

Abstract
Little is known about the neuronal voltage-gated sodium channels (NaVs) that control neurotransmission in the parasympathetic nervous system. We evaluated the expression of the α subunits of each of the nine NaVs in human, guinea pig, and mouse airway parasympathetic ganglia. We combined this information with a pharmacological analysis of selective NaV blockers on parasympathetic contractions of isolated airway smooth muscle. As would be expected from previous studies, tetrodotoxin potently blocked the parasympathetic responses in the airways of each species. Gene expression analysis showed that that NaV 1.7 was virtually the only tetrodotoxin-sensitive NaV1 gene expressed in guinea pig and human airway parasympathetic ganglia, where mouse ganglia expressed NaV1.1, 1.3, and 1.7. Using selective pharmacological blockers supported the gene expression results, showing that blocking NaV1.7 alone can abolish the responses in guinea pig and human bronchi, but not in mouse airways. To block the responses in mouse airways requires that NaV1.7 along with NaV1.1 and/or NaV1.3 is blocked. These results may suggest novel indications for NaV1.7-blocking drugs, in which there is an overactive parasympathetic drive, such as in asthma. The data also raise the potential concern of antiparasympathetic side effects for systemic NaV1.7 blockers.
Introduction
The pore-forming α subunits of voltage-gated sodium channels comprise nine distinct subtypes referred to as neuronal voltage-gated sodium channel (NaV)1.1–1.9. With respect to the peripheral nervous system, they have been extensively investigated in sensory nerves, but much less so in autonomic nerves, and virtually not at all in the parasympathetic nervous system. The parasympathetic nervous system controls the function of visceral organs. In the respiratory tract, parasympathetic nerves regulate secretions and provide the dominant control over airway smooth muscle tone and airway caliber. Dysregulation of this system can contribute to airway inflammatory diseases such as asthma and chronic obstructive pulmonary disorder (COPD) (Undem and Potenzieri, 2012).
Other than the fact that they are generally blocked by tetrodotoxin (TTX), little is known about the NaV1s involved in parasympathetic neurotransmission in visceral organs. We have addressed this question in this study by focusing on the postganglionic parasympathetic nerves controlling airway caliber. TTX potently blocks NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.6, and NaV1.7. Among these, all but NaV1.4 are expressed by peripheral neurons. Which of these channels are responsible for the TTX-sensitive neuronal cholinergic airway contractions is unknown.
We evaluated the functionality of these channels in airway cholinergic responses using selective NaV1-blocking drugs along with an evaluation of NaV1 gene expression in isolated bronchial-associated parasympathetic ganglia in guinea pigs, mice, and humans. The data support the conclusion that airway parasympathetic cholinergic neurotransmission is entirely dependent on NaV1.7 in humans and guinea pigs, but not mice. This information is significant because NaV1.7 is associated with channelopathies that can lead to a gain or loss of function (Waxman, 2013). In addition, selective NaV1.7 blockers are presently being developed aimed at controlling pain (Priest and Kaczorowski, 2007). That NaV1.7 controls parasympathetic nerve function may provide additional therapeutic targets for NaV1.7 blockers, as well as help explain potential unwanted antiparasympathetic side effects when these drugs are used systemically.
Materials and Methods
The experimental protocols for guinea pig and mouse experiments were approved by the Johns Hopkins University Animals Care and Use Committee.
Tissue Bath Studies
Guinea Pig Trachea.
Male Hartley guinea pigs (100–200 g) were obtained from Hilltop Laboratory Animals (Scottsdale, PA). The animals were killed by CO2 asphyxiation. The trachea was removed and placed in Kreb’s bicarbonate buffer composed of (in mM): 118 sodium chloride, 5.4 KCl, 1.0 NaH2PO4, 1.2 MgSO4, 1.9 CaCl2, 25 NaHCO3, and 11.1 glucose equilibrated with 95% O2–5% CO2 (pH 7.4). After trimming the trachea free of extraneous tissue, it was cut into segments comprising ∼4 cartilagenous rings. The preparations were connected to force-displacement transducers for measurements of isometric tension responses. They were equilibrated under 1 g resting tension for 90 minutes in water-jacketed tissue baths containing Kreb’s buffer maintained at 37°C, with the buffer replaced at 15-minute intervals. Platinum wires, connected to a Grass S48 stimulator, were positioned on either side of the tissue segments. Propranolol (1 µM) and indomethacin (3 µM) were added to the buffer at the beginning of the experiment to reduce the influence of β adrenergic responses and prostaglandins. The guinea pig bronchi were treated with capsaicin (10 µM) 60 minutes before the electrical field stimulation (EFS) studies. Capsaicin results in a strong tachykinin-dependent contraction that returns to baseline in about 30 minutes. This capsaicin desensitization treatment prevents subsequent tachykinergic responses to EFS, leaving a neuronal response that is entirely cholinergic in nature (Undem et al., 1990).
The nerves in the tissue were stimulated using EFS. Rectangular pulses were delivered to the tissue via the platinum wire electrodes from the Grass S48 stimulator after first being processed by a constant current Med-Laboratory Stimul-Splitter (Med-Laboratory Instruments, Fort Collins, CO). The square pulses were of 0.1-ms duration with an amplitude of 12 V. The impulses were delivered at 8 Hz every 1000 seconds. This resulted in rapid and short-lived atropine-sensitive cholinergic contractions. After stable responses were obtained ∼5 minutes, the NaV blocker was added until the reduction in response amplitude reached a steady state, at which time a 10-fold larger dose concentration was added. From data obtained, a cumulative concentration response curve was obtained. At the end of the experiment, the tissue was maximally contracted with 100 µM carbamylcholine; the EFS contractions were normalized to this maximum response.
