gillespie G.Y., Love C.E., Randall S., Whitley R.J., Markert J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA. 2000;97:2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.