

Abstract
G protein-coupled receptors (GPCRs) respond to extracellular stimuli and interact with several intracellular binding partners to elicit cellular responses, including heterotrimeric G proteins. Recent structural and biophysical studies have highlighted the dynamic nature of GPCRs and G proteins and have identified specific conformational changes important for receptor-mediated nucleotide exchange on Gα. While domain separation within Gα is necessary for GDP release, opening the inter-domain interface is insufficient to stimulate nucleotide exchange. Rather, an activated receptor promotes GDP release by allosterically disrupting the nucleotide-binding site via interactions with the Gα N- and C-termini. Highlighting the allosteric nature of GPCRs, recent studies suggest that agonist binding alone poorly stabilizes an active conformation of several receptors. Rather, full stabilization of the receptor in an active state requires formation of the agonist-receptor-G protein ternary complex. In turn, nucleotide-free Gα is able to stabilize conformational changes around the receptor’s agonist-binding site to enhance agonist affinity.
G protein-coupled receptors (GPCRs) are an important family of cell surface receptors that respond to an array of chemically diverse ligands and transduce extracellular signals into intracellular responses [1]. Due to the heavy involvement of GPCRs in regulating physiological processes, these receptors are of great therapeutic importance and therefore are targeted by ~30% of currently-marketed pharmaceutical drugs [2]. Our understanding of GPCRs has progressed from the early view of receptors as binary on-off switches to the current appreciation that GPCRs are dynamic proteins able to sample multiple conformational states. This conformational plasticity allows GPCRs to interact with multiple signaling partners to produce spatially and temporally textured signals [3]. Recent structural studies coupled with biophysical measurements have enhanced our knowledge of the interactions between receptors and G proteins, GPCR kinases (GRKs), and arrestins. In this review, we will focus on the “classical” signaling output of GPCRs: activation of heterotrimeric G proteins.
Although the nature of the GPCR-activating stimulus can vary greatly (e.g. photons, ions, small-molecule hormones/neurotransmitters, lipids, peptides, etc.), canonical signaling by GPCRs proceeds by a similar mechanism. Activation of a GPCR promotes its association with a heterotrimeric G protein, which is composed of a Gα subunit and an obligate Gβγ subunit dimer. In its inactive state, Gα is bound to a molecule of GDP. Interaction with an activated GPCR promotes nucleotide exchange on Gα by accelerating the release of bound GDP, the rate-limiting step in G protein activation. The nucleotide-binding site is quickly occupied by a molecule of GTP, a reaction driven by the high intracellular concentration of GTP (~200–300 μM) [4]. GTP binding leads to conformational changes in Gα, promoting functional dissociation of the Gα and Gβγ subunits, allowing each to modulate the activity of specific effector proteins. Gα proteins are able to interact with partners such as adenylyl cyclase, phospholipase C, or RhoGEFs, in turn altering the activity of multiple downstream target proteins. Similarly, Gβγ subunits can serve to recruit proteins to the plasma membrane, such as G protein-coupled receptor kinases (GRKs), and also can directly modulate the activity of ion channels, kinases, or phospholipases to produce cellular responses. Multiple tissue- and cell-specific factors are able to influence GPCR-G protein interactions, and cell context plays a crucial role in determining the biological output of the GPCR-G protein interaction. However, here we will focus on the initial engagement of G proteins by GPCRs. Receptor-catalyzed nucleotide exchange depends heavily on transition of the receptor between different conformational states, a process that can be influenced by the cellular environment (e.g. local membrane composition, binding of sodium ions) as well as by the binding of extracellular ligands.
Agonist activation of GPCRs
The discovery that GPCRs could activate downstream signaling in the absence of agonists helped to reveal that some receptor antagonists were capable of lowering basal signaling activity [5]. The revelation that these ligands, termed inverse agonists, could suppress the activity not only of mutant receptors, but also of wild-type receptors that intrinsically display high basal activity, suggests that GPCRs natively adopt multiple conformational states. This recognition spurred the development of the extended ternary complex model of GPCR function where receptors exist basally in an equilibrium between inactive (R) and active (R*) states [6]. Depending on its intrinsic efficacy (i.e. agonist versus inverse agonist), ligand occupancy can change the distribution of receptor states to increase the proportion of receptors in an R* state for agonists, or to stabilize proportionally more receptors in the inactive conformational state in the case of inverse agonists.
The efficiency with which agonist binding is translated to intracellular conformational changes in the receptor appears to be receptor-specific and is likely tuned to the needs of the physiological system in question. For example, the sensing of photons by the prototypic photoreceptor, rhodopsin, represents a more “rigid” system with efficient allosteric coupling across the bilayer. Dark-state rhodopsin shows little basal G protein activation due to the covalently-linked inverse agonist 11-cis-retinal, which undergoes photon-induced isomerization to the agonist all-trans retinal [7]. Electron paramagnetic resonance (EPR) spectroscopy experiments showed that light activation of rhodopsin triggers conformational changes at the receptor’s intracellular face [8], most notably a ~6Å movement of the intracellular end of TM6 away from the center of the 7TM helical bundle (as determined by subsequent double electron-electron resonance (DEER) spectroscopy studies [9]). This movement of TM6 was also observed in nuclear magnetic resonance (NMR) spectroscopy experiments [10] and in the X-ray crystal structures of constitutively active opsin, either free or bound to a C-terminal peptide of Gαt, as well as the active metaII state of rhodopsin with and without Gαt peptide [11–13]. Thus it appears that activation of rhodopsin in detergent produces a relatively stable conformational change at the receptor’s cytoplasmic face, even in the absence of G protein.