The NaV blockers did not inhibit the contractions to the exogenously applied cholinergic agonist carbamylcholine (100 µM); the contractions averaged 3.6 ± 0.7 g, 5.4 ± 3 g and 4.3 ± 0.9 g, and 5.7 ± 1.2 g in vehicle (n = 9)-, TTX (1 µM, n = 6)-, compound 801 (1 µM, n = 8)-, and compound 13 (10 µM, n = 7)-treated tissues, respectively (P > 0.1 analysis of variance). In two experiments, we evaluated the concentration-response curve of carbamycholine-induced contractions in vehicle-, TTX-, compound 801-, and compound 13-treated tissues and found them to be nearly superimposable (EC5O values averaged 48 nM, 32 nM, 28 nM, and 49 nM, respectively).
Mouse Trachea.
Mouse tracheal rings, two per animal, were prepared exactly as previously described (Weigand et al., 2009). Indomethacin (3 µM) and propranolol (1 µM) were included in Kreb’s bicarbonate buffer. The field stimulation protocol was the same as described above for guinea pig bronchi, except there was a 5-minute interval between two consecutive 8 Hz stimulations (in preliminary studies, we noted that without such an interval there was a gradual rundown of the cholinergic responses over time).
Human Bronchi.
Lungs were obtained from human organ donors through the International Institute for the Advancement of Medicine program (Exton, PA). The tissue was immediately placed in chilled Lactated Ringers solution and shipped overnight, arriving in our laboratory 12–24 hours later. Once received, the lungs were placed in 4 L chilled oxygenated Kreb’s bicarbonate buffer solution (see above). Central bronchi (3–5 mm inner diameter) were dissected free, trimmed of extraneous tissue, and prepared for EFS-induced isometric study, as described for the guinea pig bronchi and in more detail in this work (Ellis and Undem, 1992).
Analysis of NaV1 Subunit Expression in Parasympathetic Ganglion Neurons.
Parasympathetic ganglia.
The animals were killed with CO2 followed by exsanguination, and the trachea, main bronchi, and lungs with attached vagus nerves were dissected in O2/CO2-gassed Krebs solution and pinned dorsal side up under stereomicroscope. Parasympathetic ganglia were identified along vagal branches, dissected and collected with the tip of sharp glass electrode (pulled with Sutter P-2000 puller), transferred into polymerase chain reaction tube containing RNAse inhibitor (1 μl RNaseOUT; Life Technologies, Carlsbad, CA), frozen on dry ice, and stored at −80°C. A sample of nonganglion tissue of similar area (∼400 µm × 400 µm) and depth of juxtaposed to each ganglion was collected in an identical manner as a negative control. This negative control tissue would be expected to contain mainly adipocytes, some axons, fibroblasts, microvasculature, but no neurons or satellite cells. First-strand cDNA was synthesized using the Super-Script III CellsDirect cDNA Synthesis System (Life Technologies, Carlsbad, CA), according to the manufacturer’s recommendations. Samples were defrosted, lysed (10 minutes, 75°C), and treated with DNase I, poly(dT), and random hexamer primers (Roche Applied Bioscience Indianapolis, IN) added and reverse transcribed by SuperscriptIII RT for cDNA synthesis. A total of 2 µl each sample was used for polymerase chain reaction amplification by the HotStar Taq Polymerase Kit (Qiagen, Germantown, MD), according to the manufacturer's recommendations, in a final volume of 20 µl. After an initial activation step of 95°C for 15 minutes, cDNAs were amplified with custom-synthesized primers (Life Technologies, Carlsbad, CA) by 50 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 1 minute, followed by a final extension at 72°C for 10 minutes. Products were visualized in ethidium bromide–stained 1.5% agarose gels with a 50-bp or 100-bp DNA ladder. Figures were prepared from multiple original gel images by using Microsoft PowerPoint and Apple Preview. The bands indicate only the presence or absence of a product (i.e., target expression), but not the intensity of expression.
Intron-spanning primers specific for each guinea pig, human, and mouse NaV α subunit were designed based on Pubmed sequences with aid of UCSC (University of California, Santa Cruz) Genome Browser using Primer3 (v.0.4.0) program (Tables 1, 2, 3) (Rozen and Skaletsky, 2000). The selectivity of each primer was evaluated by aligning all NaV subunits with the primer using ClustalW. Positive control RNA was selected based on established NaV subunit expression pattern: whole brain RNA for NaV1.1, 1.2, 1.3, and 1.6; skeletal muscle RNA for NaV1.4; heart muscle RNA for NaV1.5; and dorsal root ganglia (DRG) or vagal sensory ganglia RNA for NaV1.7, 1.8, and 1.9. The guinea pig and mouse RNA was isolated by using RNAeasy mini kit (Qiagen). Human RNA was purchased from Clontech (Mountain View, CA).
TABLE 1.