In contrast, the relationship between agonist- and G protein-binding sites in GPCRs for diffusible agonists seem to be more loosely coupled, i.e. agonist binding is not necessarily translated into full outward TM6 movement. Site-directed fluorescence labeling (SDFL) and 19F-NMR studies have illustrated conformational changes in TM6 of the β2-adrenergic receptor (β2AR) in response to agonists, reminiscent of rhodopsin’s activation mechanism [14–17]. More recent SDFL and NMR experiments have also suggested that agonists alone do not fully stabilize an active β2AR conformation, as further conformational changes were observed upon addition of heterotrimeric Gs or the Gs-mimetic nanobody Nb80 [18–22]. While these experiments provide a qualitative description of the β2AR conformational ensemble, complementary DEER spectroscopy results demonstrate that the distribution of TM6 positions is different with agonist or agonist + Nb80, with Nb80 stabilizing a greater outward movement of TM6 [22]. Similar results were obtained in NMR studies of the β1-adrenergic receptor and the mu opioid receptor, where the fully active receptor conformation was stabilized only in the presence of intracellular-binding nanobodies [23,24]. Thus, agonists may serve to broaden the conformational distribution of GPCRs, increasing the probability of adopting intermediate active states able to interact with G protein. Single-molecule fluorescence studies have suggested that agonists may promote the GPCR-G protein interaction by both increasing the frequency of excursions into active intermediate conformations and by prolonging the residence time within these conformations [25].
Receptor-catalyzed nucleotide exchange
After achieving a G protein-interacting conformation and engaging GDP-bound G protein, the receptor is able to accelerate GDP dissociation from Gα by allosterically disrupting the nucleotide-binding site. Crystal structures of multiple Gα subunits demonstrated that the nucleotide is buried at the interface between the two domains of Gα, the ras-homology domain (RHD) and the alpha-helical domain (AHD), suggesting a necessity for receptor-mediated rearrangement of these domains for nucleotide entry or exit from its binding site [26–30]. Bound nucleotide is coordinated by interactions between the purine base with the β5-α4 and β6-α5 loops, as well as interactions between the nucleotide phosphates and the P-loop of the Gα RHD [31]. These regions of Gα are directly linked to receptor-interacting elements. The α5 helix (carboxy-terminus) of the G protein engages an activated GPCR by embedding into the site opened by the outward movement of TM6. Similarly, the P-loop is tied to the Gα N-terminal helix via the β1 strand, and several lines of evidence suggest that interaction of the receptor with the Gα N-terminus contributes to GDP release [32,33]. Below, we will consider each of these elements of the receptor-G protein interaction and their importance in receptor-catalyzed nucleotide exchange.
Role of RHD-AHD domain separation
Upon observing the buried nucleotide-binding site in transducin α-subunit, it was speculated that activated rhodopsin may promote movement of the AHD to open a path for nucleotide exchange [26]. Indeed, the crystal structure of β2AR in complex with nucleotide-free heterotrimeric Gs reveals a large movement of the AHD relative to the RHD [34]. Although the AHD position in this structure is stabilized by crystal contacts, several orthogonal lines of evidence suggest that the AHD is indeed mobile and support the necessity of domain movement for nucleotide exchange. NMR studies of chimeric Gαt/i showed that during receptor-catalyzed nucleotide exchange, Gα progressed through a dynamic nucleotide-free intermediate [35,36]. Moreover, analyses of Gαi by EPR spectroscopy revealed a significant rigid-body movement of the AHD away from the RHD upon rhodopsin-catalyzed GDP release [37,38]. Similarly, single particle imaging by electron microscopy (EM) of the nucleotide-free β2AR-Gs complex facilitated visualization of the motion of the Gαs AHD in solution, without the influence of interactions between complexes within the crystal lattice that occurs in X-ray diffraction analyses [39]. Furthermore, hydrogen-deuterium exchange (HDX) mass spectrometry analyses of the nucleotide-free β2AR-Gs complex suggest that the RHD-AHD interface undergoes increased exchange upon receptor-catalyzed GDP release [33]. More recently, in silico mutations that increased the probability of spontaneous RHD-AHD domain separation in MD simulations were also shown to increase the rate of basal nucleotide exchange in Gαi [40], while limiting Gαi AHD movement was shown to impair both basal [41] and receptor-stimulated [38] nucleotide exchange.