The guinea pig primer sequences for single-cell reverse-transcription polymerase chain reaction
| Target | Primer | Sequence (5′ to 3′) | Product Size (bp) | Genomic Size (bp) | NCBI Reference Sequence |
|---|---|---|---|---|---|
| β-actin | Forward | TCTCTTCCAGCCCTCCTTC | 373 | N/Aa | NT_176389.1 |
| Reverse | GTCCTCAAAGGTGCTGTGCT | ||||
| NaV1.1 | Forward | AAGGGTTTCGCTTCTCCATT | 206 | >1000 | XM_003478553.2 |
| Reverse | GTCCTCAAAGGTGCTGTGCT | ||||
| NaV1.2 | Forward | GAGACTTCAGTGGTGCTGGTG | 265 | N/A | XM_003478556.2 |
| Reverse | AGCAGAGACTGGTGTGGAGAA | ||||
| NaV1.3 | Forward | ACACAACAGAGGAGAGGCAGA | 171 | >1000 | XM_003478666.2 |
| Reverse | CGTCTTGGGGAGAATAGGG | ||||
| NaV1.4 | Forward | GGCTCTCCCTCCACCATC | 159 | N/A | XM_003465929.1 |
| Reverse | CAGCGGTTTCTTGCCATC | ||||
| NaV1.5 | Forward | GTCATTTTCCTGGGCTCCTT | 245 | >1000 | XM_005001107.1 |
| Reverse | TTGCTCCTTCTCTCGTGGTT | ||||
| NaV1.6 | Forward | CCTCCTATGGACGAAAAGACA | 222 | >1000 | XM_005006531.1 |
| Reverse | TGCAGATGGTGATAGCCAAG | ||||
| NaV1.7 | Forward | GAGGAAAAGGGAGATGATGAGA | 158 | >1000 | XM_003478661.2 |
| Reverse | AACAGGGAGCCACGAATG | ||||
| NaV1.8 | Forward | CGGAAAGGTGACAATGGAG | 187 | >1000 | XM_003464141.2 |
| Reverse | AGCAGGGACAGTAGCGAAGA | ||||
| NaV1.9 | Forward | GTTCCAGGTTCCCAAATCAA | 162 | >1000 | XM_003464142.2 |
| Reverse | GAGGCAGAAGAGGGTGAGG |
NCBI, National Center for Biotechnology Information.
N/A indicates no match in UCSC (University of California, Santa Cruz) Genome Browser in silico polymerase chain reaction.
TABLE 2.
The human primer sequences for single-cell reverse-transcription polymerase chain reaction
| Target | Primer | Sequence (5′ to 3′) | Product Size (bp) | Genomic Size (bp) | NCBI Reference Sequence |
|---|---|---|---|---|---|
| β-actin | Forward | AGAAAATCTGGCACCACACC | 188 | 629 | NM_001101.3 |
| Reverse | AGAGGCGTACAGGGATAGCA | ||||
| NaV1.1 | Forward | TCAGTTCCTACATCGCCTGTT | 174 | >1000 | NM_001165963.1 |
| Reverse | ACTCATTGCTCGTTGCCTTT | ||||
| NaV1.2 | Forward | GTGCTGGTGGGATAGGAGTT | 288 | N/Aa | NM_021007.2 |
| Reverse | TGCGTCTTGGAGAGAAAAGG | ||||
| NaV1.3 | Forward | GGAACCGAAGGAAGAAAAGAA | 252 | N/A | NM_006922.3 |
| Reverse | CCCGACCTCTGAAACTGAAA | ||||
| NaV1.4 | Forward | TCAACAACCCCTACCTGACC | 208 | >1000 | NM_000334.4 |
| Reverse | TTCTCCTCTGCCTGCTCCT | ||||
| NaV1.5 | Forward | CTAAAGGCAGGCGAGAACC | 188 | 708 | NM_198056.2 |
| Reverse | AGGTAGAAGGACCCCAGGAA | ||||
| NaV1.6 | Forward | CGTCTTGGTCATCTTTGTGG | 242 | >1000 | NM_014191.3 |
| Reverse | CCCCTCCTTCTTCACCTTCT | ||||
| NaV1.7 | Forward | TGGAGAGGAAAAGGGAGATG | 166 | >1000 | NM_002977.3 |
| Reverse | AGAAAACAAGGAGCCACGAA | ||||
| NaV1.8 | Forward | TAAGCGAGGCACTTCTGACC | 144 | >1000 | NM_006514.3 |
| Reverse | GAGAGGAAAGCCCAAGCAA | ||||
| NaV1.9 | Forward | GGCAAGAGGTTTCATTCTGG | 125 | >1000 | NM_014139.2 |
| Reverse | GGGGCAATAGTTTGATGGTG |
NCBI, National Center for Biotechnology Information.
N/A indicates no match in UCSC (University of California, Santa Cruz) Genome Browser in silico polymerase chain reaction.
TABLE 3.
The mouse primer sequences for single-cell reverse-transcription polymerase chain reaction
| Target | Primer | Sequence (5′ to 3′) | Product Size (bp) | Genomic Size (bp) | NCBI Reference Sequence |
|---|---|---|---|---|---|
| β-actin | Forward | GTGGGAATGGGTCAGAAGG | 302 | 756 | NM_007393.3 |
| Reverse | GAGGCATACAGGGACAGCA | ||||
| NaV1.1 | Forward | AGACAGCATCAGGAGGAAGG | 118 | >1000 | NM_018733.2 |
| Reverse | GGAGAACAGGGAACCACGA | ||||
| NaV1.2 | Forward | TTTTCGGCTCATTCTTCACA | 305 | >1000 | NM_001099298.2 |
| Reverse | CATCTCTTGGCTCTGGTCGT | ||||
| NaV1.3 | Forward | AGACAGAGGGAGCACTTGGA | 200 | >1000 | NM_018732.3 |
| Reverse | CTATTGCGTCTTGGGGAAAA | ||||
| NaV1.4 | Forward | TCATCTTCCTGGGTTCCTTC | 206 | 941 | NM_133199.2 |
| Reverse | ATCTGCCTCCTCTCCACCTT | ||||
| NaV1.5 | Forward | TGGGCTCCTTCTACCTTGTG | 247 | >1000 | NM_021544.4 |
| Reverse | CGTTTCCTCCTCTTGCTCCT | ||||
| NaV1.6 | Forward | AGGCAGCAAAGACAAACTGG | 157 | N/A | NM_001077499.2 |
| Reverse | GCAGCACTTGAACCTCTGG | ||||
| NaV1.7 | Forward | ATGCTCTTCTTTGCGGTTTC | 381 | >1000 | NM_001290674.1 |
| Reverse | CGGCTTCTTCCTGCTCTTTT | ||||
| NaV1.8 | Forward | CAATCCGACCCTTACAACCA | 147 | >1000 | NM_001205321.1 |
| Reverse | AAAGACCCCGTCATCCAAG | ||||
| NaV1.9 | Forward | CAGCTTTGGCTGGTCTTTTC | 228 | >1000 | NM_011887.3 |
| Reverse | TTCTCCTTGGCCTCTGTCTC |
NCBI, National Center for Biotechnology Information.
N/A indicates no match in UCSC (University of California, Santa Cruz) Genome Browser in silico polymerase chain reaction.
Electrophysiology.
Human NaV (hNaV)1.1, hNaV1.2, hNaV1.3, hNaV1.6, and hNaV1.7 stably expressed in HEK cells were obtained from Millipore, and cultured according to manufacturer’s specifications. Mouse NaV1.7, rat NaV1.7, and guinea pig NaV1.7 HEK cell lines were generated at Lilly Laboratories. The media consisted of the following: Dulbecco’s modified Eagle’s medium (high glucose), fetal bovine serum (10%) (heat inactivated), HEPES (20 mM), Glutamax (2 mM), Pen/Strep (1%), and G418 (0.3 mg/ml) (all obtained from Gibco, Gaithersburg, MD).
Whole-cell voltage clamp experiments were performed at room temperature, using a Multiclamp 700B amplifier, Digidata 1440A interface, and pClamp 10.0 software to obtain IC50 for the NaV blockers. For experiments using human NaV1.7 and mouse NaV1.7, the external solution contained the following (mM): 40 NaCl, 100 choline-Cl, 5.4 KCl, 1.8 CaCl2, 0.8 MgCl2, 10 HEPES, and 5 glucose, pH 7.4, adjusted with N-methyl d-glutamine (osmolarity, 300 mOsm). For experiments using hNav1.6 and mouse Nav1.6, the external solution contained the following (mM): 138 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgCl2, 10 HEPES, and 5 glucose, pH 7.4, adjusted with NaOH (osmolarity, 300 mOsm). Micropipettes were pulled from borosilicate glasses with a Sutter P-97 puller. Pipette solutions contained the following (mM): 110 CsF, 20 CsCl, 10 NaCl, 1.1 EGTA, and 10 HEPES, pH 7.2, adjusted with CsOH (osmolarity, 294 mOsm). Electrodes had a resistance of 1.5–2 MΩ when filled with pipette solution. After seal rupture, series resistance (<5 MΩ) was compensated (70–80%) and periodically monitored. Test compounds are diluted in dimethylsulfoxide (DMSO) at final stock concentration of 10 mM. Working dilutions were also done using DMSO, and the final concentration was tested at a 0.1% DMSO concentration. A steady-state inactivation curve is run at a holding potential of −130 mV using a 10-second prepulse ranging from −130 mV to −30 mV in 10 mV steps, followed by a test pulse to −10 mV for 10 ms at 0.06 Hz intervals. The data are then fit to a Boltzman Sigmoidal to calculate the V0.5 using Prism and the following formula:
where Y= current measured at the test pulse and X = prepulse potential.
The steady-state inactivation curve was repeated every 2 minutes until peak current baselines were stable. The holding potential is then changed to a potential near the calculated V0.5 and held for 1 minute. The protocol is then run from the new holding potential and then stepped to −10 mV for 10 ms at 0.1 Hz intervals. After a stable baseline is achieved, the test compound is superfused onto the cell until a new equilibrium is established. Percent inhibition is determined as [(control − drug)/control] × 100. Compound potencies are calculated with a logistic fit using Prism 6.03 (GraphPad) with formula: Y = bottom + (top − bottom)/(1 + 10^[(LogEC50 − X) × HillSlope)], where X is the logarithm of the test concentration and Y is the percent inhibition.