Although domain opening appears to be necessary for the dissociation of GDP from its binding site, the separation of AHD and RHD by itself is not sufficient to promote nucleotide exchange. Early studies showed that while the AHD was necessary for the GTPase activity of Gα, the isolated RHD of Gαs maintained nucleotide-binding capacity and was able to activate adenylyl cyclase [42]. This property of the RHD is reminiscent of its small GTPase brethren (e.g. Ras), which bind nucleotides tightly in the absence of any accessory domains/proteins. Molecular dynamics simulations have also suggested tight binding of GDP by the Gα RHD. In these simulations, even though the RHD-AHD interface opened spontaneously in GDP-bound Gi heterotrimer, the dissociation of GDP was dependent on disruption of the GDP-RHD interaction [41]. Therefore, it appears that the main function of an activated GPCR is not to pry the RHD and AHD apart, but to stabilize conformational changes in Gα that disrupt the nucleotide-binding pocket. Conformational changes can be propagated from the receptor to the nucleotide-binding pocket via two potential routes, discussed in the following sections.
Gα N-terminus
In the crystal structure of the β2AR-Gs complex, the N-terminal helix (αN) and αN-β1 junction of Gαs interacts with ICL2 of β2AR [34]. A similar interaction between ICL2 and the αN-β1 loop was observed in recent studies modeling the interaction between rhodopsin or cannabinoid CB2 receptors and Gi heterotrimer [43,44]. The N-terminal helix of Gα subunits proceeds into the β1-strand, followed by the β1-α1 loop - also known as the P-loop - a highly conserved feature of both small molecular weight and heterotrimeric G proteins that coordinates the β-phosphate of GDP. The P-loop is a classic Walker A motif, a well-established glycine-rich stretch of residues involved in phosphate binding of nucleotides. While the guanine ring of the nucleotide is also heavily coordinated (as discussed above), the interaction of the P-loop with the nucleotide’s β-phosphate appears to be the crucial determinant of binding, as GDP binds with ~105–106-fold higher affinity than does GMP [45]. Thus, disruption of the interaction between the P-loop and the β-phosphate of bound GDP results in a significant loss of binding energy and favors nucleotide dissociation. Indeed, exchange factors that promote GDP release from Ras act by disrupting Mg2+-bridged interaction of the P-loop with the β-phosphate of GDP [46].
An analogous disruption of the P-loop-GDP interaction, mediated by an activated receptor binding to the αN-β1 region, may trigger GDP release from the Gα subunit. This hypothesis is supported by the finding that truncation of the N-terminus of transducin impairs receptor-catalyzed nucleotide exchange, even though transducin was still capable of forming a complex with rhodopsin [47]. Similarly, analysis of the HDX profile of Gs upon interaction with β2AR reveals the largest changes in the β1-strand and P-loop of Gαs, suggesting that this region becomes highly dynamic during receptor interaction and during GDP dissociation [33].
The contribution of the Gα N-terminus to nucleotide exchange also provides a mechanism for the involvement of Gβγ in nucleotide exchange. In the heterotrimer, Gβγ helps to position the Gα N-terminus in a conformation that engages ICL2 of the receptor. This could explain how Gβγ, long known for its’ involvement in receptor-catalyzed nucleotide release, can contribute without actually making contact with the receptor (See Figure 1).
Figure 1. Structural models of receptor-catalyzed nucleotide exchange.
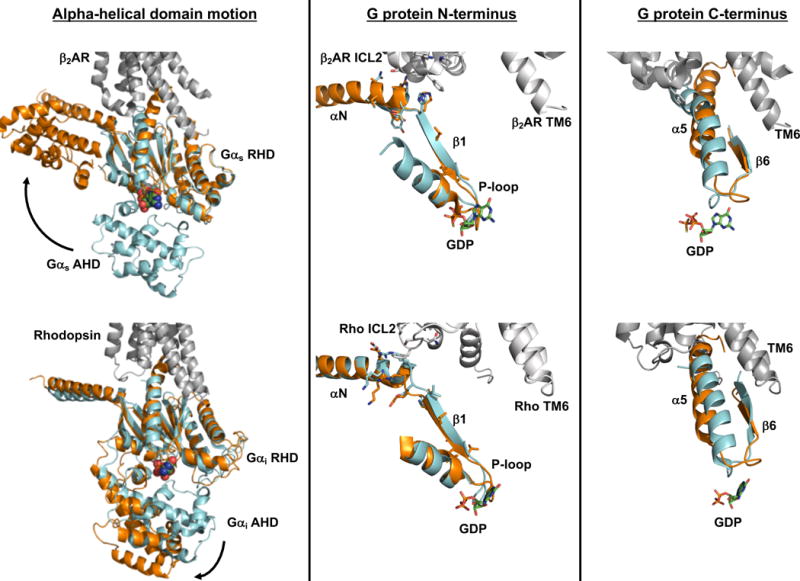
Multiple lines of evidence suggest that a separation of the G protein ras-homology domain (RHD) and alpha-helical domain (AHD) is necessary to exchange bound GDP for GTP. While motion of the alpha helical domain was observed in the crystal structure of β2AR in complex with nucleotide-free Gs heterotrimer (top left) and in a model of rhodopsin in complex with GDP-bound Gi heterotrimer (bottom left), domain separation is likely not sufficient to trigger GDP release from Gα. Rather, binding to an activated receptor stabilizes conformational changes within the G protein that disrupt nucleotide interactions with the RHD. Interaction of intracellular loop 2 (ICL2) of the receptor with the αN helix and αN-β1 junction of the G protein leads to a reorganization of the P-loop that coordinates the β-phosphate of GDP (middle column). Furthermore, the C-terminal α5 helix of Gα undergoes a rotation and translation to occupy its binding site within the hydrophobic core of the receptor that is opened upon outward movement of TM6. Movement of the α5 helix in turn alters the β6-α5 loop that directly contacts GDP (right column). Similar receptor-G protein contacts, and G protein conformational changes, are seen in the structure of β2AR-Gs complex (top panels) and the model of the rhodopsin-Gi complex (bottom panels). In both scenarios, receptor is shown in gray (PDB: 3SN6 for β2AR-Gs; Rho*-Gi is from Alexander et al. [43]). For comparison of receptor-bound and unbound Gαs in the top panels, the RHD of GTPγS-bound Gαs (PDB: 1AZT, cyan) was aligned to the RHD of Gαs in the β2AR complex (PDB: 3SN6, orange). A similar alignment was used in the bottom panels to overlay GDP-bound Gαi (PDB: 1GOT, cyan) with the RHD of receptor-bound Gαi (orange).