Compound 13 selectivity was assessed at Essen Bioscience (Ann Arbor, MI) using human Nav1.1, Nav1.2, hNav1.4, hNav1.5, hNav1.6, and hNav1.7 transfected in HEK cells and Ion Works Barracuda as electrophysiology platform using Population Patch Clamp mode. Compound potency was assessed by using functional selectivity protocols consisting in the application of different trains of depolarizations that cycle channels through states that are most representative of pathology for hNav 1.7 and physiology for the rest. Voltage protocols follow the same time scheduled, being applied once in the absence of compound, then in the presence of compound, 5 minutes after addition, and then five more times every 1 minute.
The external recording solution comprised the following (in mM): 137 NaCl, 4 KCl, 10 HEPES, 1 MgCl2, and 1.8 CaCl2, pH 7.3, with NaOH. The internal solution comprised the following (in mM): 90 K-gluconate, 40 CsCl, 10 NaCl, 3.2 EGTA, 5 HEPES, and 3.2 MgCl2, pH 7.3, with KOH. All buffer constituents were obtained from Sigma-Aldrich (St. Louis, MO). Electrical access was achieved using amphotericin (120 µg ml−1) in the internal solution to obtain the perforated patch-clamp configuration.
Compounds were solubilized in DMSO containing 6% pluronic F127 (Sigma-Aldrich) as 10 mM stock solutions. An 11-point 1:3 cross-plate dilution was carried out in DMSO (6% pluronic), diluted 1/300 in external solution, and then added to assay plate using dual addition mode from mid position without mixing to reduce nonspecific binding and improve compound exposure.
Peak inward currents were determined from the first and last depolarizing pulses. Percentage inhibition values were calculated according to the following equation, using peak currents obtained in the absence (pre) and presence (post) of test compound (last train): concentration response curves were fitted to a four-parameter logistic fit. Data were analyzed using IonWorks Barracuda software (v2.5.0.280), Microsoft Excel (v7.0), XLFit (IDBS, v5.2.0.0), and GraphPad Prism (v5).
Drugs and Solutions
Drugs were prepared fresh as 0.1 mM stock solutions. Compound 801 was identified in Icagen/Pfizer patent WO2010079443 and synthesized at Lilly Laboratories (Indianapolis, IN) and dissolved in DMSO.
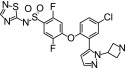
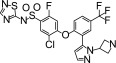
Compound 13 was first described in WO2012004706. It was synthesized at Almirall Laboratories (Barcelona, Spain) and dissolved in DMSO.
ICA121431 (Tocris Minneapolis, MN) and PF-01247324 (Sigma-Aldrich, St. Louis, MO) were dissolved in DMSO. Tetrodotoxin (Alamone Laboratories Jerusalem, Israel) and propranolol (Sigma-Aldrich) were dissolved in water; indomethacin (Sigma-Aldrich) was prepared in ethanol.
Results
In the patch-clamp analysis of various NaV1 currents, TTX inhibits NaV 1.1, 1.2, 1.3, 1.6, and 1.7 with similar potency with IC50 values in the 1–10 nM range. ICA-121431, by contrast, potently blocks NaV1.1, 1.2, and 1.3, but has little effect on 1.6 or 1.7. We noted, for example, that the guinea pig NaV1.7 current was inhibited less than 20% even at 10 µM ICA-121431. Compound 801 and compound 13 are arylsulfonamides closely related to other selective NaV1.7 blockers (Alexandrou et al., 2016). We found compound 801 to be a potent NaV1.7 blocker, being five- to 10-fold more potent at this channel than NaV1.2 and NaV1.6. Compound 801 was equally potent against human and guinea pig NaV1.7. Compound 13 was selective for blocking NaV1.7 with little affinity for NaV1.2, 1.3, or 1.6, and was found to be more potent at human versus guinea pig NaV1.7 (these data are summarized in Table 4).
TABLE 4.
Estimated IC50 values (nM) for NaV1 blockers at human NaV1 channels
| TTXa | Cmpd 801b | Cmpd 13c | ICA121431d | |
|---|---|---|---|---|
| NaV1.1 | 6 | 8 | nd | 23 |
| NaV1.2 | 12 | 6 | 900 | 240 |
| NaV1.3 | 4 | 460 | >30,000 | 13 |
| NaV1.6 | 1.6 | 8 | 6900 | 13,000 |
| NaV1.7 | 6 | 1 | 2 | 12,000 |
| NaV1.7 (GP) | 2.7 | 4.4 | 16.9 | >10,000 |
All values are for human NaV1 channels, except for the guinea pig (GP) NaV1.7 values. The values for TTX were obtained from (Catterall et al., 2005), except for the the NaV1.7, which were obtained here, as described in Materials and Methods.
The values for compound 801 were obtained, as described in Materials and Methods.
The values for compound 13 were obtained by Ion Works and provided by Dr. Silvia Fonquerna at Almirall Laboratories.
The values for ICA121431 were obtained from (McCormack et al., 2013).
nd - not determined
GP- guinea pig
Guinea Pig Bronchi.
TTX, compound 801, and compound 13 each effectively blocked the neuronally evoked cholinergic contractions of the guinea pig bronchi with IC50 of 8.4, 56, and 480 nM, respectively. In contrast, ICA121431 at concentrations that would be expected to fully block NaV1.1, 1.2, and 1.3 was virtually devoid of an inhibitory effect on the cholinergic contractions. These data are illustrated in Fig. 1. As expected, PF-01247324, a drug that selectively blocks the TTX-resistant NaV 1.8 channels, had no effect on the cholinergic contractions (34 ± 3% versus 35 ± 3% in the absence and presence of 1 µM drug, respectively; n = 6, P > 0.1) (Payne et al., 2015).