The recent structure determination of a stabilized, mutant RAS domain of Gαs in a complex with an agonist-bound adenosine A2 receptor (A2AR) reveals several compelling characteristics which support the critical role of the N-terminus-ICL2 interaction [46]. The mutant RAS domain of the Gαs, ‘mini-Gαs’, bound to A2AR shared many of the characteristics of the β2AR-Gs complex in that the G protein C-terminus interacted with the receptor core (see below) with the receptor in the active conformation. Interestingly, the structure did not reveal an ICL2 interaction with the Gα since the G protein used in this study not only lacked Gβγ, but also had a truncated N-terminus. These data further support the role of Gβγ in coordinating and positioning the N-terminus in a productive interaction with ICL2. More importantly the G protein in this complex still remained bound to GDP, despite having a disordered β5-α6 loop (see below). Taken together these data support the critical role of the P-loop in coordinating the β-phosphate of GDP and thus the contributions of the G protein N-terminus and ICL2 in receptor-catalyzed nucleotide exchange.
Gα C-terminus
Multiple lines of evidence have also implicated the C-terminal α5 helix of Gα as an important conduit that transmits information from an activated receptor to the nucleotide-binding site [47–50]. More recent structural evidence of opsin and meta-rhodopsin in complex with the C-terminal peptide derived from Gαt [12,13,51] and the β2AR-Gs complex [34] provided visualization of the interaction interface. Comparison of the interactions formed by the α5 helix in GDP-bound structures and in the β2AR-bound nucleotide-free state shows that upon receptor binding, the distal C-terminus rotates approximately 60° and translates 5 Å up into the core of the receptor, with many of the α5 interactions within the RHD reorganizing [34,41,52]. Moroever, similar interactions were observed with the C-terminus of the ‘mini-Gαs’ in the A2AR-G protein complex structure [46]. The displacement of the α5 helix seen in the crystal structure of β2AR-Gs in turn rearranges elements that contribute to GDP binding. Of particular note, contacts between the α5 and α1 helices are disrupted and the β6-α5 loop is rearranged. The α1 helix contacts both GDP and αF of the AHD, therefore disruption of the α5-α1 interaction has been proposed to facilitate domain separation and GDP release [52]. Conformational changes in α5 during GDP release have been detected by EPR spectroscopy and more recently by computational studies [43,50]. Molecular dynamics simulations have also suggested that movement of the C-terminal helix from its position in the GDP-bound Gα to its receptor-engaged conformation decreases the affinity of bound nucleotide due to rearrangement of the β6-α5 loop, which directly contacts bound GDP [41]. Mutations to this loop in Gαs can accelerate basal GDP release and cause disease due to hyperactive G protein signaling, supporting the relevance of this loop in GDP binding [53].
Mechanism of nucleotide exchange
Given the intrinsic flexibility of G proteins and their capacity to undergo spontaneous domain separation, it is conceivable that the receptor may recognize a “pre-opened” G protein, or that the G protein may spontaneously open whether pre-associated with the receptor or free, as a heterotrimer. While domain separation is required for nucleotide release, efficient promotion of GDP dissociation may require the cooperative engagement of both N and C- terminus by the receptor. Which termini engages the receptor first remains unclear; although agonist binding has been shown to promote the interaction between α5 (C-term) and receptor (and vice-versa), it has also been suggested that engagement of αN and/or Gβγ is responsible for freeing the α5 helix for insertion into the receptor core [47]. The observation that the G protein in the A2AR-mini Gαs complex structure lacked the N-terminus-ICL2 interaction and also remained GDP-bound supports this notion.
The most compelling argument in support of perhaps a larger contribution of the N-terminus, β1-strand and P loop in GDP release, concerns the large difference in the affinities between GDP over GMP. The binding energy associated with β-phosphate binding to the P-loop is quite significant, which is why guanine nucleotide exchange factors for ras-like proteins utilize P-loop disruption to promote GDP release. In this regard, perhaps in heterotrimeric Gα the N-terminus engages first, disrupting the interaction between P-loop and β-phosphate, resulting in GDP dissociation. Nucleotide loss would free the β6-α5 loop, allowing the C-terminus to rotate and insert into receptor core (as suggested by Herrmann et al.; ref. 32). This, in turn, would enhance the affinity between the receptor and G protein and stabilize the G protein in its nucleotide-free form. GTP binding and the subsequent conformational change in the switch II domain of Gα leads to the functional dissociation of Gβγ, in turn disrupting the N-terminal-ICL2 interaction on the receptor since the N-terminal helix of Gα forms such a large surface interaction with Gβγ. GTP binding would also be expected to reorder the β6-α5 loop, facilitating retraction of the Gα C-terminus from the receptor core and uncoupling the receptor-G protein complex.
G protein feedback to the agonist-binding site
The finding that nucleotides could modulate agonist affinity suggested that receptors bind ligand more tightly when engaged by nucleotide-free G protein. Nanobodies that behave as G protein mimics (e.g. Nb80, Nb39 and Nb9-8; camelid antibodies raised against agonist-bound β2AR, μ-opioid and M2 muscarinic acetylcholine receptor, respectively) also have the capacity to stabilize high-affinity agonist binding, presumably by stabilizing the same receptor conformation as the nucleotide-free G protein[19,54,55]. Indeed, the receptor in structures of the active state of β2AR bound to Nb80 or bound to the nucleotide-free Gs display an overall RMSD of only 0.6 Å.
The crystal structures of active β2AR reveal interesting conformational changes that occur in the extracellular loops above the hormone binding site. Two aromatic residues, Phe193 and Tyr308 from ECL2 and TM7, respectively, move approximately 3 Å toward each other and result in the formation of a lid-like structure over the hormone binding site. A similar conformational change involving two homologous aromatic residues in the β1AR following Nb80 binding was observed using NMR [24]. In addition, we recently reported pharmacological evidence supporting the notion that Phe193 and Tyr308 close over the hormone binding site of β2AR following G protein interaction and GDP dissociation [56]. This active, closed conformation of the receptor significantly impairs ligand dissociation, thus underlying the G protein-mediated effects on enhancing agonist binding affinity. Similar high affinity binding and the closed, active states of the receptor can be observed with Nb80 and even fragments of the G protein C-terminus. Given the diversity in GPCR structures and in the binding modes of GPCR agonists, it is unlikely that G proteins can enhance agonist affinity at all receptors by following the mechanism proposed for β2AR. However, formation of the closed, active state, as evidenced by G protein-mediated effects on agonist binding, has been observed in other class A, B, and C GPCR family [54–69]. Therefore it appears that while the specific mechanism may differ, the allosteric feedback that allows G protein binding to stabilize changes in or around the agonist-binding pocket may be conserved.
In summary, recent structural analyses have provided mechanistic insights into the allosteric coupling between agonist- and nucleotide-binding sites, and biophysical experiments have shed light on the dynamic process of GPCR activation and receptor-catalyzed nucleotide exchange. Binding of a heterotrimeric G protein to an activated receptor reorganizes the P-loop (via the Gα N-terminus and αN-β1 junction) and the β6-α5 loop (via the Gα C-terminus) to favor GDP dissociation, and the receptor likely acts to stabilize an ““open” conformation of nucleotide-free Gα to allow for GDP release and subsequent GTP binding. In turn, nucleotide-free G protein stabilizes the receptor in an active state, characterized by a “closed” conformation of the receptor’s agonist-binding pocket, slowing agonist dissociation and thereby increasing affinity for agonists.
Highlights.
Both GPCRs and G proteins sample multiple conformations in their basal states.
Stabilizing a fully active state of several GPCRs requires agonist and G protein.
GPCRs engage both the N- and C-termini of Gα to promote GDP release.
Domain separation in Gα is necessary but not sufficient for nucleotide exchange.
Nucleotide-free Gα stabilizes structural changes around the agonist-binding site.
Acknowledgments
JPM and RKS were supported by the National Institute of General Medical Sciences Grants RO1-GM083118 and U19-GM106990 (RKS) and RO1-GM068603 (RKS); National Institutes of Drug Abuse R21-031418 (RKS); Michigan Diabetes Research and Training Center Grant, National Institute of Diabetes and Digestive and Kidney Diseases, P60DK-20572 (RKS); University of Michigan Biological Sciences Scholars Program (RKS) and Pharmacological Sciences Training Program T32GM007767 (JPM) and AHA Midwest Affiliate Predoctoral Fellowship 13PRE17110027 (JPM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–6. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 3.Kenakin T. New concepts in pharmacological efficacy at 7TM receptors: IUPHAR review 2. Br J Pharmacol. 2013;168:554–75. doi: 10.1111/j.1476-5381.2012.02223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKee EE, Bentley AT, Smith RM, Jr, Ciaccio CE. Origin of guanine nucleotides in isolated heart mitochondria. Biochem Biophys Res Commun. 1999;257:466–472. doi: 10.1006/bbrc.1999.0489. [DOI] [PubMed] [Google Scholar]
- 5.Herz A, Costa T. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc Natl Acad Sci USA. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. This paper was the first to identify inverse agonism at a GPCR, a finding which would lead to the development of the extended ternary complex model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. Building on the work of Herz & Costa, this paper integrated observations of basal G protein activation by multiple GPCRs to propose the extended ternary complex model, in which receptors undergo conformational fluctuations in the absence of ligands. [PubMed] [Google Scholar]
- 7.Choe H-W, Park JH, Kim YJ, Ernst OP. Transmembrane signaling by GPCRs: insight from rhodopsin and opsin structures. Neuropharmacology. 2011;60:52–7. doi: 10.1016/j.neuropharm.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 8.Farrens DL, Altenbach C, Yang K, Hubbell WL, Gobind H, Khoranat HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–70. doi: 10.1126/science.274.5288.768. This study identified movement of TM6 in rhodopsin as a key conformational change that occurs upon light activation of the receptor. [DOI] [PubMed] [Google Scholar]
- 9.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA. 2008;105:7439–44. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein-Seetharaman J, Getmanova EV, Loewen MC, Reeves PJ, Khorana HG. NMR spectroscopy in studies of light-induced structural changes in mammalian rhodopsin: applicability of solution (19)F NMR. Proc Natl Acad Sci USA. 1999;96:13744–9. doi: 10.1073/pnas.96.24.13744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JH, Scheerer P, Hofmann KP, Choe H-W, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–7. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 12.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe H-W, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. This paper was the first to provide high-resolution structural data about the conformational changes that occur within a GPCR upon activation, and visualized the binding site of the Gα C-terminus on the receptor. [DOI] [PubMed] [Google Scholar]
- 13.Choe H-W, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP. Crystal structure of metarhodopsin II. Nature. 2011;471:651–5. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 14.Gether U, Lin S, Ghanouni P. Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. EMBO J. 1997;16:6737–47. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR, Kobilka BK. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J Biol Chem. 2001;276:24433–6. doi: 10.1074/jbc.C100162200. One of the first studies to suggest that instead of GPCRs flipping between single “inactive” and “active” conformations, individual agonists may stabilize unique active receptor conformations. [DOI] [PubMed] [Google Scholar]
- 16.Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–10. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horst R, Liu JJ, Stevens RC, Wüthrich K. β2-adrenergic receptor activation by agonists studied with 19F NMR spectroscopy. Angew Chem Int Ed Engl. 2013;52:10762–5. doi: 10.1002/anie.201305286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao XJ, Vélez Ruiz G, Whorton MR, Rasmussen SGF, DeVree BT, Deupi X, Sunahara RK, Kobilka B. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci USA. 2009;106:9501–6. doi: 10.1073/pnas.0811437106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasmussen SGF, Choi H-J, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature. 2011;469:175–80. doi: 10.1038/nature09648. This paper presented the first structure of an agonist-bound GPCR in complex with a G protein, demonstrating conformational changes that occur in both receptor and G protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. The dynamic process of β(2)-adrenergic receptor activation. Cell. 2013;152:532–42. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim TH, Chung KY, Manglik A, Hansen AL, Dror RO, Mildorf TJ, Shaw DE, Kobilka BK, Prosser RS. The role of ligands on the equilibria between functional states of a G protein-coupled receptor. J Am Chem Soc. 2013;135:9465–9474. doi: 10.1021/ja404305k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell. 2015;161:1101–11. doi: 10.1016/j.cell.2015.04.043. Using both EPR and NMR spectroscopy experiments, this paper demonstrates the cooperative effects of agonist and G protein on stabilizing a fully active receptor conformation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Déméné H, Granier S. Propagation of conformational changes during μ-opioid receptor activation. Nature. 2015;524:375–8. doi: 10.1038/nature14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai C-J, Brueckner F, Schertler GFX, Veprintsev DB, Grzesiek S. Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature. 2016;530:237–41. doi: 10.1038/nature16577. [DOI] [PubMed] [Google Scholar]
- 25.Lamichhane R, Liu JJ, Pljevaljcic G, White KL, van der Schans E, Katritch V, Stevens RC, Wüthrich K, Millar DP. Single-molecule view of basal activity and activation mechanisms of the G protein-coupled receptor β2AR. Proc Natl Acad Sci USA. 2015;112:14254–9. doi: 10.1073/pnas.1519626112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noel JP, Hamm HE, Sigler PB. The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S. Nature. 1993;366:654–63. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 27.Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science. 1994;265:1405–12. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 28.Sunahara RK, Tesmer JJ, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gs-alpha. Science. 1997;278:1943–7. doi: 10.1126/science.278.5345.1943. [DOI] [PubMed] [Google Scholar]
- 29.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJG. Snapshot of activated G proteins at the membrane: the Galphaq-GRK2-Gbetagamma complex. Science. 2005;310:1686–90. doi: 10.1126/science.1118890. [DOI] [PubMed] [Google Scholar]
- 30.Kreutz B, Yau DM, Nance MR, Tanabe S, Tesmer JJG, Kozasa T. A new approach to producing functional G alpha subunits yields the activated and deactivated structures of G alpha(12/13) proteins. Biochemistry. 2006;45:167–74. doi: 10.1021/bi051729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sprang SR. G protein mechanisms: insights from structural analysis. Ann Rev Biochem. 1997;66:639–78. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- 32.Herrmann R, Heck M, Henklein P, Hofmann KP, Ernst OP. Signal transfer from GPCRs to G proteins: role of the G alpha N-terminal region in rhodopsin-transducin coupling. J Biol Chem. 2006;281:30234–41. doi: 10.1074/jbc.M600797200. This paper demonstrated the importance of the Gα αN-β1 junction in rhodopsin-catalyzed nucleotide exchange and predicted that rhodopsin engaged both the G protein N- and C-terminus to promote GDP release. [DOI] [PubMed] [Google Scholar]
- 33.Chung KY, Rasmussen SGF, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL, Sunahara RK. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477:611–5. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ridge KD, Marino JP, Ngo T, Ramon E, Brabazon DM, Abdulaev NG. NMR analysis of rhodopsin-transducin interactions. Vision Res. 2006;46:4482–4492. doi: 10.1016/j.visres.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 36.Abdulaev NG, Ngo T, Ramon E, Brabazon DM, Marino JP, Ridge KD. The receptor-bound “empty pocket” state of the heterotrimeric G-protein alpha-subunit is conformationally dynamic. Biochemistry. 2006;45:12986–97. doi: 10.1021/bi061088h. [DOI] [PubMed] [Google Scholar]
- 37.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mapping allosteric connections from the receptor to the nucleotide-binding pocket of heterotrimeric G proteins. Proc Natl Acad Sci USA. 2007;104:7927–32. doi: 10.1073/pnas.0702623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Eps N, Preininger AM, Alexander N, Kaya AI, Meier S, Meiler J, Hamm HE, Hubbell WL. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc Natl Acad Sci USA. 2011;108:9420–4. doi: 10.1073/pnas.1105810108. This paper clearly demonstrated the separation of RHD and AHD during the process of receptor-catalyzed nucleotide exchange, before crystallographic data were available. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westfield GH, Rasmussen SGF, Su M, Dutta S, DeVree BT, Chung KY, Calinski D, Velez-Ruiz G, Oleskie AN, Pardon E, et al. Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci USA. 2011;108:16086–91. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao X-Q, Malik RU, Griggs NW, Skjærven L, Traynor JR, Sivaramakrishnan S, Grant BJ. Dynamic coupling and allosteric networks in the α subunit of heterotrimeric G proteins. J Biol Chem. 2016;291:4742–53. doi: 10.1074/jbc.M115.702605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dror RO, Mildorf TJ, Hilger D, Manglik A, Borhani DW, Arlow DH, Philippsen A, Villanueva N, Yang Z, Lerch MT, et al. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science. 2015;348:1361–1365. doi: 10.1126/science.aaa5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Markby DW, Onrust R, Bourne HR. Separate GTP binding and GTPase activating domains of a G alpha subunit. Science. 1993;262:1895–901. doi: 10.1126/science.8266082. [DOI] [PubMed] [Google Scholar]
- 43.Alexander NS, Preininger AM, Kaya AI, Stein RA, Hamm HE, Meiler J. Energetic analysis of the rhodopsin-G-protein complex links the α5 helix to GDP release. Nat Struct Mol Biol. 2014;21:56–63. doi: 10.1038/nsmb.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mnpotra JS, Qiao Z, Cai J, Lynch DL, Grossfield A, Leioatts N, Hurst DP, Pitman MC, Song Z-H, Reggio PH. Structural basis of G protein-coupled receptor-Gi protein interaction: formation of the cannabinoid CB2 receptor-Gi protein complex. J Biol Chem. 2014;289:20259–72. doi: 10.1074/jbc.M113.539916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–77. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 46.Carpenter B, Nehmé R, Warne T, Leslie AG, Tate CG. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature. 2016;536:104–7. doi: 10.1038/nature18966. This paper describes the second structure of a GPCR-G protein complex using the adenosine A2A receptor and a truncated form of the G protein alpha subunit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science. 1988;241:832–5. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- 48.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature. 1993;363:274–6. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 49.Gilchrist A, Mazzoni MR, Dineen B, Dice A, Linden J, Proctor WR, Lupica CR, Dunwiddie TV, Hamm HE. Antagonists of the receptor-G protein interface block Gi-coupled signal transduction. J Biol Chem. 1998;273:14912–9. doi: 10.1074/jbc.273.24.14912. [DOI] [PubMed] [Google Scholar]
- 50.Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol. 2006;13:772–7. doi: 10.1038/nsmb1129. [DOI] [PubMed] [Google Scholar]
- 51.Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie G, Oprian DD, Schertler GFX. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–60. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flock T, Ravarani CNJ, Sun D, Venkatakrishnan AJ, Kayikci M, Tate CG, Veprintsev DB, Babu MM. Universal allosteric mechanism for Gα activation by GPCRs. Nature. 2015;524:173–9. doi: 10.1038/nature14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iiri T, Herzmark P, Nakamoto JM, van Dop C, Bourne HR. Rapid GDP release from Gs alpha in patients with gain and loss of endocrine function. Nature. 1994;371:164–8. doi: 10.1038/371164a0. [DOI] [PubMed] [Google Scholar]
- 54.Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Gmeiner P, Kato HE, Livingston KE, Thorsen TS, et al. Structural insights into muopioid receptor activation. Nature. 2015;524:315–21. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–6. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeVree BT, Mahoney JP, Vélez-Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E, Fung JJ, Manglik A, Masureel M, Du Y, et al. Allosteric coupling from G protein to the agonist binding pocket in GPCRs. Nature. 2016 doi: 10.1038/nature18324. in press. This paper describes the mechanism underlying the allosteric influence of G proteins on agonist binding to GPCRs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bornancin F, Pfister C, Chabre M. The transitory complex between photoexcited rhodopsin and transducin. Reciprocal interaction between the retinal site in rhodopsin and the nucleotide site in transducin. Eur J Biochem. 1989;184:687–98. doi: 10.1111/j.1432-1033.1989.tb15068.x. [DOI] [PubMed] [Google Scholar]
- 58.Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–13. [PubMed] [Google Scholar]
- 59.Lefkowitz RJ, Mullikin D, Wood CL, Gore TB, Mukherjee C. Regulation of prostaglandin receptors by prostaglandins and guanine nucleotides in frog erythrocytes. J Biol Chem. 1977;252:5295–303. [PubMed] [Google Scholar]
- 60.Grandt R, Aktories K, Jakobs KH. Guanine nucleotides and monovalent cations increase agonist affinity of prostaglandin E2 receptors in hamster adipocytes. Mol Pharmacol. 1982;22:320–6. [PubMed] [Google Scholar]
- 61.Sarau HM, Mong S, Foley JJ, Wu HL, Crooke ST. Identification and characterization of leukotriene D4 receptors and signal transduction processes in rat basophilic leukemia cells. J Biol Chem. 1987;262:4034–41. [PubMed] [Google Scholar]
- 62.Rodbell M, Krans HM, Pohl SL, Birnbaumer L. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. IV. Effects of guanylnucleotides on binding of 125I-glucagon. J Biol Chem. 1971;246:1872–6. [PubMed] [Google Scholar]
- 63.Dieterich KD, Grigoriadis DE, De Souza EB. Corticotropin-releasing factor receptors in human small cell lung carcinoma cells: radioligand binding, second messenger, and northern blot analysis data. Endocrinology. 1994;135:1551–8. doi: 10.1210/endo.135.4.7925116. [DOI] [PubMed] [Google Scholar]
- 64.Hill DR, Bowery NG, Hudson AL. Inhibition of GABAB receptor binding by guanyl nucleotides. J Neurochem. 1984;42:652–7. doi: 10.1111/j.1471-4159.1984.tb02732.x. [DOI] [PubMed] [Google Scholar]
- 65.Albasanz JL, Ros M, Martín M. Characterization of metabotropic glutamate receptors in rat C6 glioma cells. Eur J Pharmacol. 1997;326:85–91. doi: 10.1016/s0014-2999(97)00154-4. [DOI] [PubMed] [Google Scholar]
- 66.Maguire ME, Van Arsdale PM, Gilman AG. An agonist-specific effect of guanine nucleotides on binding to the beta adrenergic receptor. Mol Pharmacol. 1976;12:335–9. [PubMed] [Google Scholar]
- 67.Burgisser E, De Lean A, Lefkowitz RJ. Reciprocal modulation of agonist and antagonist binding to muscarinic cholinergic receptor by guanine nucleotide. Proc Natl Acad Sci USA. 1982;79:1732–6. doi: 10.1073/pnas.79.6.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–17. [PubMed] [Google Scholar]
- 69.Childers SR, Snyder SH. Differential regulation by guanine nucleotides or opiate agonist and antagonist receptor interactions. J Neurochem. 1980;34:583–93. doi: 10.1111/j.1471-4159.1980.tb11184.x. [DOI] [PubMed] [Google Scholar]