Fig. 1.

(A–E) Effect of NaV inhibitors on airway-efferent parasympathetic nerves in the guinea pig. (A) Representative trace of the effect of TTX on bronchial contractions evoked by activation of parasympathetic nerves with EFS (16 Hz for 10 seconds, repeated every 60 seconds, denoted by bullets). Effect of (B) TTX, (C) compound 801, (D) compound 13, and (E) ICA121431 on EFS-induced bronchial contractions, n = 6. (F and G) Collection of parasympathetic neurons for reverse-transcription polymerase chain reaction. (F) Parasympathetic ganglion in the guinea pig airway. The highlighted area is magnified in (G). Some of the parasympathetic neurons are denoted by asterisks. The ganglion was dissected and collected into polymerase chain reaction tube. A sample from the neighboring tissue was collected as a negative control. (H and I) Expression of NaV1.1–1.9 subunits in parasympathetic neurons in the guinea pig. (H) Representative polymerase chain reaction gel. G1–G6, individual parasympathetic ganglia; N1–N6, corresponding negative controls. The arrows indicate expected product size. Note: NaV1.4 in G6 sample is counted as negative because of incorrect size. (I) Percentage of NaV subunit expression in parasympathetic ganglia (n = 11).
NaV1 gene expression was detected in bronchial ganglia (n = 11), but not in the juxtaposed non-neuronal tissue. NaV1.7 was the only TTX-sensitive channel mRNA expressed in most guinea pig airway-associated parasympathetic ganglia. NaV1.7 was expressed in 10 of 11 isolated ganglia. The only other TTX-sensitive channel expressed was NaV1.3, and this was expressed in only 2 of 11 ganglia. Interestingly, NaV1.9 was also expressed in 8 of 11 ganglia. These data are shown in Fig. 1. Inasmuch as NaV1.9 is a TTX-resistant channel, these data support the hypothesis based on our functional studies that NaV1.7 provides the functional control over action potential discharge in postganglionic parasympathetic nerves in the guinea pig airways.
Mouse Trachea.
TTX potently (21 nM IC50) blocked neuronally evoked cholinergic contractions of the mouse trachea. In contrast to guinea pig airways, none of the selective NaV1 blockers when studied alone inhibited the cholinergic responses. However, in the presence of NaV1.1, 1.2, and 1.3 blockade with ICA-121431 (10 µM), either compound 13 or compound 801 was able to abolish the contractions in a concentration-dependent fashion. Thus, to block parasympathetic responses in the mouse airway, both NaV1.7 and NaV1.1, 1.2 and/or NaV1.3 had to also be blocked. These data are illustrated in Fig. 2.
Fig. 2.

(A–F) Effect of NaV inhibitors on airway-efferent parasympathetic nerves in the mouse. The effect of (A) TTX, (B) compound 801, (C) compound 13, (D) ICA121431, and ICA121431 (10 µM) in combination with (E) compound 801 and (F) compound 13 on the EFS-induced bronchial contractions, n = 6. (G) Expression of NaV1.1–1.9 subunits in parasympathetic neurons in the mouse. G1–G5, individual ganglia; +, positive control; −, negative control. The arrows indicate expected product size.
We evaluated NaV1 gene expression in five ganglia isolated from mouse airways. Consistent with the functional studies, the NaV1 expression was more extensive than in guinea pig ganglia. With respect to TTX-sensitive channels, NaV1.1, NaV1.3, NaV1.6, and NaV1.7 were expressed by neurons in 100%, 80%, 40%, and 60% of the ganglia, respectively. With respect to TTX-resistant channels, NaV1.9 was expressed in three of five ganglia (Fig. 2).
Human Bronchi.
The large differences in the NaV1-regulating airway parasympathetic responses between guinea pig and mice beg the question as to which laboratory animal is the more translatable to the human condition. Like the guinea pig and mouse, the neuronally-evoked cholinergic contractions were potently and effectively blocked by TTX. Blocking NaV1.1, 1.2, and 1.3 with ICA-121431 had no effect on the cholinergic responses, whereas either compound 801 or compound 13 mimicked the TTX response (Fig. 3). Consistent with our potency predictions obtained with patch-clamp electrophysiological studies of human versus guinea pig NaV1.7 in heterologous systems (table 1), compound 13 was more potent in blocking the parasympathetic contractions in human bronchi than guinea pig bronchi (Figs. 1 and 3).
Fig. 3.

(A–E) Effect of NaV inhibitors on airway-efferent parasympathetic nerves in the human. (A) Representative trace of the effect of TTX on bronchial contractions evoked by activation of parasympathetic nerves with EFS (16 Hz for 10 seconds, repeated every 60 seconds, denoted by bullets). Effect of (B) TTX, (C) compound 801, (D) compound 13, and (E) ICA121431 on EFS-induced bronchial contractions, n = 6. (F) An example of human parasympathetic ganglion (arrow). (G) Magnified area from the ganglion shown in (F). Some of the parasympathetic neurons are denoted by asterisks. (H) Expression of NaV1.1–1.9 subunits in human isolated parasympathetic ganglia. G1–G6, individual ganglia; +, positive control. The arrows indicate expected product size.
We evaluated the NaV1 gene expression in five ganglia isolated from the bronchi of three human donors. NaV1.7 mRNA was expressed in every parasympathetic ganglia, whereas other TTX-sensitive NaV1s were largely unexpressed, with the exception of NaV1.3, which was found in only one of five ganglia (Fig. 3).
Discussion
The functional and mRNA expression data support the hypothesis that neurotransmission in parasympathetic postganglionic neurons innervating smooth muscle in human and guinea pig airways is under the control of NaV1.7. In contrast, in the mouse, the most commonly employed laboratory animal for investigations into both NaV1 biology and respiratory physiology, the parasympathetic neurons were under redundant control of NaV1.7 and 1.1 and/or 1.3. One must therefore be cautious when drawing NaV1-related conclusions about human autonomic nerves from physiologic studies carried out exclusively in mice.
NaV1.9 was expressed along with NaV1.7, especially in guinea pig parasympathetic neurons. That TTX and the selective NaV1.7 blockers could abolish the parasympathetic response indicated that NaV1.9 is not sufficient for driving action potential discharge and acetylcholine release from the terminals. This is as would be expected based on its biophysical properties, but it may play a role in regulating the electrical excitability of the neurons.
The potency of NaV blockers in blockers in isolated tissues is generally less than that seen in patch-clamp electrophysiological studies. There are several potential reasons for this. First, the concentration of the inhibitor in the tissue bath solution may not reflect that concentration in the biophase of the nerve endings. To minimize this issue, we incubated the blocking drug for a period sufficient such that the inhibitory effect reached a steady state. Secondly, the potency of NaV blockers, including aryl sulfonamide blockers such as compounds 801 and 13, can be dramatically influenced by the biophysical state of the channel (Alexandrou et al., 2016). The state of the channel (e.g., relative extent of inactivation) can be specifically manipulated in patch-clamp conditions, but remains unknown in channels inserted within nerve membranes in tissues. Finally, the β subunit composition may potentially influence the pharmacology of the NaV blockers (Wilson et al., 2015), and this may differ in channels expressed in heterologous system versus channels in the parasympathetic nerves within tissues. In the present study, it is noteworthy that the potency of the selective NaV1.7 blockers relative to TTX was similar in the tissue assay and patch-clamp analysis.
Alterations in both the sensory and parasympathetic nervous systems most likely contribute to airway pathologies such as asthma and COPD (reviewed in Undem and Potenzieri, 2012; Mazzone and Undem, 2016). That NaV1.7 is the dominant controller of neurotransmission in human and guinea pig airway parasympathetic neurons provides for a strategy of limiting parasympathetic tone in the respiratory tract via selective NaV1.7 blockade. Topically administered antimuscarinic drugs are presently used to decrease cholinergic tone in the airways of those suffering from asthma or COPD. Blocking NaV1.7 is an orthogonal approach to diminish cholinergic contractions and may have the added benefit of not only blocking the release of acetylcholine but also other parasympathetic transmitters, including, for example, vasoactive intestinal peptide and nitric oxide that provide promucus secretory activities (Baker et al., 1985; Wine, 2007). The vasoactive intestinal peptide/nitric oxide–containing postganglionic nerves in the guinea pig airways, however, are derived from parasympathetic neurons distinct from the cholinergic neurons (Canning and Undem, 1993), and therefore may reveal distinct NaV1 channel expression. The airway parasympathetic ganglia are mainly associated with larger central airways. The neurons within these ganglia then project postganglionic nerves to the smooth muscle of bronchi and bronchioles (Undem and Potenzieri, 2012). Another speculative benefit therefore of topically applied (inhaled) NaV-blocking drugs over muscarinic receptor antagonists is that even central deposition of a NaV blocker my lead to bronchodilation of more peripheral bronchi and bronchioles. Finally, we have recently reported that blocking NaV1.7 can inhibit action potential conduction along sensory C-fibers in the respiratory tract, leading to inhibition of undesirable sensations such as dyspnea and urge to cough, an effect that antimuscarinic drugs would not share (Muroi et al., 2011, 2013). It should be kept in mind that the present study focused on airways from healthy laboratory animals and donors that did not have asthma or COPD; both the expression and function of NaV channels may potentially be altered in the diseased state (Chahine and O'Leary, 2014).
There have been intensive efforts in developing selective NaV1.7 blockers for the treatment of pain (Dib-Hajj et al., 2013). As far as we know, this is the first study on the NaV1 expression and function in parasympathetic neurons. If our observations in the respiratory tract hold true for other visceral organs, it may suggest new indications for NaV1.7 blockers. The findings may also explain potential antiparasympathetic side effects for systemically acting NaV1.7 blockers.
Individuals with loss of function and gain of mutations in NaV1.7 have been identified (Dib-Hajj et al., 2005; Goldberg et al., 2007). These people suffer from a congenital insensitivity to pain or erythomelalgia, respectively. Loss-of-function mutation also led to a decreased sensitivity of smell (Weiss et al., 2011). Based on the present data, one might predict that those with loss of function mutations may experience less reflex bronchial contractions and secretions, and perhaps less airway reactive disease such as asthma, but this hypothesis has yet to be addressed.
Abbreviations
- COPD
chronic obstructive pulmonary disorder
- DMSO
dimethylsulfoxide
- EFS
electrical field stimulation
- hNaV
human NaV
- NaV
neuronal voltage-gated sodium channel
- TTX
tetrodotoxin
Authorship Contributions
Participation in research design: Kocmalova, Kollarik, Canning, Undem.
Conducted experiments: Kocmalova, Kollarik, Ru, Herbstromer, Meeker, Fonquerna, Aparici, Miralpeix, Chi, Li, Wilenkin, McDermott, Krajewski.
Contributed new reagents or analytic tools: Krajewski Nisenbaum, Fonquerna, Miralpeix.
Performed data analysis: Kocmalova, Kollarik, Undem, Krajewski, Nisenbaum, Aparici, Miralpeix.
Wrote or contributed to the writing of the manuscript: Kocmalova, Kollarik, Krajewski, Undem.
Footnotes
This work was supported by National Institutes of Health Heart and Blood Institute [Grant R01HL122228] and Biomedical Center Martin, Slovakia [Grant ITMS 26220220187].
References
- Alexandrou AJ, Brown AR, Chapman ML, Estacion M, Turner J, Mis MA, Wilbrey A, Payne EC, Gutteridge A, Cox PJ, et al. (2016) Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS One 11:e0152405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker B, Peatfield AC, Richardson PS. (1985) Nervous control of mucin secretion into human bronchi. J Physiol 365:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning BJ, Undem BJ. (1993) Evidence that distinct neural pathways mediate parasympathetic contractions and relaxations of guinea-pig trachealis. J Physiol 471:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. (2005) International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 57:397–409. [DOI] [PubMed] [Google Scholar]
- Chahine M, O’Leary ME. (2014) Regulation/Modulation of sensory neuron sodium channels. Handb Exp Pharmacol 221:111–135. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, Marshall L, Waxman SG. (2005) Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128:1847–1854. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG. (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14:49–62. [DOI] [PubMed] [Google Scholar]
- Ellis JL, Undem BJ. (1992) Antigen-induced enhancement of noncholinergic contractile responses to vagus nerve and electrical field stimulation in guinea pig isolated trachea. J Pharmacol Exp Ther 262:646–653. [PubMed] [Google Scholar]
- Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, Fraser R, Young C, Hossain S, Pape T, et al. (2007) Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet 71:311–319. [DOI] [PubMed] [Google Scholar]
- Mazzone SB, Undem BJ. (2016) Vagal afferent innervation of the airways in health and disease. Physiol Rev 96:975–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D, et al. (2013) Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci USA 110:E2724–E2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroi Y, Ru F, Chou YL, Carr MJ, Undem BJ, Canning BJ. (2013) Selective inhibition of vagal afferent nerve pathways regulating cough using Nav 1.7 shRNA silencing in guinea pig nodose ganglia. Am J Physiol Regul Integr Comp Physiol 304:R1017–R1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroi Y, Ru F, Kollarik M, Canning BJ, Hughes SA, Walsh S, Sigg M, Carr MJ, Undem BJ. (2011) Selective silencing of Na(V)1.7 decreases excitability and conduction in vagal sensory neurons. J Physiol 589:5663–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne CE, Brown AR, Theile JW, Loucif AJ, Alexandrou AJ, Fuller MD, Mahoney JH, Antonio BM, Gerlach AC, Printzenhoff DM, et al. (2015) A novel selective and orally bioavailable Nav 1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br J Pharmacol 172:2654–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest BT, Kaczorowski GJ. (2007) Subtype-selective sodium channel blockers promise a new era of pain research. Proc Natl Acad Sci USA 104:8205–8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky J. (2000) Primer3 on the WWW for general users and for biologist programmers, in Bioinformatics Methods and Protocols: Methods in Molecular Biology (Krawetz S, Misener S. eds) pp 365–386, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Myers AC, Barthlow H, Weinreich D. (1990) Vagal innervation of guinea pig bronchial smooth muscle. J Appl Physiol (1985) 69:1336-1346. [DOI] [PubMed] [Google Scholar]
- Undem BJ, Potenzieri C. (2012) Autonomic neural control of intrathoracic airways. Compr Physiol 2:1241–1267. [DOI] [PubMed] [Google Scholar]
- Waxman SG. (2013) Painful Na-channelopathies: an expanding universe. Trends Mol Med 19:406–409. [DOI] [PubMed] [Google Scholar]
- Weigand LAMA, Myers AC, Meeker S, Undem BJ. (2009) Mast cell-cholinergic nerve interaction in mouse airways. J Physiol 587:3355–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J, Pyrski M, Jacobi E, Bufe B, Willnecker V, Schick B, Zizzari P, Gossage SJ, Greer CA, Leinders-Zufall T, et al. (2011) Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature 472:186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MJ, Zhang MM, Gajewiak J, Azam L, Rivier JE, Olivera BM, Yoshikami D. (2015) Α- and β-subunit composition of voltage-gated sodium channels investigated with μ-conotoxins and the recently discovered μO§-conotoxin GVIIJ. J Neurophysiol 113:2289–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine JJ. (2007) Parasympathetic control of airway submucosal glands: central reflexes and the airway intrinsic nervous system. Auton Neurosci 133:35–54. [DOI] [PMC free article] [PubMed] [Google Scholar